
Abstract
For a long time Nrf2 transcription factor has been attracting attention of researchers investigating phenomenon of aging. Numerous studies have investigated effects of Nrf2 on aging and cell senescence. Nrf2 is often considered as a key player in aging processes, however this needs to be proven. It should be noted that most studies were carried out on invertebrate model organisms, such as nematodes and fruit flies, but not on mammals. This paper briefly presents main mechanisms of mammalian aging and role of inflammation and oxidative stress in this process. The mechanisms of Nrf2 activity regulation, its involvement in aging and development of the senescence-associated secretory phenotype (SASP) are also discussed. Main part of this review is devoted to critical analysis of available experimental data on the role of Nrf2 in mammalian aging.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
To date, more than 1500 experimental and review papers on the Nrf2 transcription factor (nuclear factor erythroid 2-related factor 2) and aging have been published. Indeed, involvement of Nrf2 in the aging of cells and organisms is beyond doubt. Nrf2 is often referred to as the “master regulator of the aging process” [1]. If any protein is the main regulator of aging, then its activity should regulate function of the genes, expression of which changes with age and directly affects aging at the cellular and organismal levels. But is it reasonable to apply such term to Nrf2? This review is not intended to provide a detailed analysis of the effect of Nrf2 activation or inhibition on aging processes, which has been done previously [2]. Also, this review does not consider data obtained on invertebrates, since their aging mechanisms may differ from those in mammals. The purpose of this review is to critically analyze the role of Nrf2 in mammalian aging.
THEORIES AND MECHANISMS OF AGING
Aging is a complex process affecting virtually every cell of the body. The aging process includes development of mitochondrial dysfunction and oxidative stress, disruption of homeostasis, and changes in epigenome, transcriptome, and proteome of the cell [3]. These changes occur at molecular, cellular, and tissue levels. As a result, the risk of developing potentially fatal diseases and conditions rises exponentially with age.
There are many signs of aging that affect biochemical and physiological parameters of the body [4]. One of the most important signs of aging is oxidative stress, which represents an imbalance between antioxidant and prooxidant systems in favor of the latter. Reactive oxygen (ROS) and nitrogen species arising from this stress damage nucleic acids, lipids, and proteins, leading to the cell and tissue dysfunction. The widely known free-radical theory of aging postulates that free radicals are the main driving force of aging [5], although this theory has been challenged by some researchers [6]. Since mitochondria are the main sources of ROS, they are promising targets for anti-aging therapy [7]. Another important feature of aging is a condition called “inflammaging” – chronic low-grade inflammation that accompanies aging of the body [8]. It should be noted that there is a positive feedback loop between oxidative stress and inflammaging: ROS and other free radicals are important signaling molecules that trigger inflammatory responses, which, in turn, results in production of ROS and other radicals (Fig. 1).

“Vicious circle” consisting of a positive feedback loop between oxidative stress and inflammaging. Nrf2 activation as an approach to anti-aging therapy. Details are presented in the text.
Study of the causes and mechanisms of aging is an urgent task. There are many theories of aging, which, to a first approximation, can be classified into theories of programmed aging, damage accumulation theories, and combined theories [9].
The programmed aging theories postulate that aging is an ontogenetic program that provides an organism with an evolutionary advantage [10]. According to the concept of phenoptosis, death of an organism is programmed genetically [11]. This approach implies that aging is a treatable condition, which opens up new avenues for preventing age-related diseases and extending life [12].
The theories of damage accumulation challenge the postulates of the programmed aging theory, arguing that aging can be explained by the absence of natural selection in the post-reproductive life stage and that aging arises due to accumulation of mutations and metabolic byproducts, which cause damage to cells, organs, and tissues [13]. Although accumulation of damages is a spontaneous entropic process, its kinetics can be regulated both genetically and environmentally, which leads to different lifespans in different genotypes [14].
Despite the difference in definition of the driving forces and causes of aging, all theories agree that organism lifespan is determined by the dynamic interaction of two factors: (i) the process of accumulation of detrimental changes (either due to the action of the aging program, or due to damage accumulation); and (ii) counteracting mechanisms for restoring and maintaining homeostasis (either due to the antiaging program, or due to response to emerging stresses) [13, 15].
Thus, regardless of the point of view on the causes of aging, a promising direction in anti-aging therapy is targeted regulation of signaling pathways that reduce inflammation and ROS levels. One such approach is activation of the transcription factor Nrf2 (Fig. 1).
TRANSCRIPTION FACTOR Nrf2 AND ITS REGULATION
Transcription factor Nrf2 (nuclear factor (erythroid-derived 2)-like 2 (NFE2L2) or NF-E2 related factor 2) is a key player in protecting body from various stresses, including oxidative and electrophilic stress. This transcription factor belongs to the family of basic leucine zipper proteins. Nrf2 was discovered more than a quarter of century ago [16] and still attracts attention of researchers [17].
