
Abstract
Background
Small cell lung cancer (SCLC) is a highly aggressive lung cancer subtype with poor survival and limited treatment options. Sequencing results have revealed gene mutations associated with SCLC, however, the correlation between the genomic alterations and clinical prognosis of SCLC is yet unclear.
Methods
Targeted next-generation sequencing of 62 cancer related genes was performed on 53 SCLC samples. The correlations between clinical outcomes and genomic alterations were analyzed.
Results
38/62 (61.3%) candidate genes harbored some alterations, while all the SCLC samples carried at least 3 gene mutations. The most common nonsynonymous mutations included ERBB2 (95.9%), CREBBP (95.9%), and TP53 (77.6%). The median nonsynonymous tumor mutation burden (TMB) was 21.7 mutations/Mb (rang, 9.3–55.9). High TMB (> 21 mutations/Mb) was good prognostic factor in overall survival (OS) (21.7 vs. 10.4 months, P = 0.012). Multivariate analysis showed that high TMB was an independent prognostic factor. The overall survival (OS) of patients carrying KIAA1211 mutation was significantly longer than those with wild-type KIAA1211 (P < 0.001).
Conclusions
The current study highlights the potential role of genomic alterations for the prognosis of SCLC. Higher TMB was associated with a better prognosis, and KIAA1211 might be a good prognostic factor in SCLC.
Similar content being viewed by others


Background
Lung cancer is the leading cause of cancer deaths in both women and men in the China and throughout the world [1]. Small-cell lung cancer (SCLC) accounts for approximately 10–15% of all lung cancers. It is a highly aggressive malignancy frequently presenting with metastases at the time of diagnosis [2]. Most patients respond to chemotherapy, unfortunately, the majority suffer disease recurrence or progression sooner rather than later. Treatment options have remained unchanged for the past three decades. Furthermore, until now, the most reproducible prognostic factor is stage of the disease and molecular biomarkers are still lacking [3]. Therefore, to better understand the clinical outcomes, it is essential to explore the genetic alterations and identify prognostic biomarkers.
The genetic mutational landscape of SCLC is complex and heterogeneous, however, the most common genetic alterations include inactivation of tumor suppressor genes TP53 and RB1, copy number gains in MYC family members, enzymes involved in chromatin remodeling, and kinases signaling pathways [4, 5]. No targeted drug has showed significant anti-tumor activity in SCLC until now. Recently, immune checkpoint inhibitors have shown efficacy in SCLC with PD-1 inhibition. Pembrolizumab demonstrated promising antitumor activity in SCLC with an objective response rate (ORR) of 33% [6], while nivolumab had an ORR of 10% as monotherapy or 19–23% in combination with ipilimumab in patients with relapsed SCLC [7]. The combination of atezolizumab and chemotherapy (platinum + etoposide) was recently approved by the US Food and Drug Agency (FDA) and led to a new treatment paradigm for extensive SCLC [8]. Besides programmed death ligand 1 (PD-L1) expression, tumor mutation burden (TMB) is regarded as a biomarker of the efficacy of programmed death 1 (PD-1) inhibitors in various cancers. Thus, a deeper understanding of the driver alterations in SCLC, and an understanding of those patients likely to respond to immune checkpoint blockade should improve patient outcome. A few seminal genomic studies have been conducted [9,10,11], and the genomic features have been correlated with the clinical outcome. However, these studies were evaluated patients with surgically resected tumors, and there were few kinds of research on Asian populations. Moreover, the relation between TMB and prognosis in SCLC is still unclear.
Here we employed selected 62 exome sequencing in SCLC and analyzed the genomic profiling and the potential association with the clinical outcomes.
Methods
Patients and samples
From May 2014 to January 2017, a total of 53 SCLCs and matched normal lung formalin-fixed and paraffin-embedded (FFPE) tissue samples were obtained from Wuhan Union Hospital, China. All clinicopathological data were retrospectively collected. The stage of SCLC were categorized by the older Veterans Administration Lung Study Group’s 2-stage classification scheme [12], which classified into limited-stage (LS) and extensive-stage (ES).
