
Abstract—
The influence of the structure of fluoroquinolones (on the example of ciprofloxacin and levofloxacin) and their complexation with methyl-β-cyclodextrin on the interaction of the drug with human serum albumin was studied. It was found that the binding of the drug molecule with albumin is significantly affected by the structure of fluoroquinolone, as well as the presence of methyl-β-cyclodextrin. It was discovered that of the two fluoroquinolones, the more hydrophobic ciprofloxacin molecule interacts more strongly with the protein, using circular dichroism and fluorescence spectroscopy methods. It has also been shown that binding of albumin to the drug causes quenching of protein fluorescence, and this effect is more pronounced for ciprofloxacin. The complexation of fluoroquinolones with methyl-β-cyclodextrin leads to a change in the interaction of fluoroquinolones with the protein: in the case of complexes, more pronounced interactions are observed for levofloxacin. The results obtained will help to bring the use of fluoroquinolones to a new level in clinical practice, by creating new highly effective drugs with improved properties.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
INTRODUCTION
Fluoroquinolones (FQs) are one of the largest classes of antibacterial drugs [1], widely used in medicine [2]. FQs have a number of important physicochemical properties of an antibacterial drug: 1) a wide spectrum of activity, which includes gram-negative and gram-positive aerobic and anaerobic bacteria, as well as mycobacteria; 2) chemical and biological stability; 3) a variety of methods of introduction into the body [3].
The action of FQs is based on the selective inhibition of DNA gyrase (type II topoisomerase) and topoisomerase IV, which allows blocking the replication of microorganism DNA, while the DNA of mammalian cells is not affected due to the high specificity of FQs to bacterial enzymes. Blocking DNA gyrase leads to the termination of the division of bacterial cells and their death [4]. Despite the high efficiency of FQs, long-term therapy and high dosages of the drug can lead to a number of side effects, the reduction of the likelihood of which can be achieved by creating highly efficient delivery systems. A promising variant of such systems can be complexes of FQs with cyclodextrins (CDs) [5].

Whey structure human albumin and ligand binding sites [13].
CDs and their derivatives are of great interest to the pharmaceutical industry. They help to increase the solubility [6, 7], bioavailability and stability of drugs, reduce toxicity, and allow varying the pharmacokinetic properties of biologically active molecules [1, 8]. Despite the widespread use of drug formulations based on CDs, the interaction of complexes with biological substances requires more detailed consideration. For the successful use of FQs and their complexes with CDs in medical practice, it is necessary to study their interaction with blood plasma proteins, with which the formulations inevitably interact on the way to the focus of infection when administered intravenously into the body. Most drugs circulate in the bloodstream, binding reversibly to plasma proteins. The binding of drugs to other plasma proteins (e.g., high- and low-density lipoproteins) occurs to a much lesser extent [9].
Human serum albumin (HSA) is the main protein in blood plasma. HSA is a globular multifunctional protein consisting of three fragments: domains I (1–195 aa), II (196–383 aa), and III (384-585 aa) in Fig. 1. They are topologically identical and have a similar tertiary structure [10, 11], but each fragment provides specific structural and functional characteristics [12].
HSA binds and transfers various substances such as hormones, fatty acids, drug molecules, etc. [12]. Basically, it binds to acidic and neutral, anionic drugs [9, 13]. At the moment, seven binding sites are known in the structure of HSA, of which two main ones can be distinguished, in which drug molecules bind: Sudlow 1 in subdomain IIA and Sudlow 2 in subdomain IIIA [13, 14]. As a rule, each drug is characterized by one or two sites of binding to HSA: for example, acetylsalicylic acid (aspirin) is almost evenly distributed between subdomains IIA and IIIA [15]. FQs are known to bind to HSA in the IIA subdomain, near the chloroform binding region [12]. Various spectral methods are used to study the binding site of drug molecules with HSA, for example, fluorescence spectroscopy using special labels such as dansyl-L-asparagine, dansyl-L-arginine, and dansyl-L-glutamate [12].
HSA ensures the transfer of drug molecules to the kidneys, intestines, liver, and other organs [10]. For many drugs, changes in pharmacokinetic and pharmacodynamic properties, as well as biodistribution when bound to HSA, have been described [9].
Despite the fact that the interaction of HSA with FQs is actively studied, the influence of the structural features of FQs, and especially their FQ–CD complexes, on the interaction of a drug with HSA is a relevant and pioneering area for research.