Nrf2 controls expression of ~250 genes. Products of these genes are involved in antioxidant response, redox homeostasis, detoxification of toxic compounds, mitochondrial biogenesis, and many other processes (Fig. 2). Activation of these genes protects cells from oxidative stress and inflammation [18, 19]. Nrf2 targets include HMOX1, NQO1, SOD1, GCLC, and GCLM. HMOX1 encodes heme oxygenase-1 (HO-1), responsible for degradation of pro-inflammatory free hemes and formation of anti-inflammatory compounds such as carbon monoxide (CO) and bilirubin. NAD(P)H:quinone oxidoreductase-1 (the product of NQO1) and superoxide dismutase-1 (the product of SOD1) have antioxidant activities, and GCLC and GCLM encode the heavy catalytic subunit, and the light regulatory subunit of glutamate cysteine ligase, respectively. Glutamate cysteine ligase is a key enzyme in biosynthesis of glutathione, which is the main cellular thiol required for maintaining redox homeostasis.

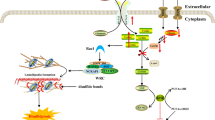
Pathways of Nrf2 activity regulation. Under normal conditions, Nrf2 in cytoplasm is in complex with its inhibitor KEAP1 [Kelch-like ECH(Nrf2)-associated protein-1], which stimulates its ubiquitinylation and proteasomal degradation. Oxidizing agents or electrophiles (E) bind to the thiol groups of the cysteine residues of KEAP1 protein. As a result, Nrf2 accumulates in the nucleus and binds to ARE (antioxidant response elements) sequences in the promoters of its target genes (HMOX1, NQO1, SOD1, GCLC, GCLM), thus stimulating their transcription. Oxidative stress also causes phosphorylation of the polyubiquitin-binding protein p62/sequestosome 1 (p62/SQSTM1), which facilitates its binding to KEAP1 and leads to ubiquitinylation and proteasome degradation of the latter. Proteasomal degradation of Nrf2 itself results from binding to the E3 ubiquitin ligases synoviolin and β-TrCP (a protein containing beta-transducin repeats). GSK3β kinase (glycogen synthase 3 beta) directly phosphorylates Nrf2 and facilitates its interaction with β-TrCP. Oxidative stress activates Akt and PI3K (phosphatidylinositol-3-kinase), thus inhibiting GSK3β and, accordingly, proteasomal degradation of Nrf2. Designations: E, electrophile; ROS, reactive oxygen species.
Induction of Nrf2 expression also leads to the decrease of inflammatory cytokine levels due to epigenetic silencing of the corresponding promoters in the immune cells [20]. The endogenous metabolite itaconate also exerts its anti-inflammatory functions through Nrf2 activation [21]. Absence of Nrf2 in mice causes an uncontrolled inflammatory response, which includes activation of innate immunity cells, high production of cytokines, chemokines, and ROS [22]. All these factors contribute to cell and tissue damage. Activation of Nrf2 is considered as a potential therapeutic approach to reduce excessive inflammatory response in sterile inflammation and some viral infections [23].
Several ways are known to regulate activity of Nrf2, which under normal conditions is a short-lived protein [24].
The first (classical) pathway is as follows: under normal conditions Nrf2 is localized in cytoplasm and associated with its inhibitor KEAP1 (Kelch-like ECH (Nrf2)-associated protein-1), which stimulates Nrf2 ubiquitinylation and its subsequent proteasomal degradation. Oxidants or electrophiles bind to KEAP1 via the -SH groups, leading to termination of the Nrf2 repression. As a result, Nrf2 avoids degradation and accumulates in the nucleus. Here, together with the set of transcription cofactors such as Maf proteins, Nrf2 interacts with specific sequences (ARE) in the gene promoters, thus stimulating their transcription [25]. It should be noted that ARE sequence is also present in the promoter of Nrf2 itself, which provides a positive feedback loop [26]. In addition to oxidants and electrophiles, Nrf2 can be activated by hydrogen sulfide and some mercaptans, which reduce disulfide bonds in the KEAP1 protein [27].
The second pathway of Nrf2 activation involves degradation of KEAP1 through the autophagy apparatus. In this case, KEAP1 interacts with the ubiquitin-binding protein p62/SQSTM1 (polyubiquitin-binding protein p62/sequestosome 1), which promotes its degradation in autophagosome and activation of Nrf2 [28]. Promoter of the gene encoding the p62/SQSTM1 protein contains the ARE sequence; its transcription is increased upon Nrf2 activation, which also contributes to its activation. Oxidative stress induces phosphorylation of Ser349 in the p62/SQSTM1 STGE motif via the mTORC1 (mammalian target of rapamycin complex 1) sensor protein. Phosphorylation leads to the increase in the affinity of p62/SQSTM1 for KEAP1 and its effective elimination [29-32]. Importantly, the p62/SQSTM1 protein is also found on the outer mitochondrial membrane and is involved in selective autophagy (mitophagy) [33]. Interaction of KEAP1 with p62 depends on the sestrin-2 protein (SESN2) [34], which is also localized in mitochondria [35].