DNA extraction
We performed DNA extraction from serial thick sections cut from tumor tissue samples and control sections. The invasive tumor content was estimated by pathologists, to ensure more than 50% of cells were tumor cells. The DNA was isolated from the FFPE and blood samples using the DNeasy Blood and Tissue Kit (69,504, QIAGEN, Venlo, Netherlands).
Next-generation sequencing
We performed targeted sequencing of 62 cancer related genes using an amplicon based sequencing panel of Ion AmpliSeq™ (Life Technologies, Carlsbad, USA), and then generated sequence data using Ion Proton™ System (Life Technologies, Carlsbad, USA).
Statistical analysis
Fisher’s exact test was used to compare the frequency data between two groups. Survival data were calculated using the Kaplan–Meier method and survival curves were compared with the log-rank test. The variables putatively associated with survival were analyzed with the Cox proportional hazards test. All tests were bilateral, with P < 0.05 indicating significant statistical difference. Statistical analysis was carried out by the statistical software package SPSS 22.0 (IBM Corp., Somers, NY, USA).
Results
Clinicopathological characteristics of SCLC patients
The present study enrolled a total of 53 patients: 49 males and 4 females. The median age of the patients was 60 years (range, 57–66 years). The Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 46 patients was ≤1, and that of 7 cases was =2; 24 cases were LS-SCLC and 29 were ES-SCLC; 42 cases presented a smoking history. Additionally, 64.2% of the patients did not have a history of chronic diseases.
All patients received first-line chemotherapy; 50 (94.3%) received platinum-based doublet chemotherapy, 2 underwent irinotecan monotherapy, and 1 had etoposide monotherapy. A total of 35 patients (66.0%) received chest radiotherapy; of these, 9 (25.7%) underwent concurrent chemoradiotherapy, while the remaining received sequential radiotherapy (Table 1).
Distribution of gene mutations
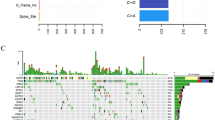
A total of 62 candidate genes were sequenced from 49/53 SCLC samples. Consequently, alterations were detected in 38 genes. All the SCLC samples carried a minimum of 3 mutations. The most common nonsynonymous mutations occurred in ERBB2 (95.9%), CREBBP (95.9%), and TP53 (77.6%) (Fig. 1a). We also analyzed the distribution of variants. A total of 156 nonsynonymous variants were identified, The most common nonsynonymous variants were ERBB2.p.L755 M (95.9%) and CREBBP.p.V1780 M (91.8%), which were primarily concentrated at one variant (> 90%)(Additional file 1: Figure S1). The variants of ERBB4, KIT and NRAS were only observed in LS-SCLC, while the variants of KDR, KRAS and PTEN were only detected in ES-SCLC. There was no significant difference in the mutation rates of the above variants between different stages (Fig. 1b) because of the small number of mutations.

Genomic alterations and TMB in SCLC. a-b Genomic alterations in the extensive stage SCLC and the limited stage SCLC; c TMB in SCLC versus lung adenocarcinoma (LUAD) and lung squamous carcinoma (LUSC) from TCGA; d TMB in extensive stage SCLC versus limited stage SCLC and TMB in early stage LUAD/LUSC versus advanced stage LUAD/LUSC
The relationship between TMB and prognosis
The median number of nonsynonymous variants carried by each sample was 7 (range, 3–18). Nonsynonymous TMB was estimated and analyses were performed to assess the effect of TMB on prognosis. The genomic coverage of the panel is 0.322 Mb. The median TMB was 21.7 mutations/Mb (rang, 9.3–55.9) (Fig. 1c). We defined 21 mutations/Mb as the cut-off value, TMB > 21 mutations/Mb as high and TMB ≤ 21 mutations/Mb as low. Univariate analysis showed longer progression free survival (PFS) with high TMB than low TMB (9.9 vs. 6.5 months) but without statistically significance (P = 0.16) (Fig. 2a), and overall survival (OS) was significantly longer in high TMB than low TMB (21.7 vs. 10.4 months, P = 0.012) (Fig. 2b). Multivariate analysis showed that high TMB was an independent prognostic factor in PFS (P = 0.004) and OS (P < 0.001) with adjustment for age, sex, smoking status, ECOG PS, and stage (Table 2). The results were consistent across the subgroups of ES-SCLC (PFS: P = 0.16, OS: P < 0.001) (Additional file 2: Figure S2).