The aim of this work is to study the effect of methyl‑β‑cyclodextrin (M‑β‑CD), which is widely used in biomedicine, for the binding of HSA to FQs (ciprofloxacin and levofloxacin), which have different architectures. For the analysis of these interactions, a complex of spectroscopic methods was used, which make it possible to determine the main binding parameters. In addition, it is important to study how HSA binds to the FQ at different FQ : protein molar ratios. One of the ways of using the drug is intravenous administration, while the dosage of the drug is ~200–400 mg, the maximum concentration in the blood reaches values of ~2.8–6.7 mg/L [16]. The normal level of HSA in an adult is 35–50 g/L. Consequently, the molar ratio of FQ to HSA after complete distribution is from 1 : 25 to 1 : 40. However, since the drug is administered locally, this ratio can change dramatically, and it is also necessary to consider large excesses of FQ in relation to HSA, in which the mechanism of drug–protein interaction may be different.
RESULTS AND DISCUSSION
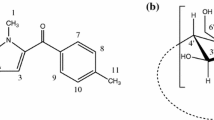
There are several fundamental differences in the structural organization of levofloxacin (LF) and ciprofloxacin (CF) molecules, which cause differences in chemical properties (Fig. 2). First, the drug possesses an additional heterocycle containing an oxygen atom, the lone electron pair of which is capable of conjugating with the entire aromatic system of the molecule, thereby increasing the delocalization of the π-electron density and, as a consequence, the hydrophilicity of the molecule. Secondly, nitrogen located in para-position to the fluorine atom, in the structure of the drug it has an environment fixed by the cycle, which prevents its inversion and increases the likelihood of the involvement of the lone electron pair in the conjugation of the aromatic backbone, which is similarly reflected in the hydrophilicity of the FQ. It should be noted that more hydrophobic drugs have a greater affinity for HSA [9]; therefore, one should expect that CF will interact more strongly with albumin. It is also important to emphasize that under conditions close to physiological (0.01-M sodium phosphate buffer solution, pH 7.4), FQs are in the solution in the form of zwitterions, because have functional groups at opposite ends of the molecules that depend on the pH of the medium [17, 18].

Structural formulas of fluoroquinolones: levofloxacin and ciprofloxacin (at pH 7.4) [21].
Complexation between FQ and CD occurs due to the immersion of the aromatic backbone of the drug molecule into the CD cavity, while the piperazine fragment, positively charged at pH 7.4, remains outside [6], and the carboxyl group becomes less accessible for interaction with other ligands. In the complexation of FQ with CD, the morpholine fragment in the drug structure promotes more complete immersion of the drug molecule into the CD cavity than in the case of CF. In this case, the anionic group of the drug is outside the host molecule. For this reason, the carboxyl groups in FQ molecules are screened in different ways. This fact can have a significant effect on the binding of FQ–M complexes‑β‑CD with HSA, because it is acidic, anionic drugs that have a greater affinity for HSA [9, 12, 13].
Interaction of HSA with fluoroquinolones and their complexes with methyl-β-cyclodextrins. To study the interaction of HSA with drugs, solutions of complexes of FQs with protein (two-component systems), as well as FQ-CD with HSA (three-component systems) were studied by fluorescence spectroscopy. This method has found wide application in the study of biological systems, because it has high sensitivity, makes it possible to study several substances in one system, allows you to work with living cells, and has a high response rate and low consumption of reagents [19].
The phenomenon of protein fluorescence quenching can be used to study the binding of a drug molecule to albumin. In this work, the processes of interaction of FQ and their complexes with M‑β‑CD (in the molar ratio of FC : M‑β‑CD 1 : 1) with HSA.
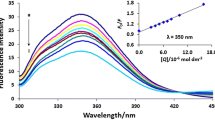
The fluorescence emission spectra of FQs at an excitation wavelength of 280 nm exhibit pronounced peaks with intensity maxima at 456 and 420 nm for drug and CF, respectively (Fig. 3a). The given spectra of fluorescence emission are due to the presence of aromatic fragments in the structure of medicinal molecules. For drug emission maxima (λmax = 456 nm) and CF (λmax = 420 nm), calibration curves were obtained (Fig. 3b) [20]. It is important to note that concentration quenching of fluorescence is observed at FQ concentrations >50 μM.

