
Abstract
Cryptochromes are a class of light-absorbing proteins that have been shown to be a part of the circadian rhythm of many animals but seem to play a central role for the magnetosensing of migratory birds. Following a documented difference in the sensitivity to an external magnetic field of cryptochrome 4a proteins from migratory and non-migratory birds, a detailed analysis of inter- and intra-protein energetics is called for. The present study relies on classical molecular dynamics simulations of cryptochrome 4a from five avian species to reveal if any of the cryptochromes feature peculiarities in their internal energetics. The five avian cryptochrome 4a proteins from pigeon, European robin, zebra finch, chicken, and Eurasian blackcap are found to be highly similar in respect of their intra-energetic behaviors, while some minor differences between the cryptochromes can be ascribed to the site of specific structural differences. Particular attention has been paid to account for the interaction of the protein with the solvent, and it has been revealed that the solvent could lead to significant stabilization of the chromophore flavin adenine dinucleotide inside of the cryptochrome 4a scaffold.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Migration of animals has been observed and has fascinated humans for hundreds of years. The tools that Nature has developed to enable birds to migrate include, among others, celestial cues, i.e., orientation related to the Sun and other celestial bodies, olfactory, and even orientation by using the Earth’s magnetic field [1, 2]. In the latter case, the inclination angle of the magnetic field can be interpreted by birds as a direction guide for a magnetic compass [3,4,5,6,7,8]. The magnetic compass inside a bird is apparently sensitive to relatively weak magnetic fields, such as the geomagnetic field [9,10,11] and its minor perturbations [12]. A possible mechanism that explains how the weak magnetic field can be perceived relies on the dynamics of intra-cellular radical pairs whose spin governs the yields of associated chemical reactions depending on the orientation to the Earth’s magnetic field. The mechanism was first proposed by Schulten et al. [13] and was later advanced by Ritz et al. [14], where a specific molecular host for the radical pairs, the protein cryptochrome (Cry), was suggested.
To this day, Cry is the only known vertebrate photoreceptor molecule that can form long-lived radical pairs upon excitation by blue light [2, 9, 15,16,17,18]. Cry is a versatile protein with diverse biophysical roles. In plant (Arabidopsis thaliana), Cry has been shown to control photomorphogenesis [19, 20], while some mammalian cryptochromes have been shown to serve as regulators of the circadian clock [21, 22]. Cry type 4a (Cry4a) has been shown to play a role in aiding migratory birds on their yearly migration [23]. In addition, Cry4a has been found in the retinae of the migratory European robin (Erithacus rubecula), where its expression level was shown to be significantly higher during the migratory season [23]. Furthermore, it has been established that the Cry4a protein from the European robin is more sensitive to magnetic fields than Cry4a proteins from non-migratory bird species [9]. It is not yet established how the differences in magnetic sensitivity occur, but the functioning of Cry4a depends on the non-covalently bound flavin adenine dinucleotide (FAD) cofactor [17, 24] (see Fig. 1) and a cascade of electron transfers that involve a chain of conserved tryptophan residues [9, 25,26,27], connecting the FAD cofactor with the protein surface at a combined transfer length of approximately 15 Å in avian Cry4a [9].

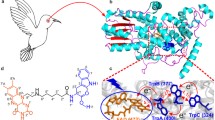
Superimposed structures of the studied avian Cry4a models aligned according to their three-dimensional structural similarity. The inset shows the FAD molecule bound inside the FAD binding cavity. The alignment was performed with the Structural Alignment of Multiple Proteins (STAMP) tool [28, 29] by using the Visual Molecular Dynamics (VMD) program [30]
While the functioning mechanism of Cry4a in different species is slowly unveiled, many questions related to intra-protein interactions are still unresolved. The present investigation focuses on elucidating the global energetical difference in different cryptochrome structures and investigating how different cryptochrome structures interact internally, with their cofactors, or with the solvent. The analyses of the intra-protein energy aims to highlight which sites are important for stabilizing the protein, stabilizing a bound cofactor and which sites are highly interacting with the solvent.
The studied models were previously homology modeled, and simulated [9, 27, 31]. Specifically we focus on the structure of the pigeon cryptochrome 4 ClCry4a derived from a crystal structure [32], along with the reconstructed structures of European robin (Erithacus rubecula) cryptochrome 4, ErCry4a [9], Chicken (Gallus gallus) cryptochrome 4a, GgCry4a, Eurasian blackcap (Sylvia atricapilla) cryptochrome 4a, SaCry4a, and Zebra finch (Taeniopygia guttata) cryptochrome 4a, TgCry4a, displayed in Fig. 1 [27]. Furthermore, the global structural differences between avian Cry4a proteins are linked to the aforementioned energetical studies and sites where an energetical difference can be ascribed to be due to a difference in sequences have been highlighted. The energetic differences of the Cry4a structures were studied through analyses of intra-protein interactions and the interactions of the FAD cofactor with the surrounding protein, with and without solvent contributions.
