
Abstract
Bispecific antibodies (bsAbs) are some of the most promising biotherapeutics due to the versatility provided by their structure and functional features. bsAbs simultaneously bind two antigens or two epitopes on the same antigen. Moreover, they are capable of directing immune effector cells to cancer cells and delivering various compounds (radionuclides, toxins, and immunologic agents) to the target cells, thus offering a broad spectrum of clinical applications. Current review is focused on the technologies used in bsAb engineering, current progress and prospects of these antibodies, and selection of various heterologous expression systems for bsAb production. We also discuss the platforms development of bsAbs for the therapy of solid tumors.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Throughout its entire history, the humankind has drawn inspiration from nature to create medicines and treatments for various diseases [1]. As an alternative to chemically produced drugs, pharmaceutical and biotechnology companies are turning their attention to biologics, such as oligonucleotides (DNA and small interfering RNAs), peptides, proteins, enzymes, and their complexes, for the development of novel medicinal products [2].
The term biotherapeutics, or biologics, refers to a broad category of macromolecular products that includes cytokines, growth factors, hormones, vaccines, proteins, peptide-based products, and antibodies, i.e., active substances obtained from biological sources. One of the most rapidly developed subclasses of biotherapeutics is monoclonal antibodies (mAbs) that are employed for treating various chronic conditions, including cancer, autoimmune disorders, and infectious diseases [3, 4]. Currently, therapeutic mAbs are used mostly, but not exclusively, for targeting tumors and in the complex cancer immunotherapy [5] in combination with other therapeutic approaches [6]. Beside their application in systemic targeted immunotherapy, mAbs serve as valuable tools in cancer diagnostics [7, 8] and imaging [9, 10].
High antigen-binding activity, stability, and low immunogenicity are the three essential physicochemical features of mAbs that determine their use in therapy [11]. Furthermore, antibodies can be easily modified by the methods of protein engineering and have an inherent ability to bind antigens and endogenous immune receptors. Therefore, the mode of action and the efficacy of therapeutic antibodies are determined by their universal features, such as antigen recognition driven by the variable regions (Fab fragments), effector activity, and extended half-life mediated by the constant regions (Fc fragments) [12].
Since monospecific IgGs may not be ideal for many therapeutic applications [15], a wide range of multispecific antibodies (msAbs) targeting multiple antigens or different epitopes of the same antigen have been developed [13, 14]. Despite the existence of numerous msAb formats, the main issue with their production is that these biomolecules sometimes lack desirable physical and chemical characteristics, which hinders their therapeutic application. Moreover, many of the innovative drugs, including bispecific antibodies (bsAbs), are composed of different antibody chains with different structure and binding properties. This requires multiple aspects of protein engineering to be taken into account during discovery and development of these compounds [16, 17]. Recent advances in genetics and protein biochemistry, as well as the accessibility of bioprocesses, have helped to overcome the above-mentioned issues, and thus far, different technologies for the industrial production of bsAbs have been well established [18-20]. In this article, we have focused on the potential of various heterologous expression systems for the production of bsAbs, which are currently tested in the late-stage clinical trials for the therapy of solid tumors, and explained the need for the further development of the recombinant antibody technology.
STRUCTURAL DESIGN AND ENGINEERING OF MONO- AND BISPECIFIC ANTIBODIES
Canonical IgGs are Y-shaped molecules composed of two identical heavy chains and two identical light chains. The heavy chain consists of four domains – three constant and one variable (CH1-CH3 and VH, respectively). The light chain consists of two domains – one constant (CL) and one variable (VL). Heavy chains are connected by disulfide bonds in the hinge region between the CH1 and CH2 domains. Each heavy chain is joined to the light chain by a disulfide bridge at the junction of CH1 and CL. The CH2 and CH3 domains of two heavy chains form the Fc fragment. The variable domains of the light and heavy chains (VH and VL, respectively) are connected to the closely located CH1 and CL domains, forming the Fab fragments.
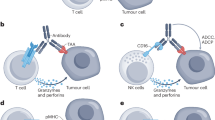
There are several approaches to the bsAb construction, including obtaining different antigen-binding fragments [Fab, single-chain variable fragment (scFv), variable domain of heavy-chain antibody (VHH)] [21] and optimizing canonical antibody structure by mutagenesis with the purpose of heavy and light chain heterodimerization [18, 22]. Out of a wide variety of approaches to bsAb classification and systematization [14, 16], we have focused on the recently developed stratification approach [18, 23] that has become widely accepted due to its simplicity. According to this classification, all types of bsAbs can be divided into three groups: (i) asymmetric, (ii) symmetric, and (iii) fragment-based (figure).

Structural arrangements of three main bsAb classes: a) canonical IgG; (b) asymmetric bsAb [Bivalent (1+1)]; c) fragment-based bsAb (tandem scFv, BiTE, diabody); and d) symmetric bsAb [Tetravalent (2+2)]
The simplest type is fragment-based bsAbs usually created by merging two or more antigen-binding fragments (scFv, VHH, Fab) with different antigen specificities via polypeptide linkers. Human serum albumin (HSA) can be added to the molecule to extend its half-life [23, 24]. The resulting molecule is a single polypeptide chain with a simple structure, which allows to avoid the problem of improper chain association [25].
Symmetric bsAbs can contain an Fc fragment, but also have two antigen-binding fragments with different specificities in the same polypeptide chain. Typically, they are obtained by addition of scFv or VHH fragments directly to canonical antibodies through the linkers or by fusion of fragment-based bsAbs with Fc fragments. Usually, such molecules have a tetravalent structure with two binding sites for each antigen and avoid the issues of improper chain association.
Asymmetric bsAbs consist of heavy and light chains obtained from different parental antibodies [26]. Their recombinant production can be complicated by the problems of proper chain association that are typically solved by introducing mutations facilitating correct heterodimerization of heavy and light chains.
Below, we provide a detailed description of main directions and advances in the development of commercially available platforms for the production of recombinant bsAbs with special focus on the progress in the clinical application of bsAbs for the treatment of solid tumors.
Asymmetric bsAbs. Xmab®. The XmAb platform developed by Xencor uses a set of amino acid substitutions in the antibody constant domains (from the hinge region to the CH3 domain) for eliminating or enhancing the effector properties of the antibody, increasing its half-life, and creating bispecific molecules [27]. Researchers from Xencor have used in silico-based rational design to find original amino acid substitutions ensuring heavy chain heterodimerization, as well as correct assembly and high thermal stability of final bsAbs [28]. The modified heavy chains were further optimized to change their pI values in order to facilitate the purification process [27].
Azymetric®. Zymeworks have developed an alternative in silico-based algorithm, the main feature of which involved unique amino acid substitutions for the heavy chain heterodimerization. The company also created a bsAb consisting of two different heavy chains and two different light chains by introducing mutations not only into the CH3 domain, but also into the CH1 and CL domains. This approach can be combined with the use of kappa and lambda light chains.
CrossMab®. The original CrossMab platform developed by Hoffman la Roche facilitated correct assembly of bsAbs with two different light chains and two different heavy chains. The main advantage of this technology is that it provided correct cross-chain dimerization that was ensured by introduction of the knob-into-hole mutations [29]. This technology can be implemented in three different variants.
The first variant (CrossMabFab format) involves incorporation of the original heavy chain VH-CH1 domains in the Fab of the second specificity as the novel “light chain”, while transferring the corresponding light chain VL-CL domains into the heavy chain for the generation of a novel “heavy chain”. The main problem in this case is formation of byproducts represented by heavy chain dimers (formed due to the knob-into-hole complementarity and complementarity of CH1 of the first heavy chain and CL of the second heavy chain) and Fab fragment (formed due to the complementarity of the CH1 and CL domains from different light chains). In the second variant (CrossMabCH1-CL), only the CH1-CL domains are exchanged between the light and heavy chains of the second specificity. Theoretically, such replacement should prevent byproduct formation. The third variant (CrossMabVH-VL) involves a similar exchange of variable domains in the light and heavy chains of the second specificity. However, such exchange can yield an undesirable byproduct with a common light chain. This variant can be optimized by introducing charged amino acid substitutions into the constant regions of the light and heavy chains of the first specificity to prevent byproduct formation.
The simplicity of the mentioned platforms allows their application for the construction of bivalent bsAbs (with one specificity per valency, 1+1), as well as tri- and tetravalent msAbs [30].
FIT-Ig®. EpiMab has developed an original platform for creating tetravalent bsAbs based on the CrossMab approach, the so-called Fabs-In-Tandem Immunoglobulin (FIT-Ig). In this method, constant and variable domains of the light chain of the second specificity are added to the N-termini of the heavy chains with the specificity to the first antigen. The resulting bsAbs consist of two canonical light chains with the specificity to the first antigen and two light chains containing constant and variable domains of the heavy chain with the second specificity. The use of the CrossMab approach allows to fully solve the problem of correct dimerization of light and heavy chains [31, 32].
cLC®. This bsAb format is similar to a canonical immunoglobulin molecule, but its dual specificity is achieved through heterodimerization of different heavy chains using the knob-into-hole mutations. The light chain is selected so that it is complementary to both heavy chains and also to avoid the loss of functional activity or changes in the antibody physicochemical properties [33, 34].