Kaplan–Meier analysis of high TMB and low TMB. a. Progression free survival; b. Overall survival
In addition, compared with lung cancer data from the Cancer Genome Atlas (TCGA), the estimates of TMB in SCLC was much higher than TMB in lung adenocarcinoma (LUAD) and lung squamous carcinoma (LUSC) (P < 0.001) (Fig. 1c). But no significant difference in TMB between LS-SCLC and ES-SCLC was observed (Fig. 1d).
Effects of genetic alterations on progression-free survival (PFS)
All SCLC patients were administered first-line chemotherapy. According to the evaluation of first-line treatment, 2 patients (4.1%) showed complete response (CR), 37 (75.5%) showed partial response (PR), 8 (16.3%) exhibited stable disease (SD), and 2 (4.1%) presented progression disease (PD). The median PFS was 8 months (range, 1.2–20.8 months). Univariate analysis revealed that patients with ERBB2mutations had a significantly prolonged PFS as compared to those with wild-type ERBB2 (P < 0.001) (Fig. 3a, Additional file 3: Table S1). On the contrary, patients with KDR or PTEN mutations had a significantly reduced PFS as compared to those with wild-type KDR (P = 0.01) or PTEN (P = 0.017) (Fig. 3b, c, Additional file 3: Table S1). The results in ES-SCLC were similar (Fig. 3d, e, f). It is worth noting that KDR and PTEN mutations were only detected in ES-SCLC. However, there were only two samples with KDR or PTEN mutation, and only two samples did not carry ERBB2 mutation, the above comparisons are meaningless. Thus, there were no significant correlation between mutations and PFS.

The univariate analysis of ERBB2, KDR, and PTEN. a-c. In total SCLC; d-f. In the extensive stage SCLC (ES-SCLC)
Among the 156 missense variants, 7 were analyzed after excluding those with low frequencies. Univariate analysis revealed that there was a significant correlation between ERBB2.p.L755 M variant and PFS (Additional file 3: Table S2). However, the ERBB2 mutation detected in our study was concentrated at ERBB2.p.L755 M variant, it is meaningless to conduct the further analysis.
Effects of genomic alterations on overall survival (OS)
The average follow-up was for 16 months, and the median OS was 16.3 months (range, 5.2–32.5 months). Univariate analysis showed that patients with mutant KIAA1211 had a longer OS than those with wild-type KIAA1211 (P = 0.009) (Fig. 4a) (Additional file 3: Table S3). The results were similar in ES-SCLC (P = 0.032) but not in LS-SCLC (P = 0.19) (Fig. 4c, e). However, for patients with NF1 mutations in LS-SCLC subgroup, their OS was significantly shorter than those with wild-type NF1 (10.4 vs. 21 months, P < 0.001) (Fig. 4f). Similar results were not observed in either SCLC (P = 0.05) (Fig. 4b) or ES-SCLC (P = 0.78) (Fig. 4d) (Additional file 3: Table S3).

The univariate analysis of KIAA1211 and NF1. a-b. In total SCLC; c-d. In the extensive stage SCLC (ES-SCLC); e-f. In the limited stage SCLC (LS-SCLC)
We established a Cox regression model using age, sex, smoking status, clinical stages, PS score, and 5 mutations as covariates for adjustment. The results demonstrated that the OS of patients carrying mutant KIAA1211 was significantly longer than those with wild-type KIAA1211 (P < 0.001) (Fig. 5), suggesting that KIAA1211 mutation predicts a positive factor for SCLC prognosis. However, there was no significant correlation between variants and OS (Additional file 3: Table S4).