Lastly, the solvent-accessible surface area (SASA) analysis has been performed to quantify the solvent exposure level of different Cry4a regions. The analysis unraveled the atomistic differences in Cry4a structures that lead to some deviations in the inter-protein energetics but also demonstrated an overall similarity of the five studied cryptochromes from the energetic perspective.
2 Methods
2.1 Molecular dynamics simulations
The analysis of Cry4a energetics was performed for proteins that were simulated dynamically in earlier studies [9, 23, 27, 31]. The simulations utilized NAMD [33,34,35] through the VIKING platform [36] to carry out the computations. CHARMM36 force fields including CMAP corrections were used for all simulations [37,38,39,40,41,42,43]. The investigated Cry4a structures were all assumed to be in the inactive state, also known as the dark state, which is characterized by the FAD cofactor being in its fully oxidized state. The corresponding parameters for the FAD cofactor were adopted from earlier studies [9, 23, 25,26,27, 31, 44]. The concentration of NaCl in all simulations was set to 0.15 mol/L. The size of the simulation boxes used for the equilibrated proteins appeared as follows: chicken Cry4a: 92.4 Å \(\times \) 94.9 Å \(\times \) 104.6 Å; blackcap Cry4a: 92.4 Å \(\times \) 95.7 Å \(\times \) 106.4 Å; zebra finch Cry4a: 93.0 Å \(\times \) 95.7 Å \(\times \) 106.4 Å; robin Cry4a: 94.4 Å \(\times \) 107.6 Å \(\times \) 102.9 Å; and pigeon Cry4a: 103.0 Å \(\times \) 104.0 Å \(\times \) 111.8 Å.
MD simulations of different Cry4a structures included 10 000 conjugate gradient steps to minimize the energy of the system prior to the actual MD simulations. Minimization was followed by structure equilibration and the production simulations [9, 27, 31]. In the present investigation, production simulations refer to those simulations that were used for the analyses. It should be noted that the simulations of the pigeon Cry4a are significantly longer than those for the other Cry4a structures because a more extensive sampling was carried out for that particular Cry4a in an earlier investigation [31]. The production simulations from the earlier studies were used here to analyze the interaction energies of the FAD cofactor with the rest of the protein and with the protein scaffold, as well as the interaction of the protein with the solvent. The calculations were done using NAMD [34, 35]. In order to ensure a better data sampling, it is required to perform replica simulations [45]. Three independent MD simulations were performed for each Cry4a presented in this study. Van der Waals and electrostatic interactions were subjected to cutoff values of 12 Å, and the particle mesh Ewald (PME) summation method was used to treat long-range electrostatic interactions [46].
2.2 Intramolecular interaction energy
To compare intra-protein, protein–solvent, and protein–FAD interactions for different avian cryptochromes, the interaction energy that includes the non-bonding energy terms has been calculated [34]. The interaction energy between the protein and the FAD cofactor, \( E_{FAD}^{P} (t)\), was calculated as [47]:
Here the subscript and superscript denote the parts of the system for which the interaction energy is calculated; P represents the protein, and FAD represents the FAD cofactor. N is the total number of residues in a given Cry4a protein, while C is the total number of atoms in the FAD cofactor. \(E_{i,c}(t)\) represents the interaction energy of the \(i\hbox {th}\) residue in the protein and the \(c\hbox {th}\) atom in the FAD cofactor, where the interaction energy contains van der Waals and Coulomb contributions. Equation (1) does not consider the cofactor’s interaction with the solvent. In that case, the interaction of the cofactor with the rest of the system, \( E_{FAD}^{PS}(t) \), can be defined as:
Here the second summation in the brackets represents the interaction between the FAD and the solvent, with k denoting a residue in the solution and K being the total number of water and ion residues. The time-averaged intra-protein interaction of a residue i with the rest of the protein can be calculated as:
where T is the total number of simulation time frames, which in the present investigation were collected every 5 ps from the production simulations; the total simulation lengths of the studied Cry4a structures are found in Table 1. The subscripts i and j denote the two different residues in a given Cry4a structure, and N is the total number of residues in the protein. The term \( E_{i,j}(t)\) denotes the intra-protein interactions arising between residues i and j. The intra-protein interactions with account for the solvent can be computed as:
Here, k enumerates the residues of the solvent, with K being the total number of solvent residues. The interaction of the individual residues inside Cry4a with the solvent can be determined by subtracting Eq. (3) from Eq. (4), which results in the residue/solvent interaction defined as:
Finally, the time-averaged interaction of a given residue i with the FAD cofactor can be calculated as:

A–E: Probability distribution of the interaction energies between FAD and the surrounding protein for the Cry4a proteins of pigeon, European robin, zebra finch, chicken, and Eurasian blackcap, respectively, with the contribution of the solvent, calculated using Eq. (2), (blue) and without the solvent, calculated using Eq. (1), (orange). F: A zoom into the exemplary zebra finch Cry4a structure, highlighting the FAD binding cavity with the surrounding protein shown as a surface
3 Results and discussion
3.1 Interaction of the protein with the FAD cofactor
The activity of proteins inside the cell is often associated with a cofactor that can enhance and specialize the protein’s role, and function [48]. In the case of the cryptochromes, FAD can absorb light of certain wavelengths which enables the creation of the radical pairs inside the Cry4a proteins that bind FAD [17, 24]. To check whether there is a significant difference in the interaction energy between protein–FAD in different avian species, an analysis has been performed by utilizing Eqs. (1)–(6).