(a) Fluorescence emission spectrum of HSA (1), ciprofloxacin (2) and levofloxacin (3) at concentrations of 20, 10, and 10 μM, respectively (pH 7.4); (b) calibration dependences for levofloxacin (1) and ciprofloxacin (2).
The fluorescence emission spectrum of HSA has a pronounced maximum intensity at a wavelength of 345 nm, due to the presence of aromatic amino acid residues (Trp, Tyr) in the protein. Since the emission spectra of FQ and HSA are almost completely resolved, the states of the analyzed substances can be simultaneously studied in a two-component system.
In order to study the complexation of FQ with HSA, fluorescence emission spectra of solutions containing the substances under study were obtained in a wide range of ratios, from 1 : 0.5 to 1 : 20 (Fig. 4a). In the presence of antibacterial drugs, a decrease in the intensity of the albumin fluorescence spectrum is observed, which is probably due to changes in the microenvironment of aromatic protein residues due to the interaction of HSA with FQ. Almost complete disappearance of the emission peak of albumin fluorescence occurs at a molar excess of CF equal to 1, for drugs the maximum quenching is observed at a molar excess of 5 (Fig. 4b) [20]. Since more significant changes are inherent in the more hydrophobic drug, CF, there is reason to believe that the main mechanism of the interaction of HSA with FQ is hydrophobic interactions. This is consistent with the literature data that the two main albumin binding sites IIA and IIIA have hydrophobic cavities that are preferred for interaction with hydrophobic molecules. For example, Ghuman et al. [21] studied the binding of oxyphenbutazone, phenylbutazone, and warfarin to HSA and found that drug molecules occupy the center of the hydrophobic cavity at the IIA binding site.

(a) Fluorescence spectra of the HSA–levofloxacin system at a molar excess of the drug equal to 0.5 (1), 1 (2) and 5 (3); (b) change in the intensity of the maximum of the fluorescence peak of HSA in the presence of levofloxacin (1) and ciprofloxacin (2), 0.01-M sodium phosphate buffer solution (pH 7.4); (c) Stern–Volmer dependences expressing the effect of quenching the emission of HSA F0/F (345 nm) on the concentration of ciprofloxacin (1), levofloxacin (2), levofloxacin complex (3) and ciprofloxacin (4) with methyl-β-cyclodextrin (ratio 1 : 1); (d) fluorescence emission spectrum of the HSA–methyl-β-cyclodextrin system (20 and 200 μM, respectively) (1) and HSA (20 μM, pH 7.4) (2).
It is important to note that the position of the HSA maximum is shifted to longer wavelengths by 10 nm in the presence of FQ. This may indicate that the binding of the FQ in the protein leads to conformational changes in the spatial structure of albumin and the formation of a more hydrophilic environment of its surface [22], which indicates the presence of electrostatic interactions between the charged HSA surface and FQ molecules present in the solution in the form of zwitterions. This process of interaction with the protein prevails in the case of drugs, the hydrophilic properties of which are higher than that of CF.
The dependence of the quenching of the fluorescence emission of HSA on the FQ concentration can be described using the Stern–Volmer plots, which express the dependence F0/F on the concentration of the extinguisher (Fig. 4c). These equations provide valuable information on the quantitative relationship between the quenching effect of protein emission and the concentration of the quencher present. Since the Stern–Volmer constant for the CF ((2.9 ± 0.2) × 105) is greater than for the LF ((2.2 ± 0.2) × 105), CF is a more effective complexing agent for HSA in comparison with drugs.
Complexation M‑β‑CD with FQ can significantly affect the efficiency of drug–protein binding by screening a part of the drug molecule; in addition, in the case of a complex, the FQ-binding site in albumin can change due to a change in the polarity of the ligand molecule. In Yan et al. [23] it was shown that free M‑β‑CD, in contrast to FQ, slightly increases the fluorescence intensity of HSA. In an independent experiment, it was found that M‑β‑CD has almost no effect on the fluorescence emission spectrum of HSA (Fig. 4d). Therefore, M‑β‑CD does not make significant changes in the microenvironment of aromatic amino acid residues of albumin, in contrast to FQ. Thus, we can talk about various types of interaction between M‑β‑CD and FQ with protein.