Triomab®. In the Triomab format, bsAbs are obtained through the somatic fusion of two hybridoma cells, each expressing a unique mAb. The resulting hybrid cell co-expresses heavy and light chains of immunoglobulins from the parental cells. The process of antibody assembly yields both parental and hybrid immunoglobulins. Although the quadroma technology has been historically used for the development of bsAb production, it has serious disadvantages, such as low productivity and high product heterogeneity. The percentage of functional bsAbs in a quadroma cell line is unpredictable, and labor-intensive purification of bsAbs from the byproducts is required. Later, this technology was optimized by using mouse and rat hybridoma cells for the fusion. The content of chimeric mouse/rat bsAbs was significantly higher due to preferential dimerization of heavy and light chains of the same species, unlike random pairing in regular mouse/mouse or rat/rat quadromas [35].
Biclonics®. Merus uses the well-known bsAb format with a common light chain, except that instead of the popular knob-into-hole mutations, they used proprietary mutations (L351D and L368E in one chain and L351K and T366Kin the other chain) to create the charge-to-charge paired construct, termed DEKK, providing heterodimerization of heavy chains [33]. Thus, the Zenocutuzumab (MCLA-128) antibody was developed using this technology.
HLE-BiTE®. The HLE-BiTE platform developed by Amgen is a bispecific T-cell engager (BiTE), a class of bsAbs directing host’s cytotoxic T cells against cancer cells. BiTEs are fusion proteins consisting of two scFvs from different antibodies. One of these fragments binds to a T cell, typically through the CD3 receptor, while the other fragments bind to a malignant cell via a tumor-specific molecule. HLE is an acronym for half-life extended, because addition of the Fc fragment to the BiTE molecule increases bsAb residence time in the patient’s body.
CRIB. Alphamab has developed its own CRIB (charge repulsion induced bispecific) platform for the heavy chains heterodimerization using the knob-into-hole mutations. Optimization of this platform by introducing one amino acid substitutions into each heavy chain increased the fraction of correctly assembled heterodimers from 74 to 94% [36].
Symmetric bsAbs. Tandem sdAb-Fc®. Alphamab uses VHH fragments genetically fused with the Fc fragment of human IgG to develop new antibodies. To obtain bsAbs, two VHH fragments were fused together and the resulting VHH dimer was directly linked to the Fc fragment. Homodimerization of the Fc fragments can yield a tetravalent bsAb with two valences.
TETRABODY – ITAB®. Akesobio had developed an advanced platform called Tetrabody, which is based on adding an scFv fragment specific to the second antigen to the C-terminus of each heavy chain, resulting in the molecule with four binding sites to two targets.
DVD-Ig®. The DVD-Ig (dual variable domain immunoglobulin) format was originally described in 2007 [37, 38]. This four-chain molecule consists of two heavy chains and two light chains and has a functional Fc region and constant domains in a configuration similar to canonical IgG. However, each arm of the molecule contains two variable domains (VDs) connected by linkers in the heavy and light chain. Similar to the Tetrabody format, DVD-Ig is a tetravalent antibody capable of interacting via its two binding sites with each of two targets. A DVD-Ig antibody can be constructed by linking together the sequences of variable fragments from any pair of mAbs. It can be composed of fully human sequences to reduce immunogenicity and its serum stability is similar to that of normal IgG. It was claimed that the DVD-Ig antibodies could be efficiently produced in standard mammalian expression systems and exhibit physicochemical properties comparable to those of regular IgG despite their larger size and non-canonical structure.
IgG + TGFβ receptor. The format utilized in this context bears a resemblance to the Tetrabody format, with a notable exception. In lieu of incorporating scFv for the second specificity, the binding domain of TGF-βRII, a receptor for transforming growth factor beta that can interact with three TGF-β isoforms, has been incorporated at the C-terminus of the anti-antibody that interacts with programmed death ligand 1 (PDL1).
Fragment-based bsAbs. TriTAC®. TriTAC (trispecific T-cell activating construct) is a new T-cell engager format developed by Harpoon Therapeutics. This new class of T cell-based immunotherapy aims to achieve a superior efficacy in solid tumors compared to the golden standard that is T-cell therapy. The TriTAC format of bsAbs consists of three antigen-binding domains, one of which binds albumin to increase the serum half-life of bsAb. TriTAC molecules have a molecular weight of ~53 kDa, which makes them smaller than most T-cell engager formats with extended half-life. One of the main advantages of small-size antibodies is faster diffusion into the tissues of human solid tumors [39].
ImmTAC®. ImmTAC is a fusion protein composed of the recombinant T-cell receptor (TCR) fused via a linker to the scFv that interacts with CD3 leading to the activation of polyclonal T cells with the following release of inflammatory cytokines and cytolytic proteins resulting in direct lysis of tumor cells. A significant difference from canonical bsAbs or fused antigen-binding fragments is that ImmTAC molecules interact with tumor antigens through the TCR. While antibodies can only target secreted molecules or cell surface antigens, TCRs can recognize peptides derived from the intracellular targets and presented by the human leukocyte antigen (HLA) molecules [40, 41].
EXPRESSION SYSTEMS AND APPROACHES TO PRODUCTION OF BISPECIFIC ANTIBODIES
A demand for highly productive expression systems has been growing with the increase in the number of new bioproducts and biosimilars. The choice of a heterologous expression system is strongly determined by the economic requirements (e.g., target protein yield and production cost), as well as by the biochemical and biophysical properties of the final product (number of chains in the protein molecule, glycosylation patterns) and characteristics of the bioprocess itself.
Relatively simple proteins, such as antibody fragments fused in a single polypeptide chain, can be produced in prokaryotic hosts, usually Escherichia coli cells [42]. More complex proteins, e.g., full-size antibodies consisting of several polypeptide chains, are typically synthesized in eukaryotic, mainly mammalian, cell lines [43]. Such systems can provide correct folding, post-translational modifications, and glycosylation according similar to those in human cells. Mammalian cell can be modified for a large-scale production to produce required amounts of therapeutic antibodies [44]. In this section, we reviewed heterologous expression systems used for bsAbs production that have already demonstrated their efficacy in the industrial manufacturing of bsAbs.
Prokaryotic cells (E. coli). E. coli strains are commonly used for the biosynthesis of recombinant proteins because of numerous advantages, including high expression levels, cost effectiveness, rapid growth, and scalability. E. coli has been used for the production of many recombinant therapeutic proteins (e.g., antibodies, cytokines, and growth hormones). Antibodies can be synthesized in E. coli cells in different formats, including scFvs, Fab fragments, and even full-length antibodies. However, production of the latter in E. coli can be challenging due to the complexity of these molecules and the need for their correct folding and assembly [45].
Production of bsAbs in E. coli is also possible; however, it can be even more challenging than production of monospecific antibodies due to the complex structure of bsAbs and the need to maintain the natural antibody architecture. According to the recent study [42], several antibody fragments and antibody fragment fusion proteins produced in E. coli have been approved for the treatment of various human diseases, while full-length mAbs and bsAbs expressed in E. coli have not yet been approved.
However, recent advances in protein engineering and expression have made possible production of different formats of bsAbs in E. coli cells using various approaches. One of them is the use of fragment-based formats, such as bispecific T-cell engagers (BiTEs) or trispecific killer engagers (TriKEs), i.e., small antibody fragments expressed in E. coli and assembled into bsAbs or trispecific antibodies. This approach was employed for the development of Tebentafusp, an agent for a novel form of immunotherapy based on the ImmTAC® platform and approved for cancer treatment. It is a soluble T-cell receptor fused to the anti-CD3 scFv. The T-cell receptor domain of Tebentafusp targets cells carrying on the surface a complex of HLA*02:01 with a peptide derived from the melanoma-associated antigen gp100 [46]. This bsAb is produced in E. coli cytoplasm as inclusion bodies and needs complex refolding procedure.
Another example of successful production and purification of bsAb from E. coli cells is a molecule composed of conventional anti-CD3 Fab fragment and HER2-targeting VHH. The purified antibody triggered the T cell-mediated cytotoxicity against HER2-positive cells both in vitro and in vivo. The authors suggested that this approach could be used for the production of bsAbs targeting other tumor antigens [47].
Another approach for the biosynthesis of bsAbs in E. coli cells involves co-expression of two different half-antibodies, each consisting of a heavy chain and a light chain, that can spontaneously assemble into an intact bsAb molecule with the natural antibody architecture [48]. The authors described a method for production of bsAbs by co-culturing two strains of bacteria, each expressing a half-antibody. Using this approach, they were able to generate 28 different bsAbs. One of these antibodies that targeted both MET and EGFR receptor tyrosine kinases was shown to inhibit the corresponding signaling pathways and suppress tumor growth driven by these kinases. This method allows quick and inexpensive production of bsAbs from any two existing antibodies in quantities sufficient for early drug development and preclinical research.
There are still challenges associated with the production of both monospecific and bispecific antibodies in E. coli cells that are related to potentially irreversible aggregation and degradation of expressed proteins, as well as the lack of certain post-translational modifications [49]. Despite these challenges, E. coli remains an attractive expression system for heterologous production of bsAbs due to the high expression levels and scalability. Large-scale production of bsAbs in E. coli is possible using fermentation technologies that provide the growth of highly dense cell cultures and production of large quantities of recombinant proteins [50].
