
Abstract
A method is proposed for the determination of some ketosteroids in human urine, including enzymatic hydrolysis using β-glucuronidase from E. coli followed by dispersive liquid-liquid microextraction, derivatization of analytes with hydroxylamine, and detection by reversed-phase ultra-HPLC–quadrupole time-of-flight mass spectrometry. Optimization of extraction and derivatization conditions of the studied compounds made it possible to find that the highest recoveries were achieved using an acetone–chloroform mixture as a dispersant and an extractant, and the completeness of the derivatization reaction was achieved by thermostating the sample at 70°C for 90 min. The proposed method has high sensitivity (limits of detection in the range of 0.1–0.25 ng/mL) and a wide linearity range.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Steroid hormones are regulators of various physiological and biochemical processes in a human body. All steroid hormones are derived from a common precursor cholesterol by hydroxylation, oxidation, and reduction reactions. Among the steroid hormones, the following classes are distinguished: estrogens, androgens, progestins, mineralocorticoids, glucocorticoids, and vitamin D and its metabolites [1, 2]. The determination of these compounds in human biological fluids is required, first of all, for clinical diagnostics because of their participation in the pathogenesis of many diseases and genetic disorders [2, 3].
Usually, steroid hormones are determined in urine (average concentrations from several hours to one day), blood and saliva (determination in real time) [4]. The results of the real-time determination of steroids can fluctuate because of the episodic hormone secretion [5, 6] and also circadian rhythms [7], while the composition of urine is not subjected to such fluctuations. Other advantages of urine as a test sample are the non-invasiveness of the procedure and the ease of obtaining large sample volumes [8].
Steroid hormones exhibit biological activity at very low concentration levels (nano- and picomolar) [3, 9, 10]; therefore, their determination requires the use of highly sensitive methods.
Various methods of the preparation of urine samples for analysis are used: liquid–liquid extraction [11–13], stir bar sorptive extraction [14], on-line in-tube solid-phase microextraction [15], automated solid-phase extraction [16], dispersive liquid–liquid microextraction (DLLME) [8], and others [17]. Among the listed methods, the most promising is DLLME because of its simplicity, high concentration factors, and compliance with the criteria of “green chemistry” [18]. If it is necessary to increase the sensitivity of the determination of steroid hormones, derivatization can be used to increase the efficiency of ionization. Thus, reviews [9, 19, 20] considered the use of various derivatizing agents for small molecules, in particular, for steroid hormones. Some of the reagents used are not commercially available, while others, e.g., dansyl chloride, lead to a decrease in the selectivity of the chromatographic separation and the nonspecific fragmentation of derivatives [21]. This imposes additional requirements on the reagent in using derivatization in sample preparation.
The aim of this work was to develop a simple, rapid, and highly sensitive method for the determination of steroid hormones of various classes, which meets the criteria of “green chemistry”.
EXPERIMENTAL
Materials and reagents. Standard samples of testosterone, dihydrotestosterone, cortisone, hydrocortisone (cortisol), estrone, progesterone, 11α-hydroxyprogesterone, and methyltestosterone (internal standard) (Sigma-Aldrich, Germany); β-glucuronidase from Escherichia coli (E. coli) (Roche Diagnostics, Germany); and a 50% aqueous solution of hydroxylamine (Sigma-Aldrich, Germany) were used. Acetonitrile (Biosolve, Israel) and methanol (J.T.Baker, Great Britain) for HPLC, formic acid (98%) (Acros Organics), water (18.2 MOhm cm, Milli-Q) were used to prepare solutions and as components of the mobile phase. Acetone, ethanol, dichloromethane, trichloromethane and tetrachloromethane (99.9%), hydrochloric acid (99%), formic acid (98%), sodium chloride, potassium dihydrogen phosphate, sodium sulfate, uric acid, urea, creatinine, sodium citrate, potassium chloride, calcium chloride, ammonium chloride, potassium oxalate, magnesium sulfate, sodium dihydrogen phosphate, sodium hydrogen phosphate, sodium azide, and sodium tetraborate (borax) (99%) were purchased from Vekton (Russia).
Preparation of standard solutions. Standard solutions of steroid hormones with a concentration of 1 mg/mL were prepared in methanol; calibration solutions and quality control solutions were prepared by diluting standard solutions with methanol. A phosphate buffer solution (50 mL, pH 6.5) was prepared from potassium dihydrogen phosphate, sodium hydrogen phosphate, and sodium azide. To optimize the conditions of sample preparation, synthetic urine was prepared in accordance with the protocol [22]. All solutions were stored at 4°C.