Also, the work of Yan et al. [23] demonstrated that in the three-component system of HSA–(aripiprazole-M‑β‑CD), less pronounced quenching was observed in the protein emission spectrum than in the two-component HSA–aripiprazole system. Study of the effect of complexation of FQ with M‑β‑CD on the fluorescence intensity of HSA showed the following results: an increase in the concentration of FQ-M complexes‑β‑CD causes quenching of albumin fluorescence; however, saturation for both FQs occurs only at a tenfold molar excess, in contrast to two-component FQ–HSA systems, for which saturation is observed already at a fivefold molar excess. A quantitative characteristic of a decrease in the strength of the interaction of FQ with protein in the presence of M‑β‑CD is a decrease in the values of the Stern–Volmer constants for three-component systems. In the case of CF, there is a significant decrease in the constant of (1.4 ± 0.3) × 105 for the HSA–(M‑β‑CD–CF) system, which is two times less than the same value for free FQ. For a drug, the change in the value of the Stern–Volmer constant ((1.8 ± 0.4) × 105 for the HSA–(M‑β‑CD-LF)) system is not so significant (Table 1). It is known that the complexation of FQ with M‑β‑CD occurs due to the immersion of the aromatic backbone of FQ in the hydrophobic cavity of CD, while the negatively charged carboxyl group of FQ becomes inaccessible for interaction with albumin, which is characterized by binding to acidic negatively charged molecules. Thus, we can say that M‑β‑CD can interfere with the interaction of FQ with HSA or lead to a change in the binding mechanism, while exerting a more pronounced effect on CF.
It is important to note that in the case of systems containing free FQ with protein, a more pronounced quenching causes CF. When M‑β‑CD is added to the solution, the picture of the effect of FQ on the fluorescence intensity of HSA is reversed: the quenching effect can be observed more clearly in a system containing drugs. As noted earlier, more complete immersion of the FQ molecule into the M‑β‑CD is realized in the case of complexation with drugs, while the carboxyl group of FQ is available for interaction with HSA. On the contrary, the CF molecule is not able to enter so deeply into the hydrophobic cavity of М‑β‑CD, which makes its acid group less accessible for external interactions and, as a consequence, leads to a decrease in the binding of CF to the protein.
CD spectroscopy. To study the changes in the secondary structure of HSA that occur during the interaction of albumin with FQs and their complexes with CD, the method of circular dichroism (CD) was used. Since the secondary structure of albumin mainly contains α-helices [10], then, as expected, negative maxima in the circular dichroism spectrum are observed at 208 and 220 nm, corresponding to n-π * -transitions of the peptide bond in α-helices [24 , 25]. The fraction of α-helices in free HSA at pH 7.4 is ~64%, which is consistent with the literature data [26, 27]. Spectra of free FQ and M‑β‑CDs in the studied range (200–260 nm) at a concentration of 20 μM have a very low intensity; therefore, changes in the HSA spectrum will be caused only by the interaction of drug formulations with a protein, and not by a “superposition” of spectra. This makes it possible to study this multicomponent system from the point of view of the effect of complexation on the HSA conformation.
The interaction of a protein with drug formulations leads to changes in the secondary structure of the protein. When FQ interacts with HSA, the proportion of α-helices in the secondary structure of the protein decreases (by ~9–12%) due to an increase in the proportion of disordered structures, and the contribution of β-sheets also increases [26]. It should be noted that such changes are expressed to a greater extent in the system containing a more hydrophobic CF molecule, which is consistent with the data obtained by the method of fluorescence spectroscopy (Table 2). These results suggest that interactions between albumin and FQs are mainly hydrophobic.
Similar changes occur in a two-component system containing HSA and M‑β‑CD, but they are less pronounced (the fraction of α-helices decreases by ~6%), this also agrees with the data obtained by the method of fluorescence spectroscopy in the work of Yan et al. [23].
In three-component systems HSA–(FQ–M‑β‑CD), there are less pronounced changes in the secondary structure of the protein (the proportion of α-helices decreased by only ~3–5% compared to free HSA). In these systems, the protein structure is closest to that observed in free HSA, i.e. M‑β‑CD stabilizes the initial conformation of albumin. Similar conclusions are reached by Yan et al. [23] and Anand et al. [28] analyzing the effect of β‑cyclodextrins on the interaction of drugs with HSA and bovine serum albumin. Thus, we can say that in the three-component system M‑β‑CD blocks the binding of FQ to HSA or, most likely, a change in the process or binding site in albumin occurs. The smallest changes in the secondary structure of the protein are observed in the system containing CF, which is consistent with the data obtained by fluorescence spectroscopy and is explained by the less complete immersion of CF in the hydrophobic cavity of the CD.