Eukaryotic cells (CHO). Expression in Chinese hamster ovary (CHO) cells is commonly used for production of recombinant proteins, in particular, canonical mAbs [51]. CHO cells are preferred for their robust cell growth, efficient post-translational modifications, and well-established standards of good manufacturing practice (GMP). These cells have been extensively studied and optimized for protein production, making them a reliable and efficient choice for the large-scale manufacturing. An important parameter for any cell line is productivity. Transient expression systems are used in early drug development due to their simplicity and speed, while later stages of drug development require the use of stable cell lines capable of producing antibodies with a consistent quality. CHO cells are currently used in the production of more than 50% of biological preparations [52].
In 1957, Theodore Puck isolated a cell from the Chinese hamster ovary tissue that was found to be immortal and had been developed into the CHO-ori cell line. Later, in 1968, CHO-ori cells were used for the creation of the CHO-K1 cell line. Both cell lines had been grown as adherent cell cultures and demonstrated limited utility for a large-scale production. In 1971, Thompson at the University of Toronto adapted another subline, now known as CHO-S, for a growth in suspension culture, thus allowing its culturing in bioreactors [53].
Currently, mAbs are produced mostly in the parental CHO cell line. As of 2017, about two third of all approved mAbs (as well as bsAbs) were produced in CHO cells [54]. By 2022, nine approved bsAbs have been produced in mammalian expression systems (excluding Catumaxomab), and six of them were synthesized in CHO cells.
Due to their advantages (rapid growth in suspension culture and high yields of recombinant proteins), CHO cells are now widely used in the production of bsAbs in both R&D and industrial settings. Also, CHO cells ensure post-translational modifications (e.g., glycosylation) important for the functioning and stability of many recombinant proteins. Some studies have shown that the same proteins, in particular, bsAbs, produced in E. coli and CHO cells demonstrate significantly different results in the functional activity tests [55, 56].
The challenge in the use of mammalian expression systems (like CHO and HEK293 cells) is the need for expensive production platforms and limited cell growth conditions to ensure cell viability and to prevent generation of undesirable byproducts. Cell culturing conditions can significantly affect post-translational modifications and functional activity of the final product [57]. To achieve the desired attributes of producing cell lines, it is essential to have a complete understanding of the biosynthetic processes and to control them. Optimizations of the production technology may be challenging during expanding the production or implementing the process in a new laboratory.
One of the challenges in obtaining pure bsAbs is correct molecule assembly. There are only a few studies that have systematically investigated the strategies for optimization of the CHO line to improving the purity and structure of the produced bsAbs. For example, it was recommended to express heavy and light chains from separate plasmids because manipulating the plasmid ratio is an easy and efficient way to optimize protein assembly. Another proposed approach is a two-stage pool selection method that increased the purity of the expressed bsAbs from 44.5 to 88.6% [58].
Another problem in the synthesis of bsAbs is formation of high-order aggregates that increase the risk of unwanted immunogenicity and should be removed, which reduces the target protein yield. It was suggested that aggregation of bsAbs in CHO cells can be caused by impaired glutathione regulation. Evaluation of the aggregate formation in the fed-batch and perfusion processes demonstrated that the use of perfusion resulted in a significantly lower bsAb aggregate content than the fed-batch process [59].
Some works have studied approaches to increasing the yield of antibodies in CHO cells. One of them is genetic engineering of cells aimed to achieve the overexpression of genes involved in the cell cycle control. It has been long known that by accelerating or slowing down the growth rate of cells at different phases of the cell cycle (G0/G1 or G2/M), it is possible to increase the yield of recombinant proteins. Thus, overexpression of CDKL3 and COX15 genes involved in the cell cycle regulation led to a significant increase in protein production [60].
An alternative way to increase cell productivity is a knockout of genes responsible for expression of host cell proteins (HCPs) secreted by the live cells or released upon cell death, as these proteins represent impurities that have to be removed from the antibody preparation. It was shown that the knockout of several HCP genes led to a significant increase in the antibody titers. The authors explained it that the blockade of HCP synthesis made available the cell resources to enhance the expression of the gene of interest, as well as helped to achieve a higher purity of the final antibody preparation [61].
Regulation of gene expression using genetic engineering is not the only way to increase cell productivity. It was shown that continuous culturing of CHO cells at low temperatures (33°C) or addition of sodium butyrate (NaBu) to the culture medium increased production of recombinant proteins presumably due to the cell cycle arrest in the G1/G0 phase. Growing the cells at low temperatures or in the presence of NaBu also led to changes in the cell transcriptome, especially, with regard to the cell cycle regulation. At the same time, the transcriptomes associated with the cell cycle and proliferation differed between the cell cultured at low temperatures and in the presence of NaBu, suggesting different mechanisms involved in the cell cycle regulation in these two approaches.
Presumably, cell culturing at low temperatures arrested cell cycle in a p53-dependent manner. p53 is a major regulator of cell cycle and apoptosis; its activation leads to the cell cycle arrest via upregulation of a series of p53-targeted genes (e.g., CDKN1A, BAX, MDM2).
On the other hand, addition of NaBu arrested cell cycle by modulating expression and activity of cyclin-dependent kinases (CDKs) and CDK inhibitors, which are regulatory proteins involved in the G-phase transition [62].
Eukaryotic cells (HEK293). HEK293 cell line has been derived from human embryonic kidney cells. HEK293 cells are another extensively used expression system for recombinant protein production, as they offer several advantages, including high proliferation rate and easy transfection procedure. Also, because of the human origin, HEK293 cells provide human-like post-translational modifications of synthesized proteins [63].
Proteins glycosylation profile is an important factor in the protein use as a biotherapeutics. Glycoproteins with proper glycosylation profiles exhibit higher potency and stability, longer half-life, and reduced immunogenicity. The main advantage of HEK293 cells as an expression platform lies in their ability to ensure specific glycosylation profiles of biotherapeutic products [64].
There are many subtypes and derivatives of HEK293 cells, including HEK293T, HEK293-F, and original HEK293 that frequently used in the production of biopharmaceuticals. For example, the HEK293E cell line (HEK293EBNA1, or 293E) is a subtype of HEK293 cells stably expressing the Epstein–Barr virus (EBV) nuclear antigen-1 (EBNA1), which allows for episomal replication of plasmids with the EBV origin of replication (oriP). Using expression vectors with oriP in HEK293-E cells can increase the yield of recombinant proteins threefold compared to analogous non-oriP vectors.
Similarly, theHEK293-6E cell line was established by expressing truncated EBNA1 lacking the Gly-Gly-Ala domain. HEK293-6E cells demonstrated improved transient expression and cell growth compared to HEK293-E cells [65].
HEK293-F cells (marketed as GIBCO brand cells) are a clonal cell line derived from HEK293 parental cells and adapted to growth in suspension cultures in serum-free media. HEK293-F cells have been used to produce several FDA-approved therapeutics, including recombinant FVIII fusion protein (rFVIIIFc) (Elosta®) and glucagon-like peptide 1 receptor agonist (Dulaglutide; Trulicity®) for the treatment of hemophilia A and type 2 diabetes, respectively.
HEK293G cells (Promega, USA) stably express cAMP GloSensor™ (20F) biosensor [66].
Commonly used HEK293T cells are derivatives of HEK293 cells established by expression of a temperature-sensitive mutant of the SV40 T-antigen. Expression of the T-antigen allows plasmids that carry the SV40 origin of replication (SV40 ori) to actively replicate in the cells [67].
HEK293 cells can also be used for a large-scale antibody production. However, there are certain challenges associated with scaling up the antibody production in HEK293 cells, including maintaining high levels of protein expression and product quality.
Some studies have shown a positive effect of the CASP8AP2 gene knockout on recombinant protein production. CASP8AP2 encodes the FLICE-associated huge protein (FLASH). During the Fas-mediated apoptosis, FLASH is a part of the death-inducing signaling complex (DISC) assembled after binding of the death receptor Fas with its ligand FasL. DISC assembly activates procaspase 8 with the formation of proteolytic caspase 8 that initiates extrinsic apoptotic cascade. FLASH also participates in the maturation of histone mRNA, regulation of histone gene transcription in the S phase of cell cycle, and activation of transcription factors cMYB and nuclear factor kappa B (NF-kB). HEK293 cells deficient by CASP8AP2 demonstrated a seven-fold increase in specific expression of recombinant luciferase and a 2.5-fold increase in expression of recombinant SEAP (secreted embryonic alkaline phosphatase) without significant effect on the cell growth and viability. Transcriptome analysis showed that disruption of the cell cycle regulation, in particular, activation of cyclin-dependent kinase inhibitor 2A (CDKN2A), contributed to the improved recombinant protein expression in cells with the CASP8AP2 deficiency. These results confirm that the CASP8AP2 gene is an important target for improving recombinant protein expression in HEK293 cells [68].