Figure 2 shows that all of the Cry4a proteins considered in this study have an attractive interaction with the FAD cofactor. This attraction is not surprising since a crystal structure of pigeon Cry4a protein [32] was shown to bind FAD, and the avian cryptochromes investigated in this paper have a high sequence similarity to pigeon Cry4a (see the sequence alignment in Fig. 3) [27]. The more pronounced attractive interaction between the FAD and the protein (without the presence of the solvent, Fig. 2 orange distributions) could be explained by a difference in the FAD binding cavity, which would cause a slight change in FAD conformation and its interactions with the surrounding residues. The results in Fig. 2 show overlapping probability distribution for all the interaction energies of the FAD with the protein scaffold in the case of all studied Cry4a structures, except for the pigeon Cry4a structure. Furthermore, the results in Fig. 2 show that there is a tendency for avian species Cry4a to experience a stabilization in FAD interaction energy of around 200 kcal/mol upon inclusion of solvent in the calculation (see Tables S2 and S3).

Solvent-assisted stabilization does not seem to be identical for all cryptochromes. Certain structural elements at the entry to the FAD binding cavity are expected to influence the amount of the surrounding solvent inside the FAD binding cavity. Particularly the different conformations of the phosphate-binding loop (residues 231-248), which is positioned at the interface of the FAD binding cavity, could partially be responsible for the noticed differences. The connection of the phosphate-binding loop and photoactivation of pigeon Cry4a protein was recently discussed in Schuhmann et al. [31] where an extensive MD simulation of the pigeon Cry4a structures showed that there is a clear difference in the position of the phosphate-binding loop for the inactive state of the Cry4a protein and its activated variant. Although only the dark state (inactive) simulations were analyzed in the present study, a similar behavior could be expected for all investigated cryptochromes due to similarities in the Cry4a structures. Furthermore, in earlier studies, water has been shown to be instrumental in facilitating the electron transfers in plant Cry and could play a similar role in avian cryptochromes [25].

Time-averaged interaction energy of every residue in different Cry4a structures with the FAD cofactor \(\left( E_{i,FAD} \right) \) calculated using Eq. (6). The data were obtained for the simulated crystal structure of pigeon Cry4a, panel A, and the reconstructed Cry4a models for European robin, zebra finch, chicken, and Eurasian blackcap, panels B–E, respectively. Each line in the subplots depicts the results obtained for a single replica simulation (see Table 1) with an offset of 100 kcal/mol for clarity. The color scheme is consistent with the nomenclature introduced in Fig. 1, while the blue-shaded area highlights the FAD binding domain. Several residues of interest were labeled (see text)
A more detailed analysis of FAD interaction energy with individual residues of the studied Cry4a proteins was also performed, and the results are shown in Fig. 4. The figure shows that there is little to no difference in the results between the individual replica simulation. Some specific residues seem to contribute to protein–FAD interaction in some avian Cry4a structures but not others. For example, Arg236 in the pigeon Cry4a (see Fig. 4A) has an interaction energy with the FAD at - 77 kcal/mol, while in other bird cryptochromes this specific residue does not interact with FAD more than -13 kcal/mol (European robin: -6 kcal/mol, zebra finch: -5 kcal/mol, blackcap: -12 kcal/mol). A detailed analysis of the simulations of pigeon Cry4a, see Fig. 5, revealed that a sodium ion binds next to the FAD. The sodium ion becomes stabilized by the oxygens of the phosphate group, Pro244 backbone oxygen, and the surrounding water molecules. The Arg236 residue then forms a salt-bridge with one of the oxygens from the phosphate groups of the FAD. The supposed salt bridge has been observed for all 3 pigeon Cry4a MD trajectories though the FAD and Arg236 are only within 4 Å of each other for 0.9 % of the first simulation trajectory compared to 37.0 % and 30.6 % the second and third simulation trajectories. Furthermore, Fig. S4 reveals that the dissociation of the sodium ion for simulations 1 and 2 occurs due to a conformational change of the protein that allows the disassociation to happen. The conformational change as a consequence gives two very different interaction energy profiles between the protein and FAD cofactor, which can be seen for the pigeon Cry4a in Fig. 2A. An additional distance analysis between a specific sodium ion and FAD for all three replica simulations is available in the Supplementary Information (SI); Fig. S4. Arg236 could therefore, through an ion such as sodium, be involved in the increased stabilization of FAD interaction with the protein scaffold for pigeon Cry4a, noticed in Fig. 2. Other avian Cry4a structures presented in this work were also analyzed for sodium ion interaction with the FAD cofactor, but no significant results were found. The results in Fig. S4 suggest that FAD stabilization inside different cryptochromes could be influenced by sodium or other ions that enter the FAD binding cavity. Certain cryptochromes do not routinely bind FAD [51, 52], possibly because their structure does not allow hosting ions that could stabilize the protein–FAD interaction.
