
Abstract
In this work, a comparative analysis of the conditions of transglycosylation reactions catalyzed by E. coli nucleoside phosphorylases was carried out, and the optimal conditions for the formation of various nucleosides were determined. Under the optimized conditions of transglycosylation reaction, fluorine-containing derivatives of N6-benzyl-2'-deoxyadenosine, potential inhibitors of replication of enteroviruses in a cell, were obtained starting from the corresponding ribonucleosides.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
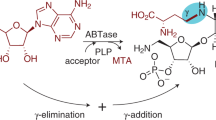
Enzymatic transglycosylation methods are widely used for obtaining drugs on the basis of nucleosides and their analogues and are based on the reaction of transferring a carbohydrate residue from one heterocyclic base to another [1–3]. Nucleoside phosphorylases (NP), which perform reversible phosphorolysis of ribonucleosides/2'-deoxyribonucleosides with the formation of the corresponding heterocyclic base and α-D-(2-deoxy)ribofuranose-1-phosphate ((d)Rib-P). The equilibrium of the phosphorolysis reaction is shifted towards the formation of nucleosides, and in the case of purines it is more significant [1–6], which makes it possible to use two coupled reactions of phosphorolysis, a donor nucleoside and a nucleoside containing a heterocyclic base-acceptor, to perform an enzymatic transglycosylation reaction, during which a carbohydrate residue is transferred from a pyrimidine or purine nucleoside donor to a purine heterocyclic base acceptor (Fig. 1). This general scheme makes it possible to obtain new modified nucleosides depending on the set of used starting compounds and the substrate specificity of NP.

Enzymatic transglycosylation. Designations: NP, nucleoside phosphorylase; Nuc-1, nucleoside donor; Nuc-2, product; B1 and B2, heterocyclic bases; Pi, phosphate anion, X = H or OH. Reaction conditions: NP—E. coli PNP, E. coli UP, E. coli TP, 50 mM Tris-HCl (pH 7.5), 20°C. Compiled from [6].
Earlier, approaches to optimizing the transglycosylation reaction using 7-methyl-2'-deoxyguanosine as the starting substrate for the production of α-D-2-deoxyribose-1-phosphate (dRib-1-P), 5-substituted derivatives of 2'-deoxyuridine, cladribine, and allopurinol-riboside [5, 7], were studied in the Laboratory of Design and Synthesis of Biologically Active Compounds (DSBAC) of the Engelhard Institute of Molecular Biology, Russian Academy of Sciences. A mathematical model of the transglycosylation process was proposed, which can be used to quantify the effect of initial conditions on the result of transglycosylation [8]. This work is a continuation of earlier studies aimed at expanding knowledge about the substrate specificity of NP and obtaining modified analogues of natural nucleosides by enzymatic transglycosylation.
DISCUSSION
In this work, a comparative analysis of the transglycosylation reaction involving purine (PNP) and pyrimidine (UP, TP) E. coli nucleoside phosphorylases was performed (Table 1). The selection of enzymes of bacterial origin as catalysts was determined by their wide substrate specificity, pH optimum in neutral/weakly alkaline media, and a fairly wide operating temperature range, which allows the reaction to be carried out under mild conditions without noticeable nonspecific cleavage of the N-glycosidic bond with yields of target products close to the theoretically predicted ones [8–13].
According to Fig. 1, the transglycosylation reaction proceeds through the formation of α-D-ribose-1-phosphate (Rib-1-P) or α-D-2-deoxyribose-1-phosphate (dRib-1-P). The synthesis of purine nucleosides from pyrimidine nucleosides and vice versa requires the participation of two enzymes: PNP and UP (or TP). The equilibrium constants of phosphorolysis of natural pyrimidine nucleosides in the presence of UP and TP are higher than the equilibrium constants of phosphorolysis of natural purine nucleosides in the presence of PNP [2, 5, 8, 11, 14]. Therefore, it is more reasonable to use Rib-1-P and dRib-1-P (or pyrimidine, but not purine, nucleosides) as donors [8, 15, 16].