Despite that HEK293 cells provide a human-like glycosylation profile, their use for the production of therapeutic proteins is still problematic because of the high levels of terminal N-acetylgalactosamine (GalNAc) modification and low levels of sialylation. Modification of therapeutic proteins with N-glycans often triggers their rapid clearance from the patient’s bloodstream via binding to the asialoglycoprotein (ASGPR) and mannose (MR) receptors. However, recently developed glycoengineered HEK293-F cell line eliminates terminal GalNAc glycosylation and increases sialylation due to the knockout of GalNAc transferases B4GALNT3 and B4GALNT4 and overexpression of sialyltransferases ST6GAL1 and ST3GAL6. This glycoengineered cell line is well-suited for the production of therapeutic proteins with fully human N-glycosylation profile and superior pharmacokinetic properties [69].
The use of HEK293 cells for the biosynthesis of therapeutics, including bsAbs, is somewhat limited. While many studies have reported production of bsAbs by transient expression in HEK293 cells [70, 71], stable CHO cell lines are more commonly used for manufacturing biopharmaceuticals. Since 2015, seven products synthesized in HEK cells have been approved the FDA. HEK293 cells and their derivatives have been used in six cellular and gene therapy procedures for production of viral vectors.
Despite this, many bsAbs generated by transient expression are currently at the clinical development stage, which demonstrates a potential of this expression system in bsAb production. In addition to traditional approaches to the bsAb development, when each fragment is tested separately and then one or several best variants are combined into a particular bsAb format, new methods have been developed. One of them involves testing various bsAbs (up to 22,000 candidates) in a cellular assay [72]. It was reported that HEK293 cells are the most suitable system for the transient expression due to the high protein titers – the yield of antibodies expressed in HEK293 cells ranged from tens to hundreds of milligrams per liter [73-75]. However, we disagree with this opinion, as it was possible to obtain up to 150 mg/liter of antibodies (which is sufficient for the lead characterization) by transient expression in CHO cells. Nevertheless, the task of increasing antibody titers, both in the case of transient expression or synthesis in stable cell lines, remains relevant. Low production of a specific antibody can be a reason for discontinuing its further development.
It has also been shown that transient overexpression of individual subunits of the eukaryotic translation factor eIF3 (eIF3i and eIF3c) in CHO and HEK293 cells led to a moderate increase in the total amount of eIF3i (maximum increase, 40% above control) and approximately 10% increase in the rate of global protein synthesis in CHOK1 cells. Engineered HEK293 cells overexpressing eIF3i demonstrated a higher growth rate associated with the increased c-Myc expression, resulting in greater biomass, and increased recombinant protein yields. Engineered CHOK1 cells expressing eIF3c showed higher proliferation rates and improved synthesis of recombinant proteins [76].
Summarizing the above information, HEK293 cells are a versatile expression system for the production of recombinant proteins, including bsAbs. While there are certain challenges associated with using HEK293 cells for antibody production, numerous advantages offered by these cells make them a valuable tool for researchers and biotechnology companies.
CONCLUSION
Here, we provide a quick summary of bsAbs formats, platforms for their production, and successful cases of bsAbs development, from research to clinical trials. We selected therapeutic bsAb that have been successful against solid tumors in phase II and later clinical trials (table). It is hard to define the universal “silver bullet” based on the bsAb molecular features. Out of numerous eukaryotic (mammalian, plant, and insect) and prokaryotic (E. coli, Bacillus subtilis) expression platforms, only three cell-based systems are used. Often, antibody production using the state-of-the-art production platforms results in candidates with compromised physicochemical features. The use of prokaryotic expression systems for the biosynthesis of bsAbs with their complex structure is impractical and risky, as they offer only limited modifications. Therefore, it is worth searching for alternative platforms allowing improvement of the biosynthesis process, such as eukaryotic CHO and HEK293 cells. Cell optimization and engineering of biochemical pathway, utilization of various cell line derivatives, investigation of transcriptomic and genomic profiles might improve the efficiency of protein production. Tailoring a chosen platform for required protein yields is a promising direction in the use of systems for heterologous expression [77, 78].
bsAbs targeting solid tumors: expression systems, structural formats, and applications
Type of expression system | Expression system | Advantages | Disadvantages | Antibody (patent holder), clinical development stage | bsAb structure |
|---|---|---|---|---|---|
Eukaryotic | CHO | robust cell growth; effective and correct post-translational modification; well-established standards (GMP); correct folding; proper glycosylation; high product quality and yields | high cost; high risk of contamination with viruses; slow growth rate and low productivity; unstable cell lines; extensive production timeframe; complex technology | HPN424 (Harpoon Therapeutics) discontinued: prostate cancer (phase I/II) | fragment-based bsAb (TriTac) |
Zanidatamab (Zymeworks) phase III: esophageal cancer phase II: biliary cancer, colorectal cancer, endometrial cancer, gastric cancer; HER2 positive breast cancer phase I/II: solid tumors | asymmetric bsAb (Azymetric) | ||||
Navicixizumab (Mereo BioPharma/OncXerna Therapeutics) phase II: breast cancer, colorectal cancer, fallopian tube cancer, gastric cancer, ovarian cancer, peritoneal cancer | symmetric bsAb (cLC, common light chain) | ||||
HEK293 | robust cell growth; easy transfection; effective and correct human-like post-translational modifications; well-established standards (GMP); correct folding; proper glycosylation; high product quality and yield | high cost; slow growth rate and low productivity; unstable cell lines; extensive production timeframe; complex technology; risk of contamination with pathogenic human viruses; need for additional measures to provide biosafety; difficulties during approval stage (FDA, EMA); absence of established metabolic selection markers (glutamine synthetase, dihydrofolate reductase); necessity to use antibiotics | Erfonrilimab (Alphamab) phase III: non-small cell lung cancer, pancreatic cancer phase II/III: malignant thymoma phase II: gastric cancer, liver cancer, esophageal cancer, solid tumors phase I/II: triple negative breast cancer | symmetric bsAb [Tandem single-domain (sd) Ab-Fc] | |
Retlirafusp alfa (Jiangsu Hengrui Medicine) phase III: cervical cancer; gastric cancer; non-small cell lung cancer, esophageal cancer phase II/III: colorectal cancer phase II: biliary cancer, pancreatic cancer, rectal cancer, squamous cell cancer phase I/II: solid tumors | symmetric bsAb (IgG + TGFβ receptor) | ||||
HEK293 EBNA | Cibisatamab (Roche) phase I/II: non-small cell lung cancer phase I: colorectal cancer, solid tumors | asymmetric bsAb (CrossMab) | |||
RG 7827 (Roche Innovation Center Zurich) phase I/II: colorectal cancer, urogenital cancer phase I: solid tumors | asymmetric bsAb (CrossMab) | ||||
RG 6232 (Roche) phase I: malignant melanoma; uveal melanoma | asymmetric bsAb (CrossMab) | ||||
HEK293E | Vudalimab (Xencor) phase II: biliary cancer, gynecological cancer, prostate cancer, urogenital cancer | asymmetric bsAb (CrossMab) | |||
Bafisontamab (EpimAb Biotherapeutics) phase I/II: gastrointestinal cancer, non-small cell lung cancer, solid tumors | symmetric bsAb (CrossMab) | ||||
HEK293F | Ivonescimab (Akeso Biopharma) phase III: non-small cell lung cancer phase II: colorectal cancer, gynecological cancer, triple negative breast cancer phase I/II: ovarian cancer, solid tumors phase I: small cell lung cancer | symmetric bsAb (IgG-scFv) | |||
Cadonilimab (Akeso Biopharma) marketed: cervical cancer phase III: gastric cancer, liver cancer, esophageal cancer phase II: nasopharyngeal cancer; non-small cell lung cancer; renal cell carcinoma, solid tumors phase I/II: malignant melanoma; peripheral T-cell lymphoma, small cell lung cancer | symmetric bsAb (ITab) | ||||
RO 7121661 (Roche) phase II: esophageal cancer | symmetric bsAb (CrossMab) | ||||
Vanucizumab (Roche) phase I: solid tumors | symmetric bsAb (CrossMab) | ||||
RO 7247669 (Roche) phase II: non-small cell lung cancer, esophageal cancer, urogenital cancer phase I/II: malignant melanoma phase I: solid tumors | symmetric bsAb (CrossMab) | ||||
RO 7300490 (Roche) phase I: solid tumors | asymmetric bsAb (CrossMab) | ||||
Zenocutuzumab (Merus) phase II: breast cancer, non-small cell lung cancer, prostate cancer phase I/II: solid tumors | asymmetric bsAb (biclonies/biclonix) | ||||
HEK293G | Dilpacimab (AbbVie) phase II: colorectal cancer phase I: solid tumors | symmetric bsAb (DVD-Ig) | |||
Prokaryotic | E. coli | high cost-effectiveness; rapid production; rapid growth rate; scalability; well-characterized strains; established regulatory approvals; high expression levels | lack of intron removal machinery; lack of post-translational modifications; significant protein misfolding; nonhuman-like glycosylation; endotoxin accumulation; codon bias | Tebentafusp (Immunocore) marketed: uveal melanoma phase II/III: malignant melanoma | fragment-based bsAb (ImmTac) |
Unfortunately, the above-described bsAbs formats and numerous expression systems, such as B. subtilis, Streptomyces, yeast, transgenic plants, transgenic animals, and insect cells, are still ineffective in producing large quantities of recombinant proteins and are rarely used in antibody production [79].