Release kinetics of fluoroquinolones. An important characteristic of a drug is its pharmacokinetics, and interaction with a protein can have a significant effect on this parameter of the drug. Therefore, the kinetics of the release of the drug molecule from the complex with HSA was investigated by equilibrium dialysis using UV spectroscopy. In the experiment, the pore size of the membrane (cutoff weight 3.5 kDa) was selected so that only low-molecular-weight FQ was released into the external solution.
Solutions of individual substances (FQ and HSA) were studied by UV spectroscopy. The FQ spectra have a pronounced absorption band with an intensity maximum at 287 and 271 nm for the drug and CF, respectively, which is due to the π–π* transitions of the FQ aromatic backbone (Fig. 5). These peaks are analytically significant for studying the content of drugs in solution. The main absorption band of HSA in the UV range has a maximum at a wavelength of 278 nm, corresponding to π–π* transitions [29] of aromatic amino acid residues of the protein.

UV spectra of ciprofloxacin (1), HSA (2) and levofloxacin (3) at concentrations of 20 μM (pH 7.4).
It was found that under conditions close to physiological (pH 7.4, 37°C), more than 80% of FQ is released in 45 min. Complexation of FQ with M‑β‑CD (1 : 1) leads to a slight decrease in the rate of release of drug molecules at the initial time interval.
Since, as shown earlier, the most efficient binding of FQ is observed at high molar excesses of the drug relative to albumin (in the case of a high local concentration), the interaction of the FQ with the protein under these conditions should lead to a significant slowdown in drug release. Indeed, when considering the release profile of CF (Fig. 6), it was found that binding of CF with protein leads to the release of 80% CF in 120 min (2.5 times longer than free CF). A less pronounced effect of HSA has on the kinetics of drug release: less than 80% is released in 60 min, the likely reason for this is the lower affinity of HSA for drugs in comparison with CF, which was discussed above (Table 2). Since absorption by the liver and glomerular filtration are directly proportional to the content of free drug present in plasma [30], the formation of complexes of albumin with FQ will affect the metabolism and the rate of drug excretion. The interaction with the protein causes the gradual release of FQ into the blood plasma. The most pronounced manifestation of this effect will be observed at high concentrations of FQ, since saturation of the binding sites of albumin occurs at a molar excess of the drug equal to five. Plasma protein-bound drugs cannot undergo glomerular filtration, as a result of which the half-life of drugs that are poorly secreted by the renal tubules and are slowly metabolized in the liver increases, which causes prolonged action of such drugs [9, 30, 31].

(a) Kinetics of levofloxacin release (1), levofloxacin bound in a complex with HSA (molar ratio of LF: protein 10: 1) (2), as well as levofloxacin complexed with M‑β‑CD and HSA (molar ratio of LF : M‑β‑CD : protein 10 : 10 : 1) (3); (b) kinetics of ciprofloxacin release (1), ciprofloxacin bound in a complex with HSA (molar ratio of CF : protein 10 : 1) (2), as well as ciprofloxacin complexed with M‑β‑CD and HSA (molar ratio of CF : M‑β‑CD : protein 10 : 10 : 1) (3), 0.01-M sodium phosphate buffer solution (pH 7.4), 37°C.
The curves of FQ release from three-component systems (ratio of FQ components : M‑β‑CD : HR is 10 : 10 : 1) have profiles similar to those described above for two-component systems (Table 3). A more pronounced effect of protein on CF is observed: after 120 min, less than 80% of the drug is released (2.75 times longer than free CF). In the case of drugs, complexation with M‑β‑CD in the presence of HSA leads to a less significant deceleration of the kinetics of FQ release, which may be due to a change in type of interaction of FQ with HSA due to the presence of M‑β‑CD.
The presented results confirm the data obtained by the method of fluorescence and circular dichroism: even with a small molar excess of HSA (1 : 1), there is a stronger change in the physicochemical parameters of CF than the drug.