Multivariate analysis between mutations and overall survival
Discussion
SCLC differs from other lung cancers irrespective of the pathological, molecular, or clinical manifestations. For nearly 30 years, no significant progress or effective targeted therapy for SCLC has been established, leading to still a poor prognosis [13]. Therefore, selection of an appropriate biomarker to assess the disease severity, monitor tumor progression, and evaluate the response to therapy is indispensable. Targeted therapy and immunotherapy aiming at specific genes improved the survival of SCLC patients; thus, identifying the genomic alterations of the patient and the effects of targeted genes on patient prognosis should be elucidated. Identifying and determining the biomarkers associated with SCLC prognosis would make a more careful assessment and classification of the SCLC population and could eventually also define subgroups of patients suitable for targeted therapies, which could improve the treatment outcome for defined subtypes and life quality for SCLC patients.
Gene sequencing-based diagnosis and treatment has been widely used among NSCLC patients, which improves the patient prognosis. In 2015, Thomas et al. [11], together with other groups, carried out a genome-wide sequencing of SCLC on the largest sample size to date. The study verified the results of previous genomic studies, such as common inactivation of TP53 and RB1 for SCLC, and also confirmed the disruption of other genes and signaling pathways, such as the TP73 and Notch signaling pathway [14]. In SCLC, the inactivation of TP53 and RB1 accounted for 75–90% and 60–90%, respectively [15], which are the initial events in SCLC [11, 16, 17]. The above study also found that 13% of the SCLC samples carried TP73 mutations or rearrangements. This phenomenon inhibited the function of wild-type TP53, which might be a potential target for the treatment of SCLC. Also, 25% of the SCLC samples were found to harbor the inactivated Notch gene, and animal studies confirmed that the Notch family exerts a tumor suppressive function and regulates the neuroendocrine differentiation of SCLC. SCLC has not been considered a homogenous tumor based on morphology. Tumor heterogeneity has been recognized many years ago: Mixed SCLC- Large cell tumors [14]. Some small sample studies also revealed tumor heterogeneity regarding the genomic analysis of SCLC. In the current study, mutations in TP53, ERBB2, and CREBBP were common with > 77% frequency. Moreover, we also detected mutations in NOTCH3 and TP73, which occupied 12.2 and 6.1%, respectively, and other previously reported mutations such as KIAA1211, RGS7, and FPR1 were also detected. Nevertheless, considerable differences were detected in the frequencies of significantly gene mutations in different studies, and the inconsistency might be attributed to the source of samples as well as different ethnicity of the patients.
Another study pointed out that the gene mutations and the total number of mutations were not associated with the OS or other clinical features of SCLC [18]. However, according to our findings, the mutations in KIAA1211 prolonged the OS of patients, whereas mutations in NF1 exhibit an opposite effect in LS-SCLC subgroup. KIAA1211 was identified by the Kazusa cDNA project with uncharacterized biological functions [19]. Recently, KIAA1211 was reported to transcriptionally upregulated in breast cancer [20]. While KIAA1211 was frequently mutated or transcriptionally downregulated in colorectal cancer, furthermore, KIAA1211 was demonstrated to act as a tumor suppressor through the maintaining of epithelial cell integrity [21]. In a comprehensive genomic analysis of somatic genome alterations in SCLC, KIAA1211 was revealed to be a significantly mutated gene with a ranking of third following TP53 and RB1, and it seems to involve the tumor pathogenesis [11]. However, KIAA1211 was a newly discovered mutation in SCLC, its functional role in SCLC needs further investigation. In this study, we also observed a correlation between ERBB2 mutation and PFS, but it lacks clinical significance due to the high occurrence rate of ERBB2 mutation in SCLC. Furthermore, patients with higher TMB had a markedly prolonged OS, which indicated a better prognosis. Our results are similar to the previous results reported by Roszik et al. that patients with higher TMB had better clinical efficacy and prognosis in NSCLC [22]. However, in surgically treated NSCLC, high TMB is a poor prognostic factor [23]. The controversial results suggested that the validation of correlations of TMB with survival is needed.