The reoccurring attractive interactions between the FAD and the protein scaffold (shown as upward peaks in Fig. 4) at the residues Arg218, Arg291, and Arg356 correspond to the positive arginines. In contrast, the repulsive peaks for Glu348, Asp385, and Asp387 represent the negatively charged amino acids. This observation indicates that most of the FAD stabilization inside the FAD binding cavity comes from interaction with the positively charged amino acids, which undeniably stabilize and interact with the more negative part of the FAD molecule and its aromatic rings.
An in-depth analysis of the hydrogen bonds arising between the FAD and its surrounding amino acids was also performed, see Table S1. Table S1 and Fig. S1 show that overall for the European robin, blackcap, and chicken Cry4a structures, the number of most reoccurring hydrogen bonds is equal to 3, while for zebra finch and pigeon Cry4a, it is 2.

Pigeon Cry4a protein with a zoom into the FAD-binding region where a bound sodium ion is found (yellow sphere). The upper inset shows water molecules, as well as Arg236 and Pro244 residues that coordinate the ion binding. Arg236 forms a salt bridge with the FAD, while the backbone oxygen of Pro244 appears to stabilize the sodium ion
3.2 Intramolecular interactions within the Cry4a structures
While the interactions between FAD and the rest of the Cry4a proteins are instrumental for understanding how the cofactor can support the functioning of the protein, the Cry4a proteins have most likely developed intra-protein interactions to complete certain processes, such as the electron transfer [9] which originates at the FAD and terminates at a peripheral tryptophan residue about 15 Å away. Analysis of the interaction energies of FAD and the Cry4a protein from migratory and non-migratory avian species could reveal differences in the stabilization of FAD, i.e., which amino acid residues contribute more to the stabilization of the FAD inside the FAD binding cavity of Cry4a. These contributions could be revealed by the interaction energy analysis since this analysis includes the contributions from electrostatic and van der Walls (non-bonding interactions), which are all known to stabilize cofactors inside proteins [27].

Figure 6 shows the time-averaged interaction energies for every individual residue and the rest of the protein, computed using Eq. (3). Figure 6 shows that there is only a minor variation between the results derived from different replica simulations, which suggests that intra-protein interactions for Cry4a proteins can, in fact, be sufficiently sampled through a single replica simulation. Additionally, a series of statistical tests is performed to quantify the similarity between the inter-residue energetics among the different replica simulations (see SM). The average energies were sorted into 20 bins and tested in keeping the deviations of the original data. The results show that the interaction energy \(E_i^P\) of each residue is similar among different Cry4a structures in at least 70% of the residues. Additionally, the similarity test revealed that the European robin Cry4a is more similar to the blackcap Cry4a and less similar to the cryptochromes from non-migratory chicken and pigeon. The results containing the similarity percentages are compiled in Table S4.
Furthermore, Fig. 6 shows that each Cry4a has a noticeable number of negative (i.e., attractive) interactions between certain residues and the protein scaffold and repulsive interactions (positive interaction energy) that are conserved among the studied Cry structures, where one of the major repulsive peaks for all the considered structures is found in the phosphate-binding loop (residues 231–248) which supports earlier findings [27] of this mobile protein domain the present results show that pairwise residue–residue interactions do not stabilize the region.
The interaction energy obtained using Eq. (3) determines the intra-protein interactions. However, the equation does not provide a realistic picture of the surroundings as Cry4a proteins are usually found in an aqueous cytosolic environment [23], where the surrounding solvent is expected to play a role in general. Equation (4) can therefore be used to investigate the intra-protein and solvent interactions of the Cry4a proteins of interest.

Deviation of the average residue-specific interaction energies for each studied Cry4a models, calculated using Eq. (7). Each line represents the deviation of the interaction energy of a specific residue from the overall average value. An offset of 200 kcal/mol has been applied for clarity
Figure S5 shows that all the repulsive interactions seen in Fig. 3 become screened and, in some cases, lead to attractive interactions, suggesting that the solvent has an important stabilizing effect on the Cry4a structures. The results in Fig. S5 show that the interaction of the residues with the protein scaffold is highly similar for different replica simulations when the solvent is taken into account, which additionally supports the rationale that the energy analyses of the simulations deliver statistically significant results, which is further highlighted in Table S4. It should be noted that the protein environment in an MD simulation differs from what one would find in a cell cytosol. The only ions present in an MD simulation are those of \(\hbox {Na}^{+}\) and \(\hbox {Cl}^{-}\) with a concentration of 0.15 M, while specific amino acid residues were protonated, assuming a pH value of 7. An avian cytosol environment contains many different ions, which are important for different cellular processes. Any addition of extra ions would depend on what is used in an experimental setting or in a cell and would influence the energetics of the cryptochromes. In other studies, the ions have been shown to play a role in the stabilization of Cry proteins [53].