However, a platform that might arise as a breakthrough in protein production is cell-free protein expression (CFPE). Popularized mostly by Sutro Biopharma (USA), it has become a valuable instrument in synthetic biology [80-85] not only as a platform for selection and direct evolution of polypeptides from large libraries, but also as a valuable tool for the structural and functional characterization of integral memrane proteins [86-88] and various formats of mAbs and bsAbs. CFPE uses prokaryotic and eukaryotic cell extracts that contain ribosomes, translation factors, aminoacyl-tRNA synthetases, amino acids, tRNAs, dNTPs, ATP and its regeneration system, and other necessary factors, as well as exogenous linear or circular DNA templates [85, 87]. Prokaryotic and eukaryotic CFPE systems for has found an increasing use in the production of antibody fragments [89, 90] and full-length antibodies [91] even against complex transmembrane antigens, such as G-protein coupled receptors [92, 93] and extracellular regions of other multispanning integral memrane proteins [94].
CFPE systems deserve special attention for a number of reasons. Despite the use of pro- or eukaryotic cell lysates, which allows to avoid problems associated with expression in cells, these systems have some limitations determined by the properties of a particular system and its suitability for the synthesis of a protein of choice. However, the diversity of existing platforms and approaches helps to overcome possible obstacles. Cell-free (CF) synthesis is performed mostly in two popular formats: batch synthesis and continuous exchange cell-free synthesis. The latter requires a relatively stable supply of fresh components to the reaction chamber, which extends the duration of synthetic process and, therefore, increases protein yield. The benefits of CF systems can be used in many areas, from antibody engineering to vaccine production.
However, despite the versatility and flexibility of CF systems, one of the cornerstones in the protein production is economic feasibility, and CF system require some improvements in this regard to become more competitive on the antibody manufacturing market in the future. Comparison of economic feasibility of a bacterial cell extract-based CF system to the commonly used CHO cells for the commercial protein production revealed that the production costs for the proteins synthesized in a CF system were significantly higher than for the production in CHO cells (the most expensive component was bacterial cell extract). Nonetheless, CF systems might offer greater benefits in a long-term prospect [95].
In general, eukaryotic CF systems allow protein production in amounts sufficient for direct functional analysis and, if optimized, industrial or therapeutical needs. They allow to perform rapid screening, separate the processes, and save time in the production of common and hard-to-synthesize proteins. CF systems, as well as proteins produced using this platform, are more flexible and susceptable to modifications, which is an important feature valued by the drug developers and facilitates a large-scale protein production for therapeutic demands [96, 97].
An example of successful commercial CF platform is the XpressCF+™ technology by Sutro Biopharma. This platfrom is fast, precise, and highly efficient and can generate “meet-all-the-requirements” proteins in less than 24 h. The problem of energy supply for the protein biosythesis was solved by manufacturing an E. coli-based cell extract containing required elements in a necessary form, which allowed to improve the overall quality of the system. The XpressCF+ platform enables easier incorporation of non-natural amino acids and conjugates, as well as reduces protein heterogenicity and facilitates biosynthesis of structurally complex precisely assembled products. At present, Sutro has four conjugates and bsAbs in its portfolio that have been produced with the XpressCF+™ technology and continues to expand its influence by collaborating with other companies [98, 99].
In conclusion, each platform or format have their own strengths and limitations. The choice should depend on the goals of a research project or industrial development.
Abbreviations
- BiTE:
-
bispecific T-cell engager
- bsAb:
-
bispecific antibody
- CF:
-
cell-free
- DVD-Ig:
-
dual variable domain immunoglobulin
- ImmTAC:
-
immune-mobilizing monoclonal T-cell receptor against cancer
- mAb:
-
monoclonal antibody
- msAb:
-
multispecific antibody
- TriTAC:
-
trispecific T-cell activating construct
- scFv:
-
scFv, single-chain variable fragment
- VHH:
-
variable domain of heavy-chain antibody
References
Fougner, C., Cannon, J., The, L., Smith, J. F., and Leclerc, O. (2023) Herding in the drug development pipeline, Nat. Rev. Drug Discov., 22, 617-618, https://doi.org/10.1038/d41573-023-00063-3.
Vargason, A. M., Anselmo, A. C., and Mitragotri, S. (2021) The evolution of commercial drug delivery technologies, Nat. Biomed. Eng., 5, 951-967, https://doi.org/10.1038/s41551-021-00698-w.
Wang, Z., Wang, G., Lu, H., Li, H., Tang, M., and Tong, A. (2022) Development of therapeutic antibodies for the treatment of diseases, Mol. Biomed., 3, 35, https://doi.org/10.1186/s43556-022-00100-4.
Bayer, V. (2019) An overview of monoclonal antibodies, Semin. Oncol. Nurs., 35, 150927, https://doi.org/10.1016/j.soncn.2019.08.006.
Mach, J. P. (2017) Recombinant monoclonal antibodies, from tumor targeting to cancer immunotherapy: a critical overview [in Russian], Mol. Biol. (Mosk.), 51, 1024-1038, https://doi.org/10.7868/S0026898417060131.
Jin, H., Wang, L., and Bernards, R. (2023) Rational combinations of targeted cancer therapies: background, advances and challenges, Nat. Rev. Drug Discov., 22, 213-234, https://doi.org/10.1038/s41573-022-00615-z.
Schmidt, G., Guhl, M. M., Solomayer, E. F., Wagenpfeil, G., Hammadeh, M. E., Juhasz-Boess, I., Endrikat, J., Kasoha, M., and Bohle, R. M. (2022) Immunohistochemical assessment of PD-L1 expression using three different monoclonal antibodies in triple negative breast cancer patients, Arch. Gynecol. Obstet., 306, 1689-1695, https://doi.org/10.1007/s00404-022-06529-w.
Karbyshev, M. S., Grigoryeva, E. S., Volkomorov, V. V., Kremmer, E., Huber, A., Mitrofanova, I. V., Kaigorodova, E. V., Zavyalova, M. V., Kzhyshkowska, J. G., Cherdyntseva, N. V., and Choynzonov, E. L. (2018) Development of novel monoclonal antibodies for evaluation of transmembrane prostate androgen-induced protein 1 (TMEPAI) expression patterns in gastric cancer, Pathol. Oncol. Res., 24, 427-438, https://doi.org/10.1007/s12253-017-0247-x.
Parakh, S., Lee, S. T., Gan, H. K., and Scott, A. M. (2022) Radiolabeled antibodies for cancer imaging and therapy, Cancers (Basel), 14, 1454, https://doi.org/10.3390/cancers14061454.
Wei, W., Rosenkrans, Z. T., Liu, J., Huang, G., Luo, Q. Y., and Cai, W. (2020) ImmunoPET: concept, design, and applications, Chem. Rev., 120, 3787-3851, https://doi.org/10.1021/acs.chemrev.9b00738.
Goulet, D. R., and Atkins, W. M. (2020) Considerations for the design of antibody-based therapeutics, J. Pharm. Sci., 109, 74-103, https://doi.org/10.1016/j.xphs.2019.05.031.
Chiu, M. L., Goulet, D. R., Teplyakov, A., and Gilliland, G. L. (2019) Antibody structure and function: the basis for engineering therapeutics, Antibodies (Basel), 8, 55, https://doi.org/10.3390/antib8040055.
Elshiaty, M., Schindler, H., and Christopoulos, P. (2021) Principles and current clinical landscape of multispecific antibodies against cancer, Int. J. Mol. Sci., 22, 5632, https://doi.org/10.3390/ijms22115632.
Ma, J., Mo, Y., Tang, M., Shen, J., Qi, Y., Zhao, W., Huang, Y., Xu, Y., and Qian, C. (2021) Bispecific antibodies: from research to clinical application, Front. Immunol., 12, 626616, https://doi.org/10.3389/fimmu.2021.626616.
Husain, B., and Ellerman, D. (2018) Expanding the boundaries of biotherapeutics with bispecific antibodies, BioDrugs, 32, 441-464, https://doi.org/10.1007/s40259-018-0299-9.
Gera, N. (2022) The evolution of bispecific antibodies, Expert. Opin. Biol. Ther., 22, 945-949, https://doi.org/10.1080/14712598.2022.2040987.
Du, Y., and Xu, J. (2021) Engineered bifunctional proteins for targeted cancer therapy: prospects and challenges, Adv. Mater., 33, e2103114, https://doi.org/10.1002/adma.202103114.
Chen, S. W., and Zhang, W. (2021) Current trends and challenges in the downstream purification of bispecific antibodies, Antib. Ther., 4, 73-88, https://doi.org/10.1093/abt/tbab007.
Tripathi, N. K., and Shrivastava, A. (2019) Recent developments in bioprocessing of recombinant proteins: expression hosts and process development, Front. Bioeng. Biotechnol., 7, 420, https://doi.org/10.3389/fbioe.2019.00420.
Kunert, R., and Reinhart, D. (2016) Advances in recombinant antibody manufacturing, Appl. Microbiol. Biotechnol., 100, 3451-3461, https://doi.org/10.1007/s00253-016-7388-9.
Blanco, B., Dominguez-Alonso, C., and Alvarez-Vallina, L. (2021) Bispecific immunomodulatory antibodies for cancer immunotherapy, Clin. Cancer Res., 27, 5457-5464, https://doi.org/10.1158/1078-0432.CCR-20-3770.