Instruments and equipment. Analytes were determined using a system consisting of a Bruker Elute ultra-high-performance liquid chromatograph and a Bruker MaXis Impact quadrupole time-of-flight mass spectrometer with an electrospray ionization source (Table 1) under the control of the Bruker Compass HyStar 4.1 software. To separate analytes, a Phenomenex Kinetex C18 column (100 × 2.1 mm, 1.7 μm) with an appropriate precolumn was used. The mobile phase was a mixture of a 0.1% solution of formic acid in water (eluent A) and a 0.1% solution of formic acid in methanol (eluent B) in the gradient elution mode (1.00 min—95% A, 2.70 min—40% A, 4.00 min—40% A, 5.00 min—10% A, 7.50 min—10% A, 7.51 min—95% A, 9.00 min—95% A) at a mobile phase flow rate of 0.4 mL/min and a thermostat temperature of 40°C. The samples were kept in an autosampler at 5°C; 10 μL of a sample were used for analysis.
Test samples. Urine samples were obtained from healthy volunteers aged 18 to 45 years, preserved with sodium azide, and stored at –20°C.
Selection of a derivatizing agent. Hydroxylamine was chosen as a derivatizing agent, as it provides high detection sensitivity, and the derivatization reaction is simple and does not require special conditions. In addition, it was possible to inject the reaction mixture into the chromatograph without additional purification steps after obtaining the derivatives. Hydroxylamine was previously used to determine steroid hormones in human biological fluids with high sensitivity [3, 23, 24]. Scheme 1 shows the derivatization reaction using hydroxylamine as a derivatizing agent.

Scheme 1 . Formation of steroid hormone oximes by derivatization with hydroxylamine.
The main disadvantage of using hydroxylamine is the possible splitting of peaks in the chromatograms because of the formation of several stereoisomers. However, when the optimization of gradient elution conditions did not allow to achieve the formation of a single peak without a decrease in selectivity, it was possible to integrate the split peak without deteriorating the analytical characteristics of the method.
Optimization of conditions for derivatization. To achieve the completeness of the reaction, parameters such as hydroxylamine concentration, temperature, and incubation time were optimized. The reaction time was varied in the range from 30 to 120 min, temperature was from room temperature to 70°C, and hydroxylamine concentration was between 0.4 and 3.2 M. The completeness of the reaction was assessed by the presence of peaks of derivatives and the absence of peaks of the initial compounds in the chromatograms (Table 2).
Optimization of conditions for dispersive liquid–liquid microextraction. To find an optimal combination of an extractant and a dispersant, the following experiment was carried out: 300 μL of a phosphate buffer solution (pH 6.5) was added to 1 mL of synthetic urine containing the test compounds and an internal standard in the concentrations 100 ng/mL. Then, using a syringe, a mixture of an extractant (100 μL) and a dispersant (500 μL) was injected into the sample, after which the mixture was vortexed for 15 s and centrifuged for 10 min at 10 000 rpm. The lower phase was evaporated to dryness in a stream of nitrogen, and a hydroxylamine solution (methanol–water (1 : 1)) was added to obtain derivatives, followed by thermostating and sample analysis.
Acetone, methanol, ethanol, and acetonitrile, which dissolve well both in the sample and in the extractant and contribute to the formation of a large phase contact surface, were considered as dispersants. Di-, tri-, and tetrachloromethane were used as dispersants, because they are poorly soluble in an aqueous sample and also have a higher density, thereby meeting the criteria for these solvents.
To achieve the highest recoveries, a multifactorial approach was used—the Box-Behnken design [25–28], which was used to assess the effects of various factors (independent variables) on the analytical signal (e.g., peak area is a dependent variable), and also allowed to assess the interaction between the factors.
Four factors were considered: the volume of the extractant (50, 100, and 150 μL), the volume of the dispersant (450, 500, and 550 μL), the amount of sodium chloride to assess the salting-out effect (0, 50, and 100 mg), and the amount of sodium tetraborate (0, 5, and 10 mg) to study the effect of pH on the recovery. The results were processed using the STATISTICA 10 software (Statsoft).
The experiment was carried out as follows: 0.3 mL of a phosphate buffer solution was added to 1 mL of synthetic urine containing the compounds under study and an internal standard (100 ng/mL), sodium chloride (0, 50, and 100 mg), and sodium tetraborate (0, 5, and 10 mg); then a mixture of an extractant and a dispersant was injected with a syringe. The mixture was vortexed for 15 s (without stirring, the recovery of the target compounds was unsatisfactory) and centrifuged for 10 min at 10 000 rpm. The lower phase was evaporated to dryness in a stream of nitrogen, and a hydroxylamine solution (methanol–water (1 : 1)) was added to obtain derivatives, followed by thermostating and sample analysis.