This expediency is well confirmed by the experimental data presented in Table 1.
When adenosine was obtained from uridine, the reaction proceeded with a high yield, which was determined as the ratio of the equilibrium concentration of the product to the initial concentration of the starting base or glycosyl donor, depending on what was taken in deficiency (Table 1, lines 1–3, 77–94% according to HPLC). When uridine was obtained from adenosine, the reaction yield was significantly reduced (Table 1, lines 4–6, 22–27% according to HPLC). In the series of purine nucleosides, inosine was a more productive glycosyl donor than adenosine; therefore, the reaction for obtaining adenosine from inosine proceeded with a higher yield (Table 1, line 8, 76% according to HPLC data) than the reverse reaction (Table 1, line 7, 53% by HPLC). With an increase in the amount of the donor nucleoside in the reaction mixture, it is naturally possible to increase the yield of the target nucleoside (and, accordingly, the conversion of the base); however, this also increases the number of unreacted components, which increases the complexity of the subsequent processing of the reaction mixture and further purification (Table 1, lines 3, 6, 13, 15).
The transglycosylation reaction can be simplified in two ways:
(1) exclusion of the stage of phosphorolysis of the nucleoside that serves as a glycosyl donor in the transglycosylation reaction (Fig. 1, stage 1) by introducing a ready-to-use Rib-1-P or dRib-1-P into the reaction [7];
(2) transformation of stage 1 into an irreversible one using 7-methyl-(2'-deoxy)guanosine (7-Me(d)Guo) as a source of ribose residue due to its almost irreversible phosphorolysis [17, 18].
The use of (d)Rib-1-P reduces the number of components in the reaction mixture, facilitates the isolation of target compounds, and makes it possible to significantly shift the equilibrium of the glycosylation reaction towards the formation of nucleosides (Table 1, line 14, 98% according to HPLC; the yield was calculated from the reagent taken in deficiency (Rib-1-P)). The replacement of (d)Rib-1-P with the more accessible 7-Me(d)Guo leads to comparable yields of the reaction products (Table 1).
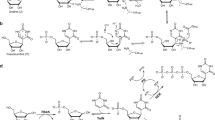
E. coli PNP was used to prepare less available deoxyribonucleosides from commercially available ribonucleosides, N6-pentafluorophenylmethyl-2'-deoxyadenosine (PFPh-dAdo, 5b) and N6-(3-trifluoromethylbenzyl)-2'-deoxyadenosine (TFMBn-dAdo, 5a) (Fig. 2), potential inhibitors of enterovirus replication in the cell [19].

Synthesis of fluoro substituted 2'-deoxyribonucleosides from ribonucleosides. Reagents and conditions: (1) 1a–1b (0.212–0.235 mmol), E. coli PNP (0.25 units), KH2AsO4 (0.212–0.235 mmol), 50 mM Tris-HCl buffer (pH 7.5, 10 mL), 50°C, 20 h—2a (91%), 2b (85%). (2–3) 2a (20 mg, 0.068 mmol), 2b (20 mg, 0.064 mmol), 3 (0.102 mmol), E. coli PNP (0.98 U), КH2PO4 (0.064–0.068 mmol), 50 mM Tris-HCl (pH 7.5, 63 mL)—DMSO (10%, 7 mL), 20°C, 24 h—86% (5a), 47% (5b).