Labrijn, A. F., Janmaat, M. L., Reichert, J. M., and Parren, P. (2019) Bispecific antibodies: a mechanistic review of the pipeline, Nat. Rev. Drug Discov., 18, 585-608, https://doi.org/10.1038/s41573-019-0028-1.
Muller, D., Karle, A., Meissburger, B., Hofig, I., Stork, R., and Kontermann, R. E. (2007) Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin, J. Biol. Chem., 282, 12650-12660, https://doi.org/10.1074/jbc.M700820200.
Bern, M., Nilsen, J., Ferrarese, M., Sand, K. M. K., Gjolberg, T. T., Lode, H. E., Davidson, R. J., Camire, R. M., Baekkevold, E. S., Foss, S., Grevys, A., Dalhus, B., Wilson, J., Hoydahl, L. S., Christianson, G. J., Roopenian, D. C., Schlothauer, T., Michaelsen, T. E., Moe, M. C., Lombardi, S., et al. (2020) An engineered human albumin enhances half-life and transmucosal delivery when fused to protein-based biologics, Sci. Transl. Med., 12, eabb0580, https://doi.org/10.1126/scitranslmed.abb0580.
Zhang, J., Yi, J., and Zhou, P. (2020) Development of bispecific antibodies in China: overview and prospects, Antib. Ther., 3, 126-145, https://doi.org/10.1093/abt/tbaa011.
Klein, C., Sustmann, C., Thomas, M., Stubenrauch, K., Croasdale, R., Schanzer, J., Brinkmann, U., Kettenberger, H., Regula, J. T., and Schaefer, W. (2012) Progress in overcoming the chain association issue in bispecific heterodimeric IgG antibodies, mAbs, 4, 653-663, https://doi.org/10.4161/mabs.21379.
Moore, G. L., Bernett, M. J., Rashid, R., Pong, E. W., Nguyen, D. T., Jacinto, J., Eivazi, A., Nisthal, A., Diaz, J. E., Chu, S. Y., Muchhal, U. S., and Desjarlais, J. R. (2019) A robust heterodimeric Fc platform engineered for efficient development of bispecific antibodies of multiple formats, Methods, 154, 38-50, https://doi.org/10.1016/j.ymeth.2018.10.006.
Moore, G. L., Bautista, C., Pong, E., Nguyen, D. H., Jacinto, J., Eivazi, A., Muchhal, U. S., Karki, S., Chu, S. Y., and Lazar, G. A. (2011) A novel bispecific antibody format enables simultaneous bivalent and monovalent co-engagement of distinct target antigens, mAbs, 3, 546-557, https://doi.org/10.4161/mabs.3.6.18123.
Merchant, A. M., Zhu, Z., Yuan, J. Q., Goddard, A., Adams, C. W., Presta, L. G., and Carter, P. (1998) An efficient route to human bispecific IgG, Nat. Biotechnol., 16, 677-681, https://doi.org/10.1038/nbt0798-677.
Surowka, M., Schaefer, W., and Klein, C. (2021) Ten years in the making: application of CrossMab technology for the development of therapeutic bispecific antibodies and antibody fusion proteins, mAbs, 13, 1967714, https://doi.org/10.1080/19420862.2021.1967714.
Gong, S., and Wu, C. (2019) Generation of Fabs-in-tandem immunoglobulin molecules for dual-specific targeting, Methods, 154, 87-92, https://doi.org/10.1016/j.ymeth.2018.07.014.
Gong, S., Ren, F., Wu, D., Wu, X., and Wu, C. (2017) Fabs-in-tandem immunoglobulin is a novel and versatile bispecific design for engaging multiple therapeutic targets, mAbs, 9, 1118-1128, https://doi.org/10.1080/19420862.2017.1345401.
De Nardis, C., Hendriks, L. J. A., Poirier, E., Arvinte, T., Gros, P., Bakker, A. B. H., and de Kruif, J. (2017) A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G(1), J. Biol. Chem., 292, 14706-14717, https://doi.org/10.1074/jbc.M117.793497.
Klein, C., Moessner, E., Hosse, R., Bruenker, P., Umana, P., and Neumann, C. (2017) Common Light Chains and Methods of Use, Hoffmann La Roche Inc., USA.
Wu, Y., Yi, M., Zhu, S., Wang, H., and Wu, K. (2021) Recent advances and challenges of bispecific antibodies in solid tumors, Exp. Hematol. Oncol., 10, 56, https://doi.org/10.1186/s40164-021-00250-1.
Wei, H., Cai, H., Jin, Y., Wang, P., Zhang, Q., Lin, Y., Wang, W., Cheng, J., Zeng, N., Xu, T., and Zhou, A. (2017) Structural basis of a novel heterodimeric Fc for bispecific antibody production, Oncotarget, 8, 51037-51049, https://doi.org/10.18632/oncotarget.17558.
Wu, C., Ying, H., Grinnell, C., Bryant, S., Miller, R., Clabbers, A., Bose, S., McCarthy, D., Zhu, R. R., Santora, L., Davis-Taber, R., Kunes, Y., Fung, E., Schwartz, A., Sakorafas, P., Gu, J., Tarcsa, E., Murtaza, A., and Ghayur, T. (2007) Simultaneous targeting of multiple disease mediators by a dual-variable-domain immunoglobulin, Nat. Biotechnol., 25, 1290-1297, https://doi.org/10.1038/nbt1345.
DiGiammarino, E., Ghayur, T., and Liu, J. (2012) Design and generation of DVD-Ig molecules for dual-specific targeting, Methods Mol. Biol., 899, 145-156, https://doi.org/10.1007/978-1-61779-921-1_9.
Wesche, H., Aaron, W., Austin, R. J., Baeuerle, P. A., Jones, A., Lemon, B., Sexton, K., and Yu, T. (2018) Abstract 3814: TriTACs are novel T cell-engaging therapeutic proteins optimized for the treatment of solid tumors and for long serum half-life, Cancer Res., 78, 3814-3814, https://doi.org/10.1158/1538-7445.Am2018-3814.
Oates, J., Hassan, N. J., and Jakobsen, B. K. (2015) ImmTACs for targeted cancer therapy: Why, what, how, and which, Mol. Immunol., 67, 67-74, https://doi.org/10.1016/j.molimm.2015.01.024.
Holland, C. J., Crean, R. M., Pentier, J. M., de Wet, B., Lloyd, A., Srikannathasan, V., Lissin, N., Lloyd, K. A., Blicher, T. H., Conroy, P. J., Hock, M., Pengelly, R. J., Spinner, T. E., Cameron, B., Potter, E. A., Jeyanthan, A., Molloy, P. E., Sami, M., Aleksic, M., Liddy, N., et al. (2020) Specificity of bispecific T cell receptors and antibodies targeting peptide-HLA, J. Clin. Invest., 130, 2673-2688, https://doi.org/10.1172/JCI130562.
Rashid, M. H. (2022) Full-length recombinant antibodies from Escherichia coli: production, characterization, effector function (Fc) engineering, and clinical evaluation, mAbs, 14, 2111748, https://doi.org/10.1080/19420862.2022.2111748.
Lalonde, M. E., and Durocher, Y. (2017) Therapeutic glycoprotein production in mammalian cells, J. Biotechnol., 251, 128-140, https://doi.org/10.1016/j.jbiotec.2017.04.028.
Dangi, A. K., Sinha, R., Dwivedi, S., Gupta, S. K., and Shukla, P. (2018) Cell line techniques and gene editing tools for antibody production: a review, Front. Pharmacol., 9, 630, https://doi.org/10.3389/fphar.2018.00630.
Sandomenico, A., Sivaccumar, J. P., and Ruvo, M. (2020) Evolution of Escherichia coli expression system in producing antibody recombinant fragments, Int. J. Mol. Sci., 21, 6324, https://doi.org/10.3390/ijms21176324.
Damato, B. E., Dukes, J., Goodall, H., and Carvajal, R. D. (2019) Tebentafusp: T cell redirection for the treatment of metastatic uveal melanoma, Cancers (Basel), 11, 971, https://doi.org/10.3390/cancers11070971.
Lin, L., Li, L., Zhou, C., Li, J., Liu, J., Shu, R., Dong, B., Li, Q., and Wang, Z. (2018) A HER2 bispecific antibody can be efficiently expressed in Escherichia coli with potent cytotoxicity, Oncol. Lett., 16, 1259-1266, https://doi.org/10.3892/ol.2018.8698.
Spiess, C., Merchant, M., Huang, A., Zheng, Z., Yang, N. Y., Peng, J., Ellerman, D., Shatz, W., Reilly, D., Yansura, D. G., and Scheer, J. M. (2013) Bispecific antibodies with natural architecture produced by co-culture of bacteria expressing two distinct half-antibodies, Nat. Biotechnol., 31, 753-758, https://doi.org/10.1038/nbt.2621.
Patil, R. S., Anupa, A., Gupta, J. A., and Rathore, A. S. (2022) Challenges in expression and purification of functional Fab fragments in E. coli: current strategies and perspectives, Fermentation, 8, 175, https://doi.org/10.3390/fermentation8040175.