It should be noted that, in the analysis of real samples before the microextraction procedure, glucuronides were deconjugated under the conditions optimized by us earlier [8]: 30 min at 50°C in the presence of a phosphate buffer solution (pH 6.5) and enzyme β‑glucuronidase from E. coli.
After choosing the optimal conditions for sample preparation, the analytical characteristics of the procedure were evaluated.
RESULTS AND DISCUSSION
Optimization of derivatization conditions. It was found that the use of hydroxylamine concentration of at least 1.6 M ensures the completeness of the reaction at an incubation temperature of 70°C and reaction time of at least 90 min; therefore, these conditions were chosen for the further experiments. At a lower concentration of the derivatizing agent and a shorter time and lower temperature, the starting compounds or mono- and disubstituted derivatives, e.g., for cortisone, cortisol, progesterone, and 11α-hydroxyprogesterone, were observed in the chromatograms.
Optimization of conditions for dispersive liquid–liquid microextraction. In optimizing the types of the extractant and dispersant, it was found that two combinations of solvents, namely acetone–trichloromethane and acetonitrile–trichloromethane, provided sufficient recoveries. In using acetonitrile, the recoveries of the studied glucocorticoids (cortisone and cortisol) were slightly higher, while acetone led to an increase in the recoveries of other compounds; therefore, it was chosen for the further experiments as a dispersant and trichloromethane as an extractant.
The use of the Box-Behnken design made it possible to establish factor levels that ensure the quantitative recovery of all analytes. All the models obtained were statistically significant (\(R_{{{\text{adj}}}}^{{\text{2}}}\) > 0.9), which indicated a high adequacy of the results obtained. Because of the optimization of the conditions for compounds with different physicochemical properties, in some cases compromise values were chosen. As a result, it was found that the addition of sodium chloride and sodium tetraborate had little effect on the recoveries values, while the volume of the extractant had a strong effect—with an increase in the volume, the recoveries increased; therefore, we chose the value 150 μL. The effect of dispersant volume on recove values was weaker: the volume of 500 μL ensures the highest recove values. Under these conditions, the recoveries of analytes were 79–98%, while the recovery values of glucuronides were the lowest compared to the rest of the compounds.
Thus, the following optimal conditions for sample preparation were chosen: 1 mL of a sample containing the internal standard methyltestosterone (100 ng/mL) and 0.3 mL of a phosphate buffer solution (pH 6.5) containing the enzyme β-glucuronidase E. coli was added to a 2-mL microcentrifuge tube, the sample was incubated for 30 min at 50°C. After cooling the sample to room temperature, a mixture of chloroform and acetone (150 and 500 μL, respectively) was injected into it using a syringe, vortexed for 15 s and centrifuged for 10 min at 10000 rpm. The lower layer was transferred to a vial, evaporated to dryness in a stream of nitrogen, the dry residue was dissolved in 100 μL of a 1.6 M hydroxylamine solution (methyl alcohol–water (1 : 1)) and the vial was thermostated for 90 min at 70°C, followed by analysis.
Validation of the developed method. The proposed method was validated in accordance with the FDA criteria for the validation of bioanalytical procedures [29]. The procedure was validated using synthetic urine because of the difficulty with obtaining a reproducible matrix with steroid hormone concentrations below the limits of detection.
Calibration curves were plotted in the concentration range of 0.1–100 ng/mL (0.1, 0.25, 0.5, 1.0, 2.5, 5, 10, 25, 50, and 100 ng/mL). The limit of detection was set as the analyte concentration providing a signal to noise ratio of at least 3 : 1, and the limit of quantification corresponded to the concentration found with an error of less than 15%. The results are presented in Table 3.
Cross-contamination was assessed by analyzing a blank solution after analyzing a 100 ng/mL sample solution; no peaks were observed in the blank solution chromatogram corresponding to the compounds being determined.
Accuracy and reproducibility were established by analyzing quality control solutions of low (2.5 ng/mL), medium (10 ng/mL), and high (50 ng/mL) concentrations on the same and different days. Reproducibility was expressed as relative standard deviation (RSD, %), and accuracy was found using Eq. (1):
The results are presented in Table 4.