EXPERIMENTAL
Reagents. We used methyl-β-cyclodextrin, ciprofloxacin, levofloxacin, human serum albumin (Sigma-Aldrich, United States); HCl (Reakhim, Russia); phosphate buffer (EKO-Service, Russia).
Obtaining complexes of HSA with fluoroquinolones. The required amount of FQ solution with the same pH was added to a solution of HSA in phosphate buffer (pH 7.4) and the volume was adjusted to 1 mL. The HSA concentration (60 µM) was kept constant in all samples, and the molar excess of FQ was varied in the range 0.5–20.0. The complexes were incubated with constant stirring at 37°C using a thermostatically controlled shaker (Shaker-Incubator ES-20, BioSan, Latvia). To study the interaction of HSA and CF using UV and fluorescence spectroscopy, solutions with a HSA concentration of 20 μM were prepared by diluting the initial samples three times.
Preparation of complexes of fluoroquinolones with methyl-β-cyclodextrin. To a 34 mM FQ solution (pH 4.0), the required amount of a 30 mM solution of M‑β‑CD in HCl (pH 4.0) to obtain complex 1 : 1. The complex was incubated for 1 h at 37°C. When the complex was added to the HSA solution, the system was converted to pH 7.4 with phosphate buffer.
Obtaining HSA systems with a FQ complex with M-β-CD. To a solution of HSA in phosphate buffer (pH 7.4) the required amount of a solution of the FQ–M complex‑β‑CD was added, which was diluted with a phosphate buffer solution to obtain the required concentration. A series of complexes was prepared, the HSA concentration (60 µM) was kept constant in all samples, and the molar excess of FQ was varied in the range 0.5–20.0 for each series. The complexes were incubated with stirring at 37°C. In the same way, a similar series of complexes with an HSA concentration of 20 μM was obtained by diluting the initial solutions three times.
Release kinetics of ciprofloxacin. The sample under study (1 mL, concentration of complexes 20 μM) was placed in a dialysis bag (MWCO 3.5 kDa, Orange Scientific, Belgium), released into an external solution of phosphate buffer (pH 7.4) with a volume of 1 mL. The system was incubated at 37°C and 150 rpm, periodically taking aliquots (50 μL) to record UV spectra.
UV spectra were recorded on an Ultrospec 2100 pro UV and visible spectrometer (Amersham Biosciences, United States) three times in the range 200–400 nm in a quartz cuvette (Hellma Analytics, Germany). The initial samples were diluted with a buffer solution (pH 7.4) to a FQ concentration of 20 μM.
Fluorescence spectroscopy. Fluorescence was measured on a Varian Cary Eclipse spectrofluorometer (Agilent Technologies, United States); fluorescence spectra were recorded at a temperature of (25 ± 0.1)°C in a 1 cm cuvette. Emission spectra were measured at an excitation wavelength of 280 nm in the range of 290–555 nm with a step of 1 nm. The protein concentration was kept constant (20 µM, phosphate buffer pH 7.4), the FQ concentration was varied in the range of 10–200 µM.
Circular dichroism spectra were recorded using a J‑815 spectrometer (Jasco, Japan) equipped with a thermostated cell. The measurements were carried out in the wavelength range 200–260 nm at 25°C in a quartz cell (length 1 mm). The spectra were acquired by five-fold scanning with a step of 1 nm. The spectra were processed using CD Spectroscopy Deconvolution Software (CDNN program version 2, Dr. Gerald Böhm, Magdeburg), using 33 base spectra for deconvolution and analysis.
CONCLUSIONS
The effect of the structure of fluoroquinolones and the complexation of FQ with M‑β‑CD for the interaction of the dosage formulation with HSA was studied for the first time. It was found that the chemical structure of FQ has a significant effect on the binding of a drug molecule to HSA, and also affects the different effects of M‑β‑CD for these interactions.
It was shown by CD and fluorescence spectroscopy that a more hydrophobic CF molecule interacts more strongly with HSA than a similar drug molecule, which has a morpholine fragment in its structure instead of cyclopropanyl in the para-position of the fluorine atom. Thus, by the method of fluorescence spectroscopy, it was found that the presence of CF causes a more pronounced quenching of the protein emission spectrum (KSV (CF–HSA) = (2.9 ± 0.2) × 105) than LF (KSV (LF–HSA) = (2.2 ± 0.2) × 105). Using the method of circular dichroism spectroscopy, it was shown that drug has less effect on the secondary structure of the protein than CF. This suggests that the binding of FQ with HSA also occurs due to hydrophobic interactions.