The synthesis method consisted of three separate stages, each stage was catalyzed by E. coli PNP. The starting bases N6-(3-trifluoromethylbenzyl)adenine (TFMBn-Ade, 2a) and N6-pentafluorophenylmethyladenine (PFPh-Ade, 2b) were obtained from ribonucleosides under the conditions of enzymatic arsenolysis (Fig. 2, stage 1). Enzymatic arsenolysis is based on the cleavage of ribonucleoside in the presence of potassium dihydroorthoarsenate (KH2AsO4) into a purine base and the highly labile α-D-ribofuranose-1-arsenate (Rib-1-As), which is irreversibly hydrolyzed; this shifts the equilibrium of ribonucleoside cleavage towards the formation of a base [20]. The poor solubility of heterocyclic bases 2a and 2b in water and in Tris-HCl buffer also leads to a shift in equilibrium towards the formation of products. To prevent the formation of a mixture of ribo- and deoxyribonucleosides during further stages, stage 1 should be performed in a separate flask. Then, bases 2a and 2b were filtered off and introduced into the transglycosylation reaction with 7-Me-dGuo in the presence of potassium dihydroorthophosphate and E. coli PNP (Fig. 2, stages 2–3). In the reaction mixture, 7-Me-dGuo was converted into dRib-1-P (Fig. 2, stage 2), which then reacted with a fluorine-containing base (Fig. 2, stage 3). Stages 2 and 3 were performed in the same flask. To increase the solubility of the base, the reaction was carried out in a buffer solution with the addition of 10 vol % dimethyl sulfoxide.
The concentration of dimethyl sulfoxide in the reaction mixture did not significantly affect the enzymatic activity of PNP, which is consistent with the literature data [18]. The transglycosylation reaction was carried out at different glycosyl donor : base : phosphate ratios (Table 1). Carrying out the reaction with a slight excess of the glycosyl donor in the presence of an equimolar amount of phosphate (1.5 : 1 : 1) or its deficiency (1.5 : 1 : 0.5, 1.5 : 1 : 0.25) led to high yields of target nucleoside products with a slight decrease in the reaction rate compared to the reaction rate in the presence of equimolar amounts of phosphate. An increase in the amount of phosphate in the reaction mixture (starting from an equimolar amount and above) leads to an increase in the rate of formation of dRib-1-P, the hydrolysis of which can reduce the yields of target nucleosides (Table 1, lines 16–17). Therefore, the highest yields were achieved at a glycosyl donor : base : phosphate ratio of 1.5 : 1 : 0.25 (Table 2). The yield of the preparative method for obtaining the 5a product was 100% by HPLC (86% after purification by reverse-phase chromatography on silica gel-C18), and the yield of the 5b product was 92% by HPLC (47% after similar purification, the low yield was due to the sorption of the compound on silica gel-C18).
The structure of the obtained compounds was confirmed by NMR spectroscopy. The 13С-NMR spectrum of the trifluoromethyl-substituted deoxynucleoside 5а contained a resonance signal of the trifluoromethyl group in the form of a low-intensity quartet with a spin–spin coupling constant (SSCC) 1JC–F = 31 Hz. The 13С-NMR spectrum of the pentafluoro-substituted deoxynucleoside 5b contained resonance signals of the 13С nuclei of the phenyl group in the form of three wide doublets with SSCC 1JC–F of approximately 250 Hz. In the 19F-NMR spectrum, the signal of the trifluoromethyl group of compound 5а was resolved as a singlet with a chemical shift δ = 61.04 ppm. The 19F-NMR spectrum of the pentafluoro-substituted nucleoside 5b shows a complex spin–spin interaction: a doublet of doublets for the 19F nuclei in the ortho position of the phenyl substituent with SSCC 3JF–F = 22 Hz, 4JF–F = 6 Hz, a triplet of doublets for the 19F nuclei in the meta position with SSCC 3JF–F = 22 Hz, 4JF–F = 6 Hz and a triplet for the 19F nuclei in the para position of the phenyl ring with SSCC 3JF–F = 22 Hz. The presence of a pentafluoro-substituted fragment in the 5b structure was also confirmed by the 1H-NMR spectrum, in which no resonance signals of the phenyl group protons were observed.
The antiviral activity of the obtained compounds is currently being studied.