The stability of quality control solutions in an autosampler passed through all stages of sample preparation was evaluated for 48 h at 5°C. It was found that the deviations of the results did not exceed 15%. Long-term stability was assessed over a month by carrying out three cycles of freezing (–20°C) and thawing of quality control samples to room temperature. There were no significant differences in the results obtained.
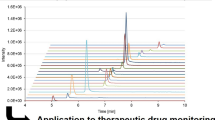
Analysis of real samples. The proposed method was used to analyze real samples using the standard addition method. For this, mixtures of analytes of three concentrations (10, 50, and 100 ng/mL) were added to a urine sample. As an example, chromatograms of a model sample and a urine sample with a testosterone addition (10, 50, and 100 ng/mL) are shown in Fig. 1. The recoveries were 76–95%, the relative standard deviation did not exceed 15%, which indicates the suitability of the procedure for the analysis of real samples.

Chromatograms of (a) extracted ion (m/z 304.2271) of a model sample and (b) urine sample (b) with additives of (1) 0, (2) 10, (3) 50, (4) 100 ng/mL of testosterone (I).
CONCLUSIONS
Thus, a sensitive, simple, and reproducible method was developed for the determination of ketosteroids. Derivatization with hydroxylamine made it possible to significantly increase the sensitivity of the method for most compounds in comparison with our previous work [8], especially for estrone. An analysis of real samples showed a possibility of using the proposed method for the determination of ketosteroids.
REFERENCES
Norman, A.W., Encyclopedia of Food Sciences and Nutrition, New Delhi: Academic, 2003, 2nd ed. https://doi.org/10.1016/B0-12-227055-X/00606-4
Biason-Lauber, A., in Physician’s Guide to the Laboratory Diagnosis of Metabolic Diseases, Blau, N., Duran, M., Blaskovics, M.E., and Gibson, K.M, Eds., Heidelberg: Springer, 2003, p. 551. https://doi.org/10.1007/978-3-642-55878-8_35
Häkkinen, M.R., Murtola, T., Voutilainen, R., Poutanen, M., Linnanen, T., Koskivuori, J., Lakka, T., Jääskeläinen, J., and Auriola, S., J. Pharm. Biomed. Anal., 2019, vol. 164, p. 642. https://doi.org/10.1016/j.jpba.2018.11.035
Márta, Z., Bobály, B., Fekete, J., Magda, B., Imre, T., Meszaros, K.V., Bálint, M., and Szabó, P.T., J. Pharm. Biomed. Anal., 2018, vol. 150, p. 258. https://doi.org/10.1016/j.jpba.2017.12.014
West, C.D., Mahajan, D.K., Chavre, V.J., Nabors, C.J., and Tyler, F.H., J. Clin. Endocrinol. Meta-b., 1973, vol. 36, no. 6, p. 1230. https://doi.org/10.1210/jcem-36-6-1230
Oerter, K.E., Guardabasso, V., and Rodbard, D., Comput. Biomed. Res., 1986, vol. 19, no. 2, p. 170. https://doi.org/10.1016/0010-4809(86)90014-5
Chung, S., Son, G.H., and Kim, K., Biochim. Biophys. Acta, 2011, vol. 1812, no. 5, p. 581. https://doi.org/10.1016/j.bbadis.2011.02.003
Dmitrieva, E., Temerdashev, A., Azaryan, A., and Gashimova, E., J. Chromatogr. B: Anal. Technol. Biomed. Life Sci., 2020, vol. 1159, 122390. https://doi.org/10.1016/j.jchromb.2020.122390
Higashi, T. and Ogawa, S., J. Steroid. Biochem. Mol. Bi-ol., 2016, vol. 162, p. 57. https://doi.org/10.1016/j.jsbmb.2015.10.003
Higashi, T., Chem. Pharm. Bull. (Tokyo), 2006, vol. 54, no. 11, p. 1479. https://doi.org/10.1248/cpb.54.1479
He, G., Wu, Y., and Lu, J., Steroids, 2018, vol. 131, p. 1. https://doi.org/10.1016/j.steroids.2017.12.011
Pitarch-Motellón, J., Sancho, J.V., Ibañéz, M., Pozo, O., and Roig-Navarro, A.F., J. Chromatogr. A, 2017, vol. 1515, p. 172. https://doi.org/10.1016/j.chroma.2017.08.006