Figure S6 supports the conclusion on the importance of water for Cry4a stabilization as it shows that the interactions of residues with the solvent for different Cry4a structures are very alike and, in general, mostly attractive, since the interaction energies shown in Fig. S6 are mainly negative with only a few minor positive peaks. Furthermore, different sub-plots in Fig. S5 once again resemble each other in shape, and the only difference arises from the energetic magnitudes while the results obtained for the different replica simulations yet again are almost identical.
It is not straightforward to conclude from Fig. 6, Fig. S5, and Fig. S6 how much the interaction energies of the different Cry4a simulations deviate from each other. In order to investigate how the changes of sequences seen in Table 2 influence the interaction energy deviation per residue from the total average energy the interaction energy deviation per residue has been calculated for each Cry4a model as follows:
Here s denotes a simulation of one of the Cry4a models considered in this study, S is the total number of Cry4a models (in this study \(S=5\)), r denotes a replica simulation, and R is the total number of replica simulations (\(R=3\)). \(E_i^P\) is defined in Eq. (3).
Figure 7 shows the deviation of the intra-protein interactions per residue from the average value. The deviations can now be compared to gauge whether there is a correlation with the difference in the Cry4a sequences shown in Fig. 3.
Table 2 lists the sites where a notable difference in \(\Delta E_i\) in Fig. 7 was recorded. Interestingly, the largest deviations correlate with the sequence differences mostly, where charged amino acids appear to be the outliers, this is especially prominent for site 40, where pigeon Cry4a has an arginine that deviates with 123 kcal/mol while the rest have an alanine, all deviating with energies of \(-\) 31 kcal/mol. Another prominent example is site 306, where Eurasian blackcap Cry4a has a lysine while the other studied proteins have a glutamine. Table 2 indicates that especially the charged amino acids (e.g., arginines) in the Cry4a proteins have a notable role in facilitating interactions with other residues. Arginines are amphipathic amino acid residues often found on the protein surface and are included in hydrogen bonding, salt bridges, and/or interactions with other proteins [54]. This has also been shown to be the case for a Drosophila melanogaster Cry, where a salt bridge is created between Arg381 and Asp410, which acts as an allosteric switch that is regulated by the change in the redox state of FAD [55].

Pigeon Cry4a protein A: Shown with averaged solvent exposure per residue (cyan) highlighting the FAD binding cavity. The red transparent surface shows the volume with a 5 Å radius surrounding the FAD cofactor. B: Shown with averaged solvent interaction per residue (orange) and the inset showing the FAD binding cavity C: Pigeon Cry4a solvent exposure (cyan) superimposed with averaged solvent interaction per residue (orange)
3.3 Solvent-accessible surface area calculations
Solvent accessibility of the protein can reveal the role of protein’s subunits since solvent accessibility can predict hydrophilic and hydrophobic areas of the protein, which contribute more to interaction with the outside, or stabilization of the structure from the inside, respectively. The solvent-accessible surface area (SASA) is a useful characteristic that defines how much a protein or a residue is exposed to the solvent. The SASA values were calculated using VMD [30, 56, 57], assuming the radius of the solvent equal to 1.4 Å. To compare the SASA values for different amino acids, the absolute SASA values were normalized with the maximum SASA (MSA) values for the corresponding amino acids (X). MSA was computed for the Gly-X-Gly tripeptide (glycine was chosen to introduce the least steric hindrance and allow the largest solvent accessibility). For each of the 20 naturally occurring amino acid residues X, the Gly-X-Gly peptide was constructed using the Pep McConst [58] software and later simulated for 1 ns in water. The SASA values for residue X in the tripeptide and every amino acid residue in the avian cryptochromes were calculated using VMD. The solvent exposure for every residue, i, in the studied Cry4a structures was calculated as:
Figure S3 shows the SASA values per residue calculated for all the simulated avian Cry4a proteins and is presented as an averaged value obtained from the three replica simulations. Figure S3 indicates that solvent accessibility, like the other studied energetic characteristics, is highly similar among studied avian Cry4a proteins. While the results in Fig. S3 show residues accessible to the solvent, it does not mean that those amino-acid residues necessarily interacted with the solvent. In order to identify the difference between those residues that are accessible to the solvent and those that, in fact, do not interact with the solvent, the correlation of the solvent exposure with the solvent–protein interaction energy per residue (see Eq. (5)) has been established by visualizing both properties onto the protein’s 3D structure as shown in Fig. 8. Figure 8 indicates that solvent exposure per residue is represented by a considerably larger area than the area that represents the interaction with the solvent. The reason why the solvent exposure area is larger could be due to the fact that aliphatic amino acid residues might be present on the surface of the protein but do not noticeably interact with the aqueous surrounding. A similar analysis was done for all of the studied cryptochromes, see Fig. S2
Furthermore, it can also be observed that some of the non-surface residues interact with water but are not characterized as such with the solvent accessibility analysis. Mainly, it can be seen that there seem to be protein–solvent interactions happening inside the FAD binding cavity, as indicated earlier by the results shown in Fig. 2. These interactions are potentially important for Cry4a functioning since it is known that during the signaling process, the FAD inside Cry4a becomes reduced [9, 59,60,61] to FADH. FADH is presumed to be important for the signaling state of the avian Cry4a. In different species, different amino acids are responsible for FAD protonation to FADH. However, the increased presence of water inside the FAD-cavity could influence the stability and/or presence of FADH inside the protein. Since the proton is ultimately expected to arrive from the solvent [53], one concludes that the hydrogen bonding network, combined with the solvent exposure, could influence the presence of FADH inside the protein.