Baumgarten, T., Ytterberg, A. J., Zubarev, R. A., and de Gier, J. W. (2018) Optimizing recombinant protein production in the Escherichia coli periplasm alleviates stress, Appl. Environ. Microbiol., 84, e00270-18, https://doi.org/10.1128/AEM.00270-18.
Jyothilekshmi, I., and Jayaprakash, N. S. (2021) Trends in monoclonal antibody production using various bioreactor syst, J. Microbiol. Biotechnol., 31, 349-357, https://doi.org/10.4014/jmb.1911.11066.
Jain, N. K., Barkowski-Clark, S., Altman, R., Johnson, K., Sun, F., Zmuda, J., Liu, C. Y., Kita, A., Schulz, R., Neill, A., Ballinger, R., Patel, R., Liu, J., Mpanda, A., Huta, B., Chiou, H., Voegtli, W., and Panavas, T. (2017) A high density CHO-S transient transfection system: comparison of ExpiCHO and Expi293, Protein Express. Purif., 134, 38-46, https://doi.org/10.1016/j.pep.2017.03.018.
Wurm, F. M. (2014) 2.2 CHO History, CHO evolution and CHO genomics – an unsolvable enigma? in Animal Cell Biotechnol., 38, https://doi.org/10.1515/9783110278965.38.
Dhara, V. G., Naik, H. M., Majewska, N. I., and Betenbaugh, M. J. (2018) Recombinant antibody production in CHO and NS0 cells: differences and similarities, BioDrugs, 32, 571-584, https://doi.org/10.1007/s40259-018-0319-9.
Naddafi, F., Shirazi, F. H., Talebkhan, Y., Tabarzad, M., Barkhordari, F., Aliabadi Farahani, Z., Bayat, E., Moazzami, R., Mahboudi, F., and Davami, F. (2018) A comparative study of the bispecific monoclonal antibody, blinatumomab expression in CHO cells and E. coli, Prep. Biochem. Biotechnol., 48, 961-967, https://doi.org/10.1080/10826068.2018.1525562.
Jain, S., Aresu, L., Comazzi, S., Shi, J., Worrall, E., Clayton, J., Humphries, W., Hemmington, S., Davis, P., Murray, E., Limeneh, A. A., Ball, K., Ruckova, E., Muller, P., Vojtesek, B., Fahraeus, R., Argyle, D., and Hupp, T. R. (2016) The development of a recombinant scFv monoclonal antibody targeting canine CD20 for use in comparative medicine, PLoS One, 11, e0148366, https://doi.org/10.1371/journal.pone.0148366.
Ha, T. K., Kim, D., Kim, C. L., Grav, L. M., and Lee, G. M. (2022) Factors affecting the quality of therapeutic proteins in recombinant Chinese hamster ovary cell culture, Biotechnol. Adv., 54, 107831, https://doi.org/10.1016/j.biotechadv.2021.107831.
Ding, M., Shen, L., Xiao, L., Liu, X., and Hu, J. (2021) A cell line development strategy to improve a bispecific antibody expression purity in CHO cells, Biochem. Engin. J., 166, 107857, https://doi.org/10.1016/j.bej.2020.107857.
Sinharoy, P., Aziz, A. H., Majewska, N. I., Ahuja, S., and Handlogten, M. W. (2020) Perfusion reduces bispecific antibody aggregation via mitigating mitochondrial dysfunction-induced glutathione oxidation and ER stress in CHO cells, Sci. Rep., 10, 16620, https://doi.org/10.1038/s41598-020-73573-4.
Jaluria, P., Betenbaugh, M., Konstantopoulos, K., and Shiloach, J. (2007) Enhancement of cell proliferation in various mammalian cell lines by gene insertion of a cyclin-dependent kinase homolog, BMC Biotechnol., 7, 71, https://doi.org/10.1186/1472-6750-7-71.
Kol, S., Ley, D., Wulff, T., Decker, M., Arnsdorf, J., Schoffelen, S., Hansen, A. H., Jensen, T. L., Gutierrez, J. M., Chiang, A. W. T., Masson, H. O., Palsson, B. O., Voldborg, B. G., Pedersen, L. E., Kildegaard, H. F., Lee, G. M., and Lewis, N. E. (2020) Multiplex secretome engineering enhances recombinant protein production and purity, Nat. Commun., 11, 1908, https://doi.org/10.1038/s41467-020-15866-w.
Avello, V., Torres, M., Vergara, M., Berrios, J., Valdez-Cruz, N. A., Acevedo, C., Molina Sampayo, M., Dickson, A. J., and Altamirano, C. (2022) Enhanced recombinant protein production in CHO cell continuous cultures under growth-inhibiting conditions is associated with an arrested cell cycle in G1/G0 phase, PLoS One, 17, e0277620, https://doi.org/10.1371/journal.pone.0277620.
Dietmair, S., Hodson, M. P., Quek, L. E., Timmins, N. E., Gray, P., and Nielsen, L. K. (2012) A multi-omics analysis of recombinant protein production in Hek293 cells, PLoS One, 7, e43394, https://doi.org/10.1371/journal.pone.0043394.
Tan, E., Chin, C. S. H., Lim, Z. F. S., and Ng, S. K. (2021) HEK293 cell line as a platform to produce recombinant proteins and viral vectors, Front. Bioeng. Biotechnol., 9, 796991, https://doi.org/10.3389/fbioe.2021.796991.
Abaandou, L., Quan, D., and Shiloach, J. (2021) Affecting HEK293 cell growth and production performance by modifying the expression of specific genes, Cells, 10, 1667, https://doi.org/10.3390/cells10071667.
Cullum, S. A., Veprintsev, D. B., and Hill, S. J. (2023) Kinetic analysis of endogenous beta(2) –adrenoceptor-mediated cAMP GloSensor responses in HEK293 cells, Br. J. Pharmacol., 180, 1304-1315, https://doi.org/10.1111/bph.16008.
DuBridge, R. B., Tang, P., Hsia, H. C., Leong, P. M., Miller, J. H., and Calos, M. P. (1987) Analysis of mutation in human cells by using an Epstein–Barr virus shuttle system, Mol. Cell. Biol., 7, 379-387, https://doi.org/10.1128/mcb.7.1.379-387.1987.
Abaandou, L., Sharma, A. K., and Shiloach, J. (2021) Knockout of the caspase 8-associated protein 2 gene improves recombinant protein expression in HEK293 cells through up-regulation of the cyclin-dependent kinase inhibitor 2A gene, Biotechnol. Bioeng., 118, 186-198, https://doi.org/10.1002/bit.27561.
Uhler, R., Popa-Wagner, R., Kroning, M., Brehm, A., Rennert, P., Seifried, A., Peschke, M., Krieger, M., Kohla, G., Kannicht, C., Wiedemann, P., Hafner, M., and Rosenlocher, J. (2021) Glyco-engineered HEK 293-F cell lines for the production of therapeutic glycoproteins with human N-glycosylation and improved pharmacokinetics, Glycobiology, 31, 859-872, https://doi.org/10.1093/glycob/cwaa119.
Gunasekaran, K., Pentony, M., Shen, M., Garrett, L., Forte, C., Woodward, A., Ng, S. B., Born, T., Retter, M., Manchulenko, K., Sweet, H., Foltz, I. N., Wittekind, M., and Yan, W. (2010) Enhancing antibody Fc heterodimer formation through electrostatic steering effects: applications to bispecific molecules and monovalent IgG, J. Biol. Chem., 285, 19637-19646, https://doi.org/10.1074/jbc.M110.117382.
Schaefer, W., Regula, J. T., Bahner, M., Schanzer, J., Croasdale, R., Durr, H., Gassner, C., Georges, G., Kettenberger, H., Imhof-Jung, S., Schwaiger, M., Stubenrauch, K. G., Sustmann, C., Thomas, M., Scheuer, W., and Klein, C. (2011) Immunoglobulin domain crossover as a generic approach for the production of bispecific IgG antibodies, Proc. Natl. Acad. Sci. USA, 108, 11187-11192, https://doi.org/10.1073/pnas.1019002108.
Segaliny, A. I., Jayaraman, J., Chen, X., Chong, J., Luxon, R., Fung, A., Fu, Q., Jiang, X., Rivera, R., Ma, X., Ren, C., Zimak, J., Hedde, P. N., Shang, Y., Wu, G., and Zhao, W. (2023) A high throughput bispecific antibody discovery pipeline, Commun. Biol., 6, 380, https://doi.org/10.1038/s42003-023-04746-w.
Mazor, Y., Oganesyan, V., Yang, C., Hansen, A., Wang, J., Liu, H., Sachsenmeier, K., Carlson, M., Gadre, D. V., Borrok, M. J., Yu, X. Q., Dall'Acqua, W., Wu, H., and Chowdhury, P. S. (2015) Improving target cell specificity using a novel monovalent bispecific IgG design, mAbs, 7, 377-389, https://doi.org/10.1080/19420862.2015.1007816.
Fang, X. T., Sehlin, D., Lannfelt, L., Syvanen, S., and Hultqvist, G. (2017) Efficient and inexpensive transient expression of multispecific multivalent antibodies in Expi293 cells, Biol. Proced. Online, 19, 11, https://doi.org/10.1186/s12575-017-0060-7.