Rincon, A.V., Ostner, J., Heistermann, M., and Deschner, T., Gen. Comp. Endocrinol., 2019, vol. 281, p. 117. https://doi.org/10.1016/j.ygcen.2019.05.017
Huang, X., Yuan, D., and Huang, B., Talanta, 2008, vol. 75, no. 1, p. 172. https://doi.org/10.1016/j.talanta.2007.10.052
Saito, K., Yagi, K., Ishizaki, A., and Kataoka, H., J. Pharm. Biomed. Anal., 2010, vol. 52, no. 5, p. 727. https://doi.org/10.1016/j.jpba.2010.02.027
Andersen, D.W. and Linnet, K., J. Anal. Toxicol., 2014, vol. 38, no. 9, p. 637. https://doi.org/10.1093/jat/bku098
Denver, N., Khan, S., Homer, N.Z.M., Maclean, M.R., and Andrew, R., J. Steroid. Biochem. Mol. Biol., 2019, vol. 192, 105373. https://doi.org/10.1016/j.jsbmb.2019.04.022
Leong, M.-I., Fuh, M.-R., and Huang, S.-D., J. Chromatogr. A, 2014, vol. 1335, p. 2. https://doi.org/10.1016/j.chroma.2014.02.021
Higashi, T. and Shimada, K., Anal. Bioanal. Chem., 2004, vol. 378, no. 4, p. 875. https://doi.org/10.1007/s00216-003-2252-z
Zhang, T.-Y., Li, S., Zhu, Q.-F., Wang, Q., Hussain, D., and Feng, Y.-Q., TrAC, Trends Anal. Chem., 2019, vol. 119, 115608. https://doi.org/10.1016/j.trac.2019.07.019
Kushnir, M.M., Rockwood, A.L., Bergquist, J., Varshavsky, M., Roberts, W.L., Yue, B., Bunker, A.M., and Meikle, A.W., Am. J. Clin. Pathol., 2008, vol. 129, no. 4, p. 530. https://doi.org/10.1309/LC03BHQ5XJPJYEKG
Sarigul, N., Korkmaz, F., and Kurultak, I., Sci. Rep., 2019, vol. 9, 20159. https://doi.org/10.1038/s41598-019-56693-4
Keski-Rahkonen, P., Huhtinen, K., Poutanen, M., and Auriola, S., J. Steroid. Biochem. Mol. Biol., 2011, vol. 127, nos. 3–5, p. 396. https://doi.org/10.1016/j.jsbmb.2011.06.006
Liu, Q., Chi, Q., Fan, R.-T., Tian, H.-D., and Wang, X., Nat. Prod. Bioprospect., 2019, vol. 9, no. 3, p. 201. https://doi.org/10.1007/s13659-019-0204-3
Mousavi, L., Tamiji, Z., and Khoshayand, M.R., Talanta, 2018, vol. 190, p. 335. https://doi.org/10.1016/j.talanta.2018.08.002
Ferreira, S.L.C., Bruns, R.E., Ferreira, H.S., Matos, G.D., David, J.M., Brandao, G.C., Silva, E.G.P., Portugal, L.A., Reis, P.S., Souza, A.S., and Santos, W.N.L., Anal. Chim. Acta, 2007, vol. 597, no. 2, p. 179. https://doi.org/10.1016/j.aca.2007.07.011
Ebrahimi-Najafabadi, H., Leardi, R., and Jalali-Heravi, M., J. AOAC Int., 2014, vol. 97, no. 1, p. 3. https://doi.org/10.5740/jaoacint.sgeebrahimi1
Bezerra, M.A., Santelli, R.E., Oliveira, E.P., Villar, L.S., and Escaleira, L.A., Talanta, 2008, vol. 76, no. 5, p. 965. https://doi.org/10.1016/j.talanta.2008.05.019
Food and Drug Administration Gguidance. Bioanalytical Method Validation. Guidance for Industry, 2018. http://www.fda.gov/files/drugs/published/Bioanalytical-Method-Validation-Guidance-for-Industry.pdf. Accessed April 2, 2021.
ACKNOWLEDGMENTS
This work was performed using the scientific equipment of the Center for Collective Use “Ecological and Analytical Center” of Kuban State University.
Funding
The innovation project was carried out with the financial support of the Kuban Science Foundation in the framework of the Commercializable scientific and innovation projects competition no. NIP-20.1/4 and the State Assignment of the Ministry of Education.
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by V. Kudrinskaya
Rights and permissions
Open Access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dmitrieva, E.V., Temerdashev, A.Z. & Osipova, A.K. Determination of Ketosteroids in Human Urine Using Dispersive Liquid-Liquid Microextraction and Ultra High-Performance Liquid Chromatography-High Resolution Mass Spectrometry. J Anal Chem 76, 1305–1311 (2021). https://doi.org/10.1134/S1061934821110034
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S1061934821110034