EXPERIMENTAL
Commercially available reagents and solvents were used to carry out reactions and isolate compounds. The course of reactions was monitored by HPLC analysis on a Styer-M instrument (Akvilon, Russia). HPLC analysis conditions: column 4.6 × 250 mm (Nucleosil 100-5 C18, 5 µm, Macherey-Nagel GmbH&Co. KG), linear gradient of acetonitrile in 10 mM sodium acetate solution in deionized water from 2 to 60% over 25 min (with further washing in the system 60–80% acetonitrile/10 mM NaOAc in H2O for 25–25.1 min and then 80–2% for 25.1–25.9 min) at a flow rate of 1 mL/min. UV detection was performed at 265 nm, sample volume was 20 µL. NMR spectra were recorded on a Bruker AMX 400 and Bruker AMX 300 instruments (Germany). The values of the spin–spin coupling constants (SSCC, J) are measured in hertz (Hz). When describing NMR spectra, the following abbreviations are used: s—singlet, br s—broadened singlet, d—doublet, dd—doublet of doublets, ddd—doublet of doublets of doublets, t—triplet, dt—doublet of triplets, m—multiplet. The 1Н- and 13С-NMR spectra were calibrated using the residual signal of the solvent DMSO-d6 (2.50 and 39.52 ppm, respectively).
Preparation of N6-(2,3,4,5,6-pentafluorobenzyl)adenine (2b)
A mixture of N6-(2,3,4,5,6-pentafluorobenzyl)adenosine 1b (95 mg, 0.212 mmol) and KH2AsO4 (38 mg, 0.212 mmol, 1 equiv.) in 50 mM Tris-HCl buffer (pH 7.5, 10 mL) was supplemented with PNP (0.050 mL, 5 activity units), and the mixture was incubated at 50°C for 20 h. During the reaction, the initial nucleoside was dissolved and the product was crystallized. After 20 h of incubation, the reaction mixture was cooled to room temperature and incubated for 24 h at 4°C. The formed precipitate was filtered off, washed with water (5 × 5 mL), and dried in a vacuum desiccator over P2O5 for 1 day. Yield 57 mg (85%) as white powder. Rf = 0.4 (CH2Cl2 : EtOH-95 : 5 \({\text{v/v}}\)). 1H–NMR (300 МHz, DMSO-d6): δ = 12.96 (br s, 1H, N9H), 8.21 (s, 1H, H2-Ade), 8.15 (br s, 1H, N6H), 8.11 (s, 1H, H8-Ade), 4.83 (br s, 1H, N6CH2). 19F-NMR (282 МHz, DМSO-d6): δ = –142.50 (dd, 3JF–F = 24.0 Hz, 4JF–F = 7.9 Hz), –156.85 (t, 3JC–F = 22.1), –163.68 (td, 3JF–F = 23.1 Hz, 4JF–F = 7.8 Hz).
Preparation of N6-(3-trifluoromethylbenzyl)adenine (2a)
The procedure is similar to the preparation of N6-(2,3,4,5,6-pentafluorobenzyl)adenine (2b) from N6-(3-trifluoromethylbenzyl)adenosine (1a) (100 mg, 0.235 mmol). Yield 2a 63 mg (91%) as white powder. Rf = 0.42 (CH2Cl2 : EtOH – 95 : 5 \({\text{v/v}}\)). 1H–NMR (300 МHz, DMSO-d6): δ = 12.95 (br s, 1H, N9H), 8.29 (br s, 1H, N6H), 8.17 (s, 1H, H2–Ade), 8.12 (s, 1H, H8-Ade), 8.0–7.4 (м, 4H, Ph), 4.81 (br s, 1H, N6CH2). 19F-NMR (282 МHz, DМSO-d6): δ = –61.00.