The third pathway of Nrf2 regulation is mediated by two E3 ubiquitin ligases: β-TrCP and synoviolin (also known as Hrd1). Synoviolin is activated during endoplasmic reticulum stress (ER-stress) and reduces the amount of Nrf2 protein [36]. In addition, GSK3β kinase can directly phosphorylate Nrf2 protein, leading to its recognition by the β-TrCP ubiquitin ligase and subsequent degradation [37]. GSK3β kinase exhibits constitutive activity and is inhibited by PI3K/Akt kinases, activated during oxidative stress [38]. Thus, the β-TrCP-dependent degradation of Nrf2 is inhibited under oxidative stress.
In addition, GSK3β kinase can phosphorylate Fyn tyrosine kinase from the Src kinase family. It leads to Fyn translocation to the nucleus, where it phosphorylates Nrf2, causing Nrf2 export from the nucleus [39, 40].
The 5′-end of the untranslated region of the Nrf2 mRNA contains an internal ribosome entry site (IRES), which allows rapid translation under stress. Oxidative stress, viral infection, heat shock, nutrient deprivation, and alkylating agents all lead to activation of the cap-independent translation through IRES thus increasing the amount of Nrf2 [41, 42].
Nrf2 IN CELLULAR AGING
At the cellular level, aging is accompanied by the changes in extracellular matrix, cellular composition and morphology, and accumulation of macromolecule damage, as well as by the appearance of “senescent cells.” Cellular senescence (CS) is a cell division arrest that occurs in normally functioning tissues upon reaching the Hayflick limit. CS can also be initiated by a number of factors: DNA damage, oxidative stress, mitochondrial dysfunction, and oncogene activation [43]. On the one hand, the CS phenomenon is a fundamental biological process that is beneficial for the organism, since it provides protection against oncogenic transformation. On the other hand, senescent cells synthesize pro-inflammatory cytokines and matrix metalloproteinases, which cause inflammation and local tissue dysfunction [44, 45]. This condition is referred to as the senescence-associated secretory phenotype (SASP). Although SASP contributes markedly to inflammaging, its effect is not limited to this, but is of a complex pleiotropic nature, which has been explained in detail in the recent review by Birch and Gil [46]. Destruction of cells with SASP leads to restoration of organ and tissue functions, and is a promising anti-aging approach [47].
Nrf2 is widely known as a factor inhibiting CS processes. Nrf2 affects numerous signaling pathways, including antioxidant system, autophagy, maintaining genome integrity by p53, AMP-activated protein kinase (AMPK), nuclear factor-κB (NF-κB), etc. (reviewed by Yuan et al. [48]). Nrf2 activity is reduced during the aging of human fibroblasts, its silencing induces premature aging, and pharmacological activation of Nrf2 increases cell longevity [49]. In accordance with this, the negative Nrf2 regulator, caveolin-1, also causes premature aging of fibroblasts [50]. Normally, Nrf2 is partially localized in caveolae, invaginations of the plasma membrane, where it is in complex with its endogenous inhibitor, caveolin-1. Under oxidative stress, caveolin-1 restricts Nrf2 migration into the nucleus, thereby preventing activation of the Nrf2 target genes. Under oxidative stress the caveolin-1 overexpression leads to inhibition of Nrf2 signaling and development of premature cell aging [50]. Paradoxically, in mice with the deletion of the Nrf2 site responsible for binding to KEAP1, permanent Nrf2 activation leads to premature aging of fibroblasts and increases the probability of cancer development [51]. Most researchers consider that Nrf2 activation prevents initial oncogenic transformation of cells, but at the same time assists survival of the existing cancer cells by protecting them from oxidative stress and chemotherapeutic drugs. The ambiguous role of Nrf2 in oncogenesis is discussed in more detail in the review by Wu et al. [52].
On the one hand, there is abundant experimental evidence that Nrf2 activation reduces oxidative stress, slows CS, and reduces the SASP phenotype [53-57]. Nevertheless, excessive Nrf2 activation may induce oxidative stress by increasing NADPH oxidase NOX4 activity [58], which may contribute to CS. Thus, duration and amplitude of Nrf2 activation are extremely important for regulation of ROS and CS levels.