Although molecular targeted therapy have not yet proven effective in SCLC, we detected some well-known oncogenic driver mutations including PIK3CA (9/49) [24], KIT (1/49) [25], and BRAF (2/49) [26], which suggested opportunities for more targeted therapeutic approaches.
Based on tumor characteristics, high-throughput sequencing of small panels underwent bioinformatics analysis. As a result, the mutation could be interpreted easily and rapidly, which reduces the economic burden of patients. This technique has many advantages, such as high targeting ability and cost-efficiency, in clinical practice. Hence, based on the previous studies, we established a panel of 62 genes closely associated with SCLC, sequenced the tumor samples from 53 SCLC patients, and analyzed the alterations of genes and the correlation with disease prognosis.
Nevertheless, the current study has several limitations. First, it is a retrospective study with a relatively small sample size, such that except for some common mutations, the others were low-frequency mutations, which could significantly affect the subsequent survival analysis. Second, because of the small panel, the estimates of TMB was higher in this study than TMB in the previous studies [27, 28]. In addition, we only detected two RB1 mutations: RB1.p.R334STOP and RB1.p.R579QfsSTOP29. Several mutations in RB1 occurred at exon-intron junctions, which caused protein-damaging splice events as confirmed by transcriptome sequencing. This phenomenon might be attributed to the sequencing of the small panel of 62 genes in this study, which might fail to detect all the deletion mutations.
Conclusion
In conclusion, targeted high-throughput sequencing can detect specific gene regions accurately and efficiently, and understanding the correlation between genomic alterations and SCLC prognosis is essential for more individualized treatment of SCLC patients. Furthermore, due to the uncharacterized function of KIAA1211, it is of great significance to investigate the biological function of KIAA1211 in SCLC.
Availability of data and materials
The datasets used and/or analyzed during the current study are available from the corresponding author on reasonable request.
Abbreviations
- ES:
-
Extensive-stage
- LS:
-
Limited-stage
- OS:
-
Overall survival
- PFS:
-
Progression free survival
- SCLC:
-
Small cell lung cancer
- TMB:
-
Tumor mutation burden
References
Torre LA, Bray F, Siegel RL, Ferlay J, Lortet-Tieulent J, Jemal A. Global cancer statistics, 2012. CA Cancer J Clin. 2015;65:87–108.
van Meerbeeck JP, Fennell DA, De Ruysscher DK. Small-cell lung cancer. Lancet. 2011;378:1741–55.
Sculier JP, Chansky K, Crowley JJ, Van Meerbeeck J, Goldstraw P, International Staging C, Participating I. The impact of additional prognostic factors on survival and their relationship with the anatomical extent of disease expressed by the 6th edition of the TNM classification of malignant tumors and the proposals for the 7th edition. J Thorac Oncol. 2008;3:457–66.
Demedts IK, Vermaelen KY, van Meerbeeck JP. Treatment of extensive-stage small cell lung carcinoma: current status and future prospects. Eur Respir J. 2010;35:202–15.
Rossi A, Maione P, Palazzolo G, Sacco PC, Ferrara ML, Falanga M, Gridelli C. New targeted therapies and small-cell lung cancer. Clin Lung Cancer. 2008;9:271–9.
Ott PA, Elez E, Hiret S, Kim DW, Morosky A, Saraf S, Piperdi B, Mehnert JM. Pembrolizumab in patients with extensive-stage small-cell lung Cancer: results from the phase Ib KEYNOTE-028 study. J Clin Oncol. 2017;35:3823–9.
Antonia SJ, Lopez-Martin JA, Bendell J, Ott PA, Taylor M, Eder JP, Jager D, Pietanza MC, Le DT, de Braud F, Morse MA, Ascierto PA, Horn L, Amin A, Pillai RN, Evans J, et al. Nivolumab alone and nivolumab plus ipilimumab in recurrent small-cell lung cancer (CheckMate 032): a multicentre, open-label, phase 1/2 trial. Lancet Oncol. 2016;17:883–95.
Diker O. Atezolizumab plus chemotherapy in small-cell lung Cancer. N Engl J Med. 2019;380:889.