Preparation of N6-(3-trifluoromethylbenzyl)-2'-deoxyadenosine (5a)
Solution (70 mL) containing 7-methyl-2'-deoxyguanosine (41.82 mg, 0.102 mmol), meta-trifluoromethylbenzyladenine 2a (20.00 mg, 0.068 mmol), and potassium dihydrogen phosphate (2.32 mg, 0.017 mmol) in 50 mM Tris-HCl buffer (pH 7.5) with the addition of 10 vol % DMSO was supplemented with 0.98 units of E. coli PNP (10 µL of 1.0 mg/mL Sigma solution at a concentration of 98 units/mL) at room temperature. The mixture was gently stirred for 5 min and left at room temperature for 24 h. The precipitate of 7-methylguanine was filtered off through a Phenomenex nylon membrane (diameter 47 mm, pore size 0.2 µm). The filtered clear solution was evaporated in vacuo to a volume of ~7 mL, diluted with water (7 mL), and applied onto a column with a reverse-phase sorbent C18. Yield: 99% (HPLC, quantitative). The column was washed with a water : ethanol mixture (ethanol concentration gradient of 0–20%). The product was eluted in the water : ethanol mixture 60 : 40. The fractions containing the product were pooled, evaporated in vacuo, and coevaporated with ethanol. Yield after isolation and purification was 26 mg (86%) as a foam. 1H-NMR (400 МHz, DMSO-d6): δ = 8.51 (br s, 1H, N6H-Ade), 8.38 (s, 1H, H2-Ade), 8.21 (s, 1H, H8-Ade), 7.71 s (1H, о-Н, Ph), 7.65 d (1Н, 3J = 7.2, p-H, Ph), 7.60–7.49 m (2H, Ph), 6.36 dd (1H, J1'2'a= 7.6, J1'2'b = 6.2, H-1'), 5.32 br s (1H, 3'-OH), 5.19 br s (1H, 5'-OH), 4.79 br s (2H, CH2), 4.46–4.38 m (1H, H-3'), 3.89 td (1Н, J4'5'а = J4'3' = 4.2, J4'5'b = 2.6, H-4'), 3.62 br d (1Н, J5'а5'b = –11.7, H-5'a), 3.52 br d (1Н, J5'b5'a = –11.7, H-5'b), 2.73 ddd (1Н, J2'а2'b= –13.3, J2'а1'= 7.6, J2'b3' = 5.9, H-2'а), 2.27 ddd (1Н, J2'b2'a= –13.3, J2'b1'= 6.2, J2'b3' = 2.6, H-2'b). 13C-NMR (150 МHz, DMSO-d6): 154.37 (C-2), 152.32 (C-4), 141.59 (C-6), 139.74 (C-8), 131.33 (Ph), 129.29 (Ph), 128.90 q (1JC–F = 31.4, CF3), 123.65 q (3JC–F = 3.8, Ph), 123.43 q (3JC–F = 3.7, Ph), 119.68 (C-5), 88.03 (C-1'), 83.97 (C-3'), 70.94 (C-4'), 61.87 (C-5'), 42.62 (CH2), 39.47 (C-2'). 19F-NMR (282 МHz, DМSO-d6): δ = –61.04.
Preparation of N6-(2,3,4,5,6-pentafluorophenyl-1-methyl)-2'-deoxyadenosine (5b)
The procedure is similar to the previous one, starting from N6-(2,3,4,5,6-pentafluorobenzyl)adenine 2b (20.00 mg, 0.064 mmol). Purification was carried out on a column with a reversed-phase sorbent C18 using a water : ethanol mixture (ethanol concentration gradient of 0–20%) as a mobile phase. Yield 13 mg (47%) as white silver foam. 1H-NMR (400 МHz, DMSO-d6): δ = 8.37 (s, 1H, H2-Ade), 8.35 (br s, 1H, N6H-Ade), 8.25 (s, 1H, H8-Ade), 6.35 dd (1H, J1'2'a= 7.6, J1'2'b = 6.2, H-1'), 5.30 br s (1H, 3'-OH), 5.14 br t (1H, J5'OH = 4.2, 5'-OH), 4.82 br s (2H, CH2), 4.45–4.37 m (1H, H-3'), 3.87 td (1Н, J4'5'а = J4'3' = 4.3, J4'5'b = 2.8, H-4'), 3.62 br d (1Н, J5'а5'b = –11.7, H-5'a), 3.55–3.45 m (1Н, H-5'b), 2.72 ddd (1Н, J2'а2'b= –13.3, J2'а1'= 7.7, J2'b3' = 5.8, H-2'а), 2.27 ddd (1Н, J2'b2'a= –13.3, J2'b1'= 6.1, J2'b3' = 2.8, H-2'b). 13C-NMR (150 МHz, DMSO-d6): 153.85 (C-2), 152.14 (C-4), 148.57 (C-6), 145.10 dddd (1JC–F = 245.8, 2JC–F = 13.2, 2JC–F = 9.1, 3JC–F = 3.9, ortho-С6F5), 139.79 (C-8), 139.68 dddd (1JC–F = 250.5, 2JC–F = 13.7, 2JC–F = 12.4, 3JC–F = 5.6, meta-С6F5), 136.69 dtt (1JC–F = 248.8, 2JC–F = 12.6, 3JC–F = 3.9, para-С6F5), 119.65 (C-5), 113.01 t (2JC–F = 17.6, C-1-C6F5), 87.98 (C-1'), 83.88 (C-3'), 70.88 (C-4'), 61.81 (C-5'), 39.42 (C-2'), 32.22 (CH2). 19F-NMR (282 МHz, DМSO-d6): δ = –142.50 dd (3JF–F = 22, 4JF–F = 6), –156.74 t (3JF–F = 22), –163.65 td (3JF–F = 22, 4JF–F = 6).