Lu, X., Ye, Y., Wang, Y., Xu, J., Sun, J., Ji, J., Zhang, Y., and Sun, X. (2023) Rapid generation of high-quality recombinant antibodies using an Expi293F expression system for a 17 beta-estradiol immunoassay, J. Hazard. Mater., 451, 131126, https://doi.org/10.1016/j.jhazmat.2023.131126.
Roobol, A., Roobol, J., Smith, M. E., Carden, M. J., Hershey, J. W. B., Willis, A. E., and Smales, C. M. (2020) Engineered transient and stable overexpression of translation factors eIF3i and eIF3c in CHOK1 and HEK293 cells gives enhanced cell growth associated with increased c-Myc expression and increased recombinant protein synthesis, Metab. Eng., 59, 98-105, https://doi.org/10.1016/j.ymben.2020.02.001.
Malm, M., Saghaleyni, R., Lundqvist, M., Giudici, M., Chotteau, V., Field, R., Varley, P. G., Hatton, D., Grassi, L., Svensson, T., Nielsen, J., and Rockberg, J. (2020) Evolution from adherent to suspension: systems biology of HEK293 cell line development, Sci. Rep., 10, 18996, https://doi.org/10.1038/s41598-020-76137-8.
Malm, M., Kuo, C. C., Barzadd, M. M., Mebrahtu, A., Wistbacka, N., Razavi, R., Volk, A. L., Lundqvist, M., Kotol, D., Tegel, H., Hober, S., Edfors, F., Graslund, T., Chotteau, V., Field, R., Varley, P. G., Roth, R. G., Lewis, N. E., Hatton, D., and Rockberg, J. (2022) Harnessing secretory pathway differences between HEK293 and CHO to rescue production of difficult to express proteins, Metab. Eng., 72, 171-187, https://doi.org/10.1016/j.ymben.2022.03.009.
Tepap, C. Z., Anissi, J., and Bounou, S. (2023) Recent strategies to achieve high production yield of recombinant protein: A review, J. Cell. Biotechnol., 9, 25-37, https://doi.org/10.3233/jcb-220084.
Perez, J. G., Stark, J. C., and Jewett, M. C. (2016) Cell-free synthetic biology: engineering beyond the cell, Cold Spring Harb. Perspect. Biol., 8, a023853, https://doi.org/10.1101/cshperspect.a023853.
Silverman, A. D., Karim, A. S., and Jewett, M. C. (2020) Cell-free gene expression: an expanded repertoire of applications, Nat. Rev. Genet., 21, 151-170, https://doi.org/10.1038/s41576-019-0186-3.
Liu, W.-Q., Zhang, L., Chen, M., and Li, J. (2019) Cell-free protein synthesis: recent advances in bacterial extract sources and expanded applications, Biochem. Eng. J., 141, 182-189, https://doi.org/10.1016/j.bej.2018.10.023.
Jin, X., and Hong, S. H. (2018) Cell-free protein synthesis for producing ‘difficult-to-express’ proteins, Biochem. Eng. J., 138, 156-164, https://doi.org/10.1016/j.bej.2018.07.013.
Kightlinger, W., Duncker, K. E., Ramesh, A., Thames, A. H., Natarajan, A., Stark, J. C., Yang, A., Lin, L., Mrksich, M., and DeLisa, M. P. (2019) A cell-free biosynthesis platform for modular construction of protein glycosylation pathways, Nat. Commun., 10, 5404, https://doi.org/10.1038/s41467-019-12024-9.
Contreras-Llano, L. E., and Tan, C. (2018) High-throughput screening of biomolecules using cell-free gene expression systems, Synth. Biol., 3, ysy012, https://doi.org/10.1093/synbio/ysy012.
Dondapati, S. K., Stech, M., Zemella, A., and Kubick, S. (2020) Cell-free protein synthesis: a promising option for future drug development, BioDrugs, 34, 327-348, https://doi.org/10.1007/s40259-020-00417-y.
Zhang, L., Guo, W., and Lu, Y. (2020) Advances in cell-free biosensors: principle, mechanism, and applications, Biotechnol. J., 15, 2000187, https://doi.org/10.1002/biot.202000187.
Ojima-Kato, T., Hashimura, D., Kojima, T., Minabe, S., and Nakano, H. (2015) In vitro generation of rabbit anti-Listeria monocytogenes monoclonal antibody using single cell based RT-PCR linked cell-free expression systems, J. Immunol. Methods, 427, 58-65, https://doi.org/10.1016/j.jim.2015.10.001.
Hashimoto, Y., Zhou, W., Hamauchi, K., Shirakura, K., Doi, T., Yagi, K., Sawasaki, T., Okada, Y., Kondoh, M., and Takeda, H. (2018) Engineered membrane protein antigens successfully induce antibodies against extracellular regions of claudin-5, Sci. Rep., 8, 8383, https://doi.org/10.1038/s41598-018-26560-9.
Lee, K.-H., and Kim, D.-M. (2018) Recent advances in development of cell-free protein synthesis systems for fast and efficient production of recombinant proteins, FEMS Microbiol. Lett., 365, fny174, https://doi.org/10.1093/femsle/fny174.
Stech, M., Merk, H., Schenk, J. A., Stocklein, W. F., Wustenhagen, D. A., Micheel, B., Duschl, C., Bier, F. F., and Kubick, S. (2012) Production of functional antibody fragments in a vesicle-based eukaryotic cell-free translation system, J. Biotechnol., 164, 220-231, https://doi.org/10.1016/j.jbiotec.2012.08.020.
Martin, R. W., Majewska, N. I., Chen, C. X., Albanetti, T. E., Jimenez, R. B. C., Schmelzer, A. E., Jewett, M. C., and Roy, V. (2017) Development of a CHO-based cell-free platform for synthesis of active monoclonal antibodies, ACS Synth. Biol., 6, 1370-1379, https://doi.org/10.1021/acssynbio.7b00001.
Takeda, H., Ogasawara, T., Ozawa, T., Muraguchi, A., Jih, P. J., Morishita, R., Uchigashima, M., Watanabe, M., Fujimoto, T., Iwasaki, T., Endo, Y., and Sawasaki, T. (2015) Production of monoclonal antibodies against GPCR using cell-free synthesized GPCR antigen and biotinylated liposome-based interaction assay, Sci. Rep., 5, 11333, https://doi.org/10.1038/srep11333.
Sonnabend, A., Spahn, V., Stech, M., Zemella, A., Stein, C., and Kubick, S. (2017) Production of G protein-coupled receptors in an insect-based cell-free system, Biotechnol. Bioeng., 114, 2328-2338, https://doi.org/10.1002/bit.26346.
Stamatis, C., and Farid, S. S. (2021) Process economics evaluation of cell-free synthesis for the commercial manufacture of antibody drug conjugates, Biotechnol. J., 16, e2000238, https://doi.org/10.1002/biot.202000238.
Stech, M., Nikolaeva, O., Thoring, L., Stocklein, W. F. M., Wustenhagen, D. A., Hust, M., Dubel, S., and Kubick, S. (2017) Cell-free synthesis of functional antibodies using a coupled in vitro transcription-translation system based on CHO cell lysates, Sci. Rep., 7, 12030, https://doi.org/10.1038/s41598-017-12364-w.
Haueis, L., Stech, M., and Kubick, S. (2022) A cell-free expression pipeline for the generation and functional characterization of nanobodies, Front. Bioeng. Biotechnol., 10, 896763, https://doi.org/10.3389/fbioe.2022.896763.
Ratner, M. (2014) Celgene wagers on Sutro’s cell-free platform to ramp up ADCs, Nat. Biotechnol., 32, 1175, https://doi.org/10.1038/nbt1214-1175.
Hanson, J., Groff, D., Carlos, A., Usman, H., Fong, K., Yu, A., Armstrong, S., Dwyer, A., Masikat, M. R., Yuan, D., Tran, C., Heibeck, T., Zawada, J., Chen, R., Hallam, T., and Yin, G. (2023) An integrated in vivo/in vitro protein production platform for site-specific antibody drug conjugates, Bioengineering (Basel), 10, 304, https://doi.org/10.3390/bioengineering10030304.
Acknowledgments
The authors are grateful to Dr. Aisha K. Lagguerre for valuable comments and proofreading of the manuscript and to Elena K. Kokinos, Kristina O. Baskakova, and Sergei A. Niskanen for valuable suggestions and corrections.
Author information
Authors and Affiliations
Contributions
A.K.M. analyzed the data and wrote the manuscript; D.O.Ch. edited the manuscript and wrote the Conclusion section; M.S.K. conceived and supervised the study, analyzed the data, and edited the manuscript.
Corresponding author
Ethics declarations
Aleksei K. Misorin, Darya O. Chernyshova, and Mikhail S. Karbyshev are the full-time employees of BIOCAD, a company that develops therapeutic antibodies to complex membrane proteins. This article does not contain description of studies involving human participants or animals performed by any of the authors.
Rights and permissions
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Misorin, A.K., Chernyshova, D.O. & Karbyshev, M.S. State-of-the-Art Approaches to Heterologous Expression of Bispecific Antibodies Targeting Solid Tumors. Biochemistry Moscow 88, 1215–1231 (2023). https://doi.org/10.1134/S0006297923090031
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297923090031