4 Conclusion
Following a recent structural and dynamical analysis of five avian cryptochromes [27], a detailed energetic analysis of inter- and intra-protein interactions was performed. Overall, it is found that interaction energies behave very similar across all of the investigated avian Cry4a structures. The interaction of the FAD chromophore with the protein was studied in detail, and it was revealed that there are differences in the strength of the protein–FAD interaction energies; in all cases, the inclusion of the solvent clearly leads to the stabilization of the protein–FAD interaction which is further confirmed by the SASA analysis which showed that solvent can protrude into the FAD binding cavity, therefore allowing the stabilization and possibly influencing the creation of FADH, which is crucial for the signaling state of avian cryptochromes, as postulated by earlier work [9]. The interaction of the FAD with the protein with the inclusion of the solvent was shown to be the strongest for the pigeon Cry4a, which would be attributed to the presence of a sodium ion in close proximity to FAD and the subsequent creation of a salt bridge between Arg236 and FAD.
The presented analysis also included residue interactions with the protein and the solvent. Even though sequence alignment shows high sequence similarity, deviations in inter-protein interaction arise and are largely attributed to a change of charged amino acid residues, mainly arginine, to others.
The study clearly displays the similarity in energetics among cryptochromes from selected avian species studied in this paper. Interestingly, this work opens up questions about the role of ions in cryptochrome biophysics, among others: how do ions tailor FAD interactions? It would be beneficial in the future to perform a similar analysis for different activated states of cryptochromes to have a better insight into cryptochrome activation that could affect the energetic landscape of the protein.
It should be noted that the cryptochromes presented in this work are studied in the so-called dark state (FAD assumed oxidized), which implies that specific energetic peculiarities that could be associated with the magnetic-field effects in the studied proteins could not be seen in the performed analysis. A possible factor that could influence the magnetic sensing ability of different cryptochrome is spin relaxation which is related to different molecular environments inside different Cry4a proteins and differences in FAD surrounding, which could dictate the recovery rates for the radical-mechanism process. However, such simulations and analyses are planned for future investigations.
Data Availability Statement
The datasets generated during and/or analyzed during the current study are available from the corresponding author upon reasonable request. This manuscript has no associated data or the data will not be deposited.