CONCLUSIONS
In the study, a comparative analysis of the conditions of transglycosylation in the presence of E. coli nucleoside phosphorylases was carried out, which made it possible to optimize the conditions for the enzymatic synthesis of N6-pentafluorophenylmethyl-2'-deoxyadenosine and N6-(3-trifluoromethylbenzyl)-2'-deoxyadenosine, potential nucleoside inhibitors of enterovirus replication in cells.
REFERENCES
Mikhailopulo, I.A. and Miroshnikov, A.I., Biologically important nucleosides: modern trends in biotechnology and application, Mendeleev Commun., 2011, vol. 21, no. 2, pp. 57–68.
Mikhailopulo, I.A. and Miroshnikov, A.I., New trends in nucleoside biotechnology, Acta Naturae, 2010, vol. 2.2, no. 5, pp. 36–56.
Iglesias, L.E., Lewkowicz, E.S., Medici, R., Bianchi, P., and Iribarren, A.M., Biocatalytic approaches applied to the synthesis of nucleoside prodrugs, Biotechnol. Adv., 2015, vol. 33, no. 5, pp. 412–434.
Parker, W.B., Enzymology of purine and pyrimidine antimetabolites used in the treatment of cancer, Chem. Rev., 2009, vol. 109, no. 7, pp. 2880–2893.
Goldberg, R.N., Tewari, Y.B., and Bhat, T.N., Thermodynamics of enzyme-catalyzed reactions—a database for quantitative biochemistry, Bioinformatics, 2004, vol. 20, pp. 2874–2877.
Drenichev, M.S., Alexeev, C.S., Kurochkin, N.N., et al., Use of nucleoside phosphorylases for the preparation of purine and pyrimidine 2'-deoxynucleosides, Adv. Synth. Catal., 2018, vol. 360, pp. 305–312.
Kulikova, I.V., Drenichev, M.S., Solyev, P.N., Alexeev, C.S., and Mikhailov, S.N., Enzymatic synthesis of 2-deoxyribose 1-phosphate and ribose 1 phosphate and subsequent preparation of nucleosides, Eur. J. Org. Chem., 2019, pp. 6999–7004.
Alexeev, C.S., Kulikova, I.V., Gavryushov, S.A., et al., Quantitative prediction of yield in transglycosylation reaction catalyzed by nucleoside phosphorylases, Adv. Synth. Catal., 2018, vol. 360, pp. 3090–3096.
Ubiali, D., Rocchietti, S., Scaramozzino, F., Terreni, M., Albertini, A.M., Fernandez-Lafuente, R., Guisa, J.M., and Pregnolato, M., Synthesis of 2'-deoxynucleosides by transglycosylation with new immobilized and stabilized uridine phosphorylase and purine nucleoside phosphorylase, Adv. Synth. Catal., 2004, vol. 346, pp. 1361–1366.
Gordon, G.E.R., Visser, D.F., Brady, D., Raseroka, N., Moira, L., and Bode, M.L., Defining a process operating window for the synthesis of 5-methyluridine by transglycosylation of guanosine and thymine, J. Biotechnol., 2011, vol. 151, pp. 108–113.