Peifer M, Fernandez-Cuesta L, Sos ML, George J, Seidel D, Kasper LH, Plenker D, Leenders F, Sun R, Zander T, Menon R, Koker M, Dahmen I, Muller C, Di Cerbo V, Schildhaus HU, et al. Integrative genome analyses identify key somatic driver mutations of small-cell lung cancer. Nat Genet. 2012;44:1104–10.
Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, Rivers CS, Foo CK, Bhatt D, Stinson J, Gnad F, Haverty PM, et al. Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet. 2012;44:1111–6.
George J, Lim JS, Jang SJ, Cun Y, Ozretic L, Kong G, Leenders F, Lu X, Fernandez-Cuesta L, Bosco G, Muller C, Dahmen I, Jahchan NS, Park KS, Yang D, Karnezis AN, et al. Comprehensive genomic profiles of small cell lung cancer. Nature. 2015;524:47–53.
Zelen M. Keynote address on biostatistics and data retrieval. Cancer Chemother Rep 3. 1973;4:31–42.
Gazdar AF, Bunn PA, Minna JD. Small-cell lung cancer: what we know, what we need to know and the path forward. Nat Rev Cancer. 2017;17:725–37.
Bunn PA Jr, Minna JD, Augustyn A, Gazdar AF, Ouadah Y, Krasnow MA, Berns A, Brambilla E, Rekhtman N, Massion PP, Niederst M, Peifer M, Yokota J, Govindan R, Poirier JT, Byers LA, et al. Small cell lung Cancer: can recent advances in biology and molecular biology be translated into improved outcomes? J Thorac Oncol. 2016;11:453–74.
Wistuba II, Gazdar AF, Minna JD. Molecular genetics of small cell lung carcinoma. Semin Oncol. 2001;28:3–13.
Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, Berns A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell. 2003;4:181–9.
Lee JK, Lee J, Kim S, Kim S, Youk J, Park S, An Y, Keam B, Kim DW, Heo DS, Kim YT, Kim JS, Kim SH, Lee JS, Lee SH, Park K, et al. Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J Clin Oncol. 2017;35:3065–74.
Paesmans MN, Lafitte JJ, Lecomte JN. Validation and comparison of published prognostic classifi cations for small cell lung cancer. J Thorac Oncol. 2010;5:s104(abstr 265O).
Snider PL, Snider E, Simmons O, Lilly B, Conway SJ. Analysis of uncharacterized mKiaa1211 expression during mouse development and cardiovascular morphogenesis. J Cardiovasc Dev Dis. 2019;6.
Bao Y, Wang L, Shi L, Yun F, Liu X, Chen Y, Chen C, Ren Y, Jia Y. Transcriptome profiling revealed multiple genes and ECM-receptor interaction pathways that may be associated with breast cancer. Cell Mol Biol Lett. 2019;24:38.
Jung YS, Wang W, Jun S, Zhang J, Srivastava M, Kim MJ, Lien EM, Shang J, Chen J, McCrea PD, Zhang S, Park JI. Deregulation of CRAD-controlled cytoskeleton initiates mucinous colorectal cancer via beta-catenin. Nat Cell Biol. 2018;20:1303–14.
Rizvi NA, Hellmann MD, Snyder A, Kvistborg P, Makarov V, Havel JJ, Lee W, Yuan J, Wong P, Ho TS, Miller ML, Rekhtman N, Moreira AL, Ibrahim F, Bruggeman C, Gasmi B, et al. Cancer immunology. Mutational landscape determines sensitivity to PD-1 blockade in non-small cell lung cancer. Science. 2015;348:124–8.
Owada-Ozaki Y, Muto S, Takagi H, Inoue T, Watanabe Y, Fukuhara M, Yamaura T, Okabe N, Matsumura Y, Hasegawa T, Ohsugi J, Hoshino M, Shio Y, Nanamiya H, Imai JI, Isogai T, et al. Prognostic impact of tumor mutation burden in patients with completely resected non-small cell lung Cancer: brief report. J Thorac Oncol. 2018;13:1217–21.