References
H. Mouritsen, Nature 558(7708), 50 (2018)
P.J. Hore, H. Mouritsen, Annu. Rev. Biophys. 45(May), 299 (2016)
W. Wiltschko, R. Wiltschko, Science 176(4030), 62 (1972)
W. Wiltschko, R. Wiltschko, J. Comp. Physiol. A 177, 363 (1995)
N. Lefeldt, D. Dreyer, N.L. Schneider, F. Steenken, H. Mouritsen, J. Exp. Biol. 218(2), 206 (2015)
J. Wynn, O. Padget, H. Mouritsen, C. Perrins, T. Guilford, Curr. Biol. 30(14), 2869 (2020)
I.A. Solov’yov, D.E. Chandler, K. Schulten, Biophys. J . 92(8), 2711 (2007)
I.A. Solov’yov, H. Mouritsen, K. Schulten, Biophys. J . 99(1), 40 (2010)
J. Xu, L.E. Jarocha, T. Zollitsch, M. Konowalczyk, K.B. Henbest, S. Richert, M.J. Golesworthy, J. Schmidt, V. Déjean, D.J.C. Sowood et al., Nature 594(7864), 535 (2021)
L. Spiecker, B. Leberecht, C. Langebrake, M. Laurien, S.R. Apte, H. Mouritsen, G. Gerlach, M. Liedvogel, Neuroforum 27(3), 127 (2021)
B. Leberecht, D. Kobylkov, T. Karwinkel, S. Döge, L. Burnus, S.Y. Wong, S. Apte, K. Haase, I. Musielak, R. Chetverikova et al., J. Comp. Psychol. A 208, 97 (2022)
J. Wynn, O. Padget, H. Mouritsen, J. Morford, P. Jaggers, T. Guilford, Science 375(6579), 446 (2022)
K. Schulten, C.E. Swenberg, Z. Phys. Chem. (N. F.) 111(1), 1 (1978)
T. Ritz, S. Adem, K. Schulten, Biophys. J . 78(2), 707 (2000)
B. Giovani, M. Byrdin, M. Ahmad, K. Brettel, Nat. Struct. Mol. 10(6), 489 (2003)
M. Liedvogel, K. Maeda, K. Henbest, E. Schleicher, T. Simon, C.R. Timmel, P. Hore, H. Mouritsen, PLoS ONE 2(10), e1106 (2007)
C. Nielsen, M.S. Nørby, J. Kongsted, I.A. Solov’yov, J. Phys. Chem. Lett. 9(13), 3618 (2018)
I.A. Solov’yov, T. Domratcheva, K. Schulten, Sci. Rep. 4, 3845 (2014)
H. Guo, H. Yang, T.C. Mockler, C. Lin, Science 279(5355), 1360 (1998)
A.R. Cashmore, Cell 114(5), 537 (2003)
C. Lin, T. Todo, Genome Biol. 6(5), 220 (2005)
Q. Wang, C. Lin, Annu. Rev. Plant Biol. 71(1), 103 (2020)
A. Günther, A. Einwich, E. Sjulstok, R. Feederle, P. Bolte, K.W.W. Koch, I.A. Solov’yov, H. Mouritsen, Curr. Biol. 28(2), 211 (2018)
E. Sjulstok, I.A. Solov’yov, J. Phys. Chem. Lett. 11(10), 3866 (2020)
G. Lüdemann, I.A. Solov’yov, T. Kubař, M. Elstner, J. Am. Chem. Soc. 137(3), 1147 (2015)
I..A.. Solov’yov, T. Domratcheva, A.R. Moughal Shahi, K. Schulten, J. Am. Chem. Soc. 134, 18046 (2012)
M. Hanić, F. Schuhmann, A. Frederiksen, C. Langebrake, G. Manthey, M. Liedvogel, J. Xu, H. Mouritsen, I.A. Solov’yov, J. Phys. Chem. B 126(25), 4623 (2022)
R.B. Russell, G.J. Barton, Proteins 14(2), 309 (1992)
J. Eargle, D. Wright, Z. Luthey-Schulten, Bioinformatics 22(4), 504 (2006)
W. Humphrey, A. Dalke, K. Schulten, J. Mol. Gr. 14, 33 (1996)
F. Schuhmann, D.R. Kattnig, I.A. Solov’yov, J. Phys. Chem. B 125(34), 9652 (2021)
B.D. Zoltowski, Y. Chelliah, A. Wickramaratne, L. Jarocha, N. Karki, W. Xu, H. Mouritsen, P.J. Hore, R.E. Hibbs, C.B. Green et al., Proc. Natl. Acad. Sci. U. S. A. 116(39), 19449 (2019)
L. Kalé, R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, K. Schulten, J. Comput. Phys. 151(1), 283 (1999)
J.C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R.D. Skeel, L. Kalé, K. Schulten, J. Comput. Chem. 26(16), 1781 (2005)
J.C. Phillips, D.J. Hardy, J.D.C. Maia, J.E. Stone, J.V. Ribeiro, R.C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang et al., J. Chem. Phys. 153(4), 044130 (2020)
V. Korol, P. Husen, E. Sjulstok, C. Nielsen, I. Friis, A. Frederiksen, A.B. Salo, I.A. Solov’yov, ACS Omega 5(2), 1254 (2019)
N. Foloppe, A.D. MacKerell, J. Comput. Chem. 21(2), 86 (2000)
K. Hart, N. Foloppe, C.M. Baker, E.J. Denning, L. Nilsson, A.D. MacKerell, J. Chem. Theory Comput. 8(1), 348 (2012)
J. Huang, A.D. MacKerell, J. Comput. Chem. 34(25), 2135 (2013)
M. Buck, S. Bouguet-Bonnet, R.W. Pastor, A.D. MacKerell Jr., Biophys. J . 90(4), L36 (2006)
R.B. Best, X. Zhu, J. Shim, P.E. Lopes, J. Mittal, M. Feig, A.D. MacKerell, J. Chem. Theory Comput. 8(9), 3257 (2012)
J.J. Pavelites, J. Gao, P.A. Bash, J. Comput. Chem. 18(2), 221 (1996)