Serra, I., Bavaro, T., Cecchinia, D.A., Daly, S., Albertini, A.M., Terrenia, M., and Ubiali, D., A comparison between immobilized pyrimidine nucleoside phosphorylase from bacillus subtilis and thymidine phosphorylase from Escherichia coli in the synthesis of 5-substituted pyrimidine 2-deoxyribonucleosides, J. Mol. Catal. B: Enzym., 2013, vol. 95, pp. 16–22.
Cattaneo, G., Rabuffetti, M., Speranza, G., Kupfer, T., Peters, B., Massolini, G., Ubiali, D., and Calleri, E., Synthesis of adenine nucleosides by transglycosylation using two sequential nucleoside phosphorylase-based bioreactors with on-line reaction monitoring by using HPLC, ChemCatChem, 2017, vol. 9, pp. 4614–4620.
Bzowska, A., Kulikowska, E., and Shugar, D., Properties of purine nucleoside phosphorylase (PNP) of mammalian and bacterial origin, Z. Naturforsch, C: J. Biosci., 1990, vol. 45, pp. 59–70.
Alexeev, C.S., Drenichev, M.S., Dorinova, E.O., Esipov, R.S., Kulikova, I.V., and Mikhailov, S.N., Use of nucleoside phosphorylases for the preparation of 5-modified pyrimidine ribonucleosides, Biochim. Biophys. Acta, Proteins Proteomics, 2020, vol. 1868, art. 140292.
Roivainen, J., Elizarova, T., Lapinjoki, S., Mikhailopulo, I.A., Esipov, R.S., and Miroshnikov, A.I., An enzymatic transglycosylation of purine bases, Nucleosides, Nucleotides Nucleic Acids, 2007, pp. 905–909.
Zuffi, G., Ghisotti, D., Oliva, I., Capra, E., Frascotti, G., Tonon, G., and Orsini, G., Immobilized biocatalysts for the production of nucleosides and nucleoside analogues by enzymatic transglycosylation reactions, Biocatal. Biotransform., 2004, vol. 22, pp. 25–33.
Kulikowska, E., Bzowska, A., Wierzchowski, J., and Shugar, D., Properties of two unusual, and fluorescent, substrates of purine-nucleoside phosphorylase: 7-methylguanosine and 7-methylinosine, Biochim. Biophys. Acta, 1986, vol. 874, pp. 355–363.
Rabuffetti, M., Bavaro, T., Semproli, R., Cattaneo, G., Massone, M., Morelli, C.F., Speranza, G., and Ubi-ali, D., Synthesis of ribavirin, tecadenoson, and cladribine by enzymatic transglycosylation, Catalysts, 2019, vol. 9, p. 355.
Oslovsky, V.E., Drenichev, M.S., Sun, L., Kurochkin, N.N., Kunetsky, V.E., Mirabelli, C., Neyts, J., Leyssen, P., and Mikhailov, S.N., Fluorination of naturally occurring N 6-benzyladenosine remarkably increased its antiviral activity and selectivity, Molecules, 2017, vol. 22, no. 7, p. 1219.
Kline, P.C. and Schramm, V.L., Purine nucleoside phosphorylase. Catalytic mechanism and transition-state analysis of the arsenolysis reaction, Biochemistry, 1993, vol. 32, no. 48, pp. 13212–13219.
Funding
This study was supported by the Russian Science Foundation (project no. 21-14-00346).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare that they have no conflicts of interest. This article does not contain any studies involving animals or human participants performed by any of the authors.
Additional information
Translated by M. Batrukova
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Drenichev, M.S., Dorinova, E.O., Varizhuk, I.V. et al. Synthesis of Fluorine-Containing Analogues of Purine Deoxynucleosides: Optimization of Enzymatic Transglycosylation Conditions. Dokl Biochem Biophys 503, 52–58 (2022). https://doi.org/10.1134/S1607672922020053
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1607672922020053