Juric D, Rodon J, Tabernero J, Janku F, Burris HA, Schellens JHM, Middleton MR, Berlin J, Schuler M, Gil-Martin M, Rugo HS, Seggewiss-Bernhardt R, Huang A, Bootle D, Demanse D, Blumenstein L, et al. Phosphatidylinositol 3-kinase alpha-selective inhibition with Alpelisib (BYL719) in PIK3CA-altered solid tumors: results from the first-in-human study. J Clin Oncol. 2018;36:1291–9.
Dieter SM, Heining C, Agaimy A, Huebschmann D, Bonekamp D, Hutter B, Ehrenberg KR, Frohlich M, Schlesner M, Scholl C, Schlemmer HP, Wolf S, Mavratzas A, Jung CS, Groschel S, von Kalle C, et al. Mutant KIT as imatinib-sensitive target in metastatic sinonasal carcinoma. Ann Oncol. 2017;28:142–8.
Ritterhouse LL, Barletta JA. BRAF V600E mutation-specific antibody: a review. Semin Diagn Pathol. 2015;32:400–8.
Chalmers ZR, Connelly CF, Fabrizio D, Gay L, Ali SM, Ennis R, Schrock A, Campbell B, Shlien A, Chmielecki J, Huang F, He Y, Sun J, Tabori U, Kennedy M, Lieber DS, et al. Analysis of 100,000 human cancer genomes reveals the landscape of tumor mutational burden. Genome Med. 2017;9:34.
Hellmann MD, Callahan MK, Awad MM, Calvo E, Ascierto PA, Atmaca A, Rizvi NA, Hirsch FR, Selvaggi G, Szustakowski JD, Sasson A, Golhar R, Vitazka P, Chang H, Geese WJ, Antonia SJ. Tumor mutational burden and efficacy of Nivolumab Monotherapy and in combination with Ipilimumab in small-cell lung Cancer. Cancer Cell. 2018;33:853–61 e4.
Acknowledgements
Not applicable.
Funding
This work was supported in part by the National Natural Science Foundation of China (No. 81703847).
Author information
Authors and Affiliations
Contributions
All authors participated in the planning and execution of this study. MZ, JF and ZL collected the data and conducted the relevant experiments. ZL, and HQ drafted the manuscript. PL, YS, YY, and WC performed the statistical analyses. XD designed the study, and supervised the project. FRH provided critical comments and suggestions and revised the manuscript. Shanghai Tongshu Biotechnology Co., Ltd. conducted the targeted sequencing of all included samples. All authors read and approved the final version of the manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
This Study was approved by Ethics Committee of Wuhan Union Hospital. All patients signed informed consent forms before the initiation of any study-related procedure.
Consent for publication
Not applicable.
Competing interests
Fred R. Hirsch has participated in scientific advisory boards for: AstraZeneca, Genentech/Roche, Merck, Abbvie, Ventana Novartis, BMS, Pfizer, HTG Molecular and Biocept. FRH has received research funding (through University of Colorado) from: Amgen, Merck, Biodesix, Mersana, HTG Molecular, Rain Therapeutics., Abbvie.
Huiwei Qi and Weijing Cai are the employees of Shanghai Tongshu Biotechnology Co., Ltd.
All other authors declare no potential conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Additional file 1: Figure S1.
Thermal map of mutation variants in SCLC.
Additional file 2: Figure S2.
Kaplan–Meier analysis of high TMB and low TMB. a. PFS in ES-SCLC; b. OS in ES-SCLC; c. PFS in LS-SCLC; b. OS in LS-SCLCC.
Additional file 3: Table S1.
Univariate analysis between gene mutations and PFS. Table S2. Univariate analysis between variants and PFS. Table S3. Univariate analysis between gene mutations and OS. Table S4. Univariate analysis between variants and OS.
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Zhou, M., Fan, J., Li, Z. et al. Prognostic impact of tumor mutation burden and the mutation in KIAA1211 in small cell lung cancer. Respir Res 20, 248 (2019). https://doi.org/10.1186/s12931-019-1205-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12931-019-1205-9