A decrease in the Stern–Volmer constants and the effect of drug formulations on the secondary structure of albumin during the interaction of HSA with FQ bound in a complex with M‑β‑CD was demonstrated. This effect is more pronounced in the system containing CF, which is most likely explained by the immersion of the FQ molecule in the hydrophobic cavity М‑β‑CD, leading to blocking of the carboxyl group and aromatic backbone of CF for interaction with HSA. On the contrary, in the case of complexation M‑β‑CD with drugs, due to the presence of a morpholine fragment in the FQ structure, the anionic group of the drug is outside the host molecule, which makes it available for binding with HSA. From this we can conclude that it is the carboxyl groups of the molecules that provide the interaction of drugs with albumin. These conclusions are consistent with the fact that HSA predominantly interacts with anionic acid molecules [9]. Formation of the FQ–M complex‑β‑CD leads to a change in the interaction of the drug with HSA and affects the rate of release of these systems from the model dialysis membrane. Consequently, complexation can lead to a change in the pharmacokinetic and pharmacodynamic properties of FQs. Thus, the role of M‑β‑CD in the interactions of medicinal molecules with HSA.
These results are of great interest for designing drug delivery systems with a controlled release rate. Complexation with HSA allows you to vary the physicochemical properties of molecules, which makes it possible to control the pharmacokinetic and pharmacodynamic properties of the drug, help reduce the manifestation of side effects, and increase the activity of drug molecules, which is extremely important for planning competent therapy with long-term use of antibacterial drugs.
REFERENCES
Van Bambeke, F., Michot, J.-M., Van Eldere, J., and Tulkens, P.M., Clin. Microbiol. Infect., 2005, vol. 11, pp. 256–280. https://doi.org/10.1111/j.1469-0691.2005.01131.x
Talley, J.H., Postgrad. Med., 1991, vol. 89, pp. 101–113.
Wolfson, J.S. and Hooper, D.C., Clin. Microbiol. Rev., 1989, vol. 2, pp. 378–424.
Campoli-Richards, D.M., Monk, J.P., Price, A., Benfield, P., Todd, P.A., and Ward, A., Drugs, 1988, vol. 35, pp. 373–447.
Skuredina, A.A., Le-Deygen, I.M., Uporov, I.V., and Kudryashova, E.V., Colloid J., 2017, vol. 79, no. 5, pp. 668–676. https://doi.org/10.7868/s0023291217050135
Skuredina, A.A., Kopnova, T.Y., Le-Deygen, I.M., and Kudryashova, E.V., Moscow Univ. Chem. Bull., 2020, vol. 75, pp. 218–224. https://doi.org/10.3103/S0027131420040069
Skuredina, A.A., Le-Deygen, I.M., and Kudryashova, E.V., Colloid J., 2018, vol. 80, no. 3, pp. 312–319. https://doi.org/10.7868/s0023291218030102
Le-Deygen, I.M., Skuredina, A.A., Uporov, I.V., and Kudryashova, E.V., Anal. Bioanal. Chem., 2017, vol. 409, pp. 6451–6462. https://doi.org/10.1007/s00216-017-0590-5
Fanali, G., di Masi, A., Trezza, V., Marino, M., Fasano, M., and Ascenzi, P., Mol. Aspects Med., 2012, vol. 33, pp. 209–290. https://doi.org/10.1016/j.mam.2011.12.002
He, X.M. and Carter, D.C., Nature, 1992, vol. 358, pp. 209–215. https://doi.org/10.1038/358209a0
Sugio, S., Kashima, A., Mochizuki, S., and Noda, M., Protein Eng., 1999, vol. 12, pp. 439–446. https://doi.org/10.1093/protein/12.6.439
Varshney, A., Ansari, Y., Zaidi, N., Ahmad, E., Badr, G., Alam, P., and Khan, R.H., Cell Biochem. Biophys., 2014, vol. 70, pp. 93–101. https://doi.org/10.1007/s12013-014-9863-1
Varshney, A., Sen, P., Ahmad, E., Rehan, M., Subbarao, N., and Khan, R.H., Chirality, 2010, vol. 22, pp. 77–87. https://doi.org/10.1002/chir.20709
Sudlow, G., Birkett, D.J., and Wade, D.N., Mol. Pharmacol., 1975, vol. 11, pp. 824–832.