E.J. Denning, U.D. Priyakumar, L. Nilsson, A.D. MacKerell, J. Comput. Chem. 32(9), 1929 (2011)
E. Sjulstok, I.A. Solov’yov, P.L. Freddolino, in New Approaches for Flavin Catalysis, edited by B.A. Palfey (Academic Press, Cambridge (US)), Vol. 620 of Methods in Enzymology, pp. 277–314 (2019)
B. Knapp, L. Ospina, C.M. Deane, J. Chem. Theory Comput. 14(12), 6127 (2018)
T. Darden, D. York, L. Pedersen, J. Chem. Phys. 98(12), 10089 (1993)
A. Frederiksen, I.A. Solov’yov, Eur. Phys. J. D 74(3), 44 (2020)
T.D.H. Bugg, Introduction to Enzyme and Coenzyme Chemistry (Wiley, Chichester, 2012), ISBN 9781118348970
H.D. Larkin MA, G. Blackshields, N.P. Brown, R. Chenna, P.A. McGettigan, H. McWilliam, F. Valentin, I.M. Wallace, A. Wilm, R. Lopez, J.D. Thompson, T.J. Gibson, Bioinformatics 23, 2947 (2007)
R. Rose, O. Golosova, D. Sukhomlinov, A. Tiunov, M. Prosperi, Bioinformatics 35(11), 1963 (2018)
R.J. Kutta, N. Archipowa, L.O. Johannissen, A.R. Jones, N.S. Scrutton, Sci. Rep. 7(1), 1 (2017)
X. Wang, C. Jing, C.P. Selby, Y.Y. Chiou, Y. Yang, W. Wu, A. Sancar, J. Wang, Cell. Mol. Life Sci. 75(24), 4629 (2018)
S. Chandrasekaran, C.M. Schneps, R. Dunleavy, C. Lin, C.C. DeOliveira, A. Ganguly, B.R. Crane, Commun. Biol. 4(1), 1 (2021)
S.M. Frahs, J.C. Reeck, K.M. Yocham, A. Frederiksen, K. Fujimoto, C.M. Scott, R.S. Beard, R.J. Brown, T.J. Lujan, I.A. Solov’yov et al., ACS Appl. Mater. Interfaces. 11(45), 41906 (2019)
Y. Wang, G. Veglia, D. Zhong, J. Gao, Sci. Adv. 7(25), 3815 (2021)
W. Humphrey, A. Dalke, K. Schulten, J. Mol. Gr. Model. 14, 33 (1996)
A. Varshney, F.P. Brooks, W.V. Wright, I.E.E.E. Comput, Gr. Appl. 14, 19 (1994)
F. Schuhmann, V. Korol, I.A. Solov’yov, J. Comp. Chem. 42(8), 572 (2021)
B.D. Zoltowski, A.T. Vaidya, D. Top, J. Widom, M.W. Young, B.R. Crane, Nature 480(7377), 396 (2011)
I.A. Solov’yov, K. Schulten, J. Phys. Chem. B 116(3), 1089 (2012)
B.D. Zoltowski, Proc. Natl. Acad. Sci. U.S.A. 112(29), 8811 (2015)
Acknowledgements
The authors would like to thank the Volkswagen Foundation (Lichtenberg professorship awarded to I.A.S.), the Deutsche Forschungsgemeinschaft (GRK1885 Molecular Basis of Sensory Biology and SFB 1372 Magnetoreception and Navigation in Vertebrates, no. 395940726), the Ministry for Science and Culture of Lower Saxony (Simulations meet experiments on the nanoscale: opening up the quantum world to artificial intelligence (SMART)). F.S acknowledges DAAD for financial support. Computational resources for the simulations were provided by the CARL Cluster at the Carl-von-Ossietzky University, Oldenburg, supported by the DFG and the Ministry for Science and Culture of Lower Saxony. This work was also supported by the North-German Supercomputing Alliance (HLRN).
Funding
Open Access funding enabled and organized by Projekt DEAL.
Author information
Authors and Affiliations
Contributions
Conceptualization was performed by MH, AF, IAS ; data curation was done by MH, AF; formal analysis was performed by MH, AF, FS; funding acquisition was performed by IAS; investigation was carried out by MH and AF; methodology was carried out by MH, AF, FS, IAS; project administration was done by IAS; resources were provided by IAS; software was developed by MH and AF; supervision was carried out by IAS; validation was performed by MH, AF, FS; visualization was carried out by MH and AF; writing—original draft—was performed by MH and AF; writing—review & editing–was done by MH, AF, FS, IAS.
Corresponding author
Ethics declarations
Conflict of interest
The authors have no conflict of interest to declare that are relevant to the content of this article.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Hanić, M., Frederiksen, A., Schuhmann, F. et al. On the energetic differences of avian cryptochromes 4 from selected species. Eur. Phys. J. D 76, 198 (2022). https://doi.org/10.1140/epjd/s10053-022-00520-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1140/epjd/s10053-022-00520-5