Trynda-Lemiesz, L., Bioorg. Med. Chem., 2004, vol. 12, pp. 3269–3275. https://doi.org/10.1016/j.bmc.2004.03.073
Davis, R., Markham, A., and Balfour, J.A., Drugs, 1996, vol. 51, pp. 1019–1074. https://doi.org/10.2165/00003495-199651060-00010
Blokhina, S.V., Sharapova, A.V., Ol’khovich, M.V., Volkova, T.V., and Perlovich, G.L., Eur. J. Pharm. Sci., 2016, vol. 93, pp. 29–37. https://doi.org/10.1016/j.ejps.2016.07.016
Davis, R. and Bryson, H.M., Drugs, 1994, vol. 47, pp. 677–700. https://doi.org/10.2165/00003495-199447040-00008
Valeur, B., Encyclopedia of Applied Spectroscopy, Weinheim, Germany: Publishing GmbH & Co. KGaA, 2009, pp. 477–531.
Skuredina, A.A., Kopnova, T.Yu., Yakupova, L.R., Le-Deygen, I.M., and Kudryashova, E.V., Biotekhnologiya, 2020, vol. 36, no. 6, pp. 24–29. https://doi.org/10.21519/0234-2758-2020-36-6-149-154
Ghuman, J., Zunszain, P.A., Petitpas, I., Bhattacharya, A.A., Otagiri, M., and Curry, S., J. Mol. Biol., 2005, vol. 353, pp. 38–52. https://doi.org/10.1016/j.jmb.2005.07.075
Kudryashova, E.V., Gladilin, A.K., and Levashov, A.V., Usp. Biol. Khim., 2002, vol. 42, pp. 257–294.
Yan, J., Wu, D., Ma, X., Wang, L., Xu, K., and Li, H., Carbohydr. Res., 2015, vol. 131, pp. 65–74. https://doi.org/10.1016/j.carbpol.2015.05.037
Poureshghi, F., Ghandforoushan, P., Safarnejad, A., and Soltani, S., J. Photochem. Photobiol., 2017, vol. 166, pp. 187–192. https://doi.org/10.1016/j.jphotobiol.2016.09.046
Zhang, G. and Ma, Y., Food Chem., 2013, vol. 136, pp. 442–449. https://doi.org/10.1016/j.foodchem.2012.09.026
Varlan, A., Ionescu, S., and Hillebrand, M., Luminescence, 2011, vol. 26, pp. 710–715. https://doi.org/10.1002/bio.1302
Dockal, M., Carter, D.C., and Ruker, F., J. Biol. Chem., 2000, vol. 275, pp. 3042–3050. https://doi.org/10.1074/jbc.275.5.3042
Anand, U. and Mukherjee, S., Phys. Chem. Chem. Phys., 2013, vol. 15, p. 9375. https://doi.org/10.1039/c3cp50207d
Kaur, A., Khan, I.A., Banipal, P.K., and Banipal, T.S., Spectrochim. Acta Part A Mol. Biomol. Spectrosc., 2018, vol. 191, pp. 259–270. https://doi.org/10.1016/j.saa.2017.10.017
Lindup, W.E. and Orme, M.C., Br. Med. J. (Clin. Res. Ed.), 1981, vol. 282, pp. 212–214. https://doi.org/10.1136/bmj.282.6277.1707-b
Schmidt, S., Gonzalez, D., and Derendorf, H., J. Pharm. Sci., 2010, vol. 99, pp. 1107–1122. https://doi.org/10.1002/jps.21916
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interests
The authors declare they have no conflict of interest.
This article does not contain a description of research carried out by any of the authors of this work, involving humans or using animals as research objects.
Additional information
Abbreviations: LV, levofloxacin; M-β-CD, methyl-β-cyclodextrin; FQ, fluoroquinolone; CD, cyclodextrin; CF, ciprofloxacin; HSA, human serum albumin.
Corresponding autor: phone: +7 (977) 713-21-54.
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yakupova, L.R., Kopnova, T.Y., Skuredina, A.A. et al. Effect of Methyl-β-Cyclodextrin on the Interaction of Fluoroquinolones with Human Serum Albumin. Russ J Bioorg Chem 48, 163–172 (2022). https://doi.org/10.1134/S1068162022010149
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1068162022010149