AGING OF MAMMALS AND Nrf2 ACTIVITY
The pioneering work of Suh et al. [59] showed that both the amount and transcriptional activity of Nrf2 are reduced in aging rats. It leads to the decrease in the expression of glutathione biosynthesis machinery, thus glutathione level drops, which contributes to the development of oxidative stress. Subsequently, a number of studies have directly or indirectly connected the decrease of Nrf2 activity with the organism aging [60]. Nrf2 activity is reduced in the skeletal muscles of sedentary elderly people [61]. In the myocardium of old mice, Nrf2 transcriptional activity was reduced, but it could be restored by moderate exercise [62].
The amount of Nrf2 or its activity also decreased with age in the mouse spinal cord [63], vascular endothelium [64], rat tongue tissue [65], rat liver [66], and also in human bronchial epithelium [67]. However, age-related changes in Nrf2 activity in the organs and tissues of mammals have not been studied sufficiently [2]. At the same time, the study did not reveal the age-related decrease of Nrf2 in macaque arteries [68]. On the contrary, other studies detected increase of Nrf2 activity in the vascular smooth muscle cells of old rats [69]. Further efforts of researchers are required to resolve these inconsistencies.
Despite the existing experimental contradictions, it can be assumed that the age-related decrease in Nrf2 activity is of universal interspecies character: rodents with high Nrf2 activity have a longer lifespan than rodents with low activity [70]. A similar situation is observed in birds – animals with high metabolic activity and high ROS production, but paradoxically long lifespan. In the Neoaves clade, representing up to 95% of all bird species, the Nrf2 system is constantly activated, which is supposed to facilitate bird adaptation to high levels of ROS and, as a result, high longevity [71]. Activation of this system occurred due to mutation of the KEAP1 gene in the Neoavian ancestor, which disrupted repression of Nrf2 by KEAP1 in the tissues and cells of wild Neoaves [71]. However, among the members of the Neoaves clade, proportion of the species with a long lifespan is not very high [72]. At the same time, other bird species that do not belong to this clade, such as swans, are characterized by high life expectancy [73]. Thus, increased activation of Nrf2 in birds cannot be the only explanation for high longevity phenomenon.
Genetic knockout of Nrf2 usually leads to the increased senescent phenotype in a variety of animal organs and tissues: hippocampus [74], skeletal muscles [75, 76], retina [77], auditory system [78], and also reduces lifespan of the female mice [79]. As a rule, these changes are accompanied by inflammatory reactions and increased levels of total and mitochondrial ROS. Nevertheless, there are also opposite examples of the effect of Nrf2 knockout on oxidative stress and signs of aging. For example, Nrf2 knockout in the aging mice reduced iron ions deposition in the brain, lowered the level of oxidative damage in the striatum, and also alleviated the age-related motor dysfunction [80]. The authors attribute this effect to the decrease in expression of ferropontin 1 in the brain endothelium. Ferropontin 1 gene expression is controlled by Nrf2: in the Nrf2-deficient animals, decrease of the level of ferropontin in the brain leads to decrease in iron deposition and, consequently, reduced oxidative burden. Therefore, it would be incorrect to consider the effect of Nrf2 transcription factor at the level of the organism as exclusively antioxidant, anti-inflammatory, and, ultimately, anti-aging. Nrf2 controls many genes, products of which have complex, pleiotropic effects on the body. For instance, excessive Nrf2 activation in the mouse liver leads to the development of hepatomegaly due to activation of the Akt signaling pathway [81].
Genetic or chemical activation of Nrf2 may lead to the reduction of signs of aging. Unfortunately, no experiments that use Nrf2 chemical inducers as anti-aging drugs have been performed so far. At the same time, Nrf2 activation by electrophiles from plant extracts (Protandim) increased median lifespan of the mice, however, only of male mice [82].
Rapamycin is one of the few compounds ability of which to prolong life in model animals has been confirmed in many independent studies [83]. Although the main target of rapamycin is mTOR, rapamycin is also capable of activating Nrf2 [84], but this property was not required to prevent CS in vitro.
Another well-known candidate for an “anti-aging drug” is the anti-diabetic drug metformin, effect of which on longevity has been verified in numerous experiments [85]. Interestingly, metformin can also activate Nrf2 in Caenorhabditis elegans [86], but apparently, in mice metformin has the opposite effect, inhibiting Nrf2 in the brain [87].
Permanent activation of Nrf2 by the genetic knockout of its negative regulator KEAP1 is extremely unfavorable for the organism: newborn knockout mice die quickly from starvation, most likely due to increased expression of α-keratins and subsequent gastrointestinal keratosis [88]. In the mice with reduced KEAP1 expression, the level of Nrf2 activation is not increased as much as in the knockout animals, providing an opportunity to investigate contribution of Nrf2 to age-related changes. These mice develop delayed aging features: progression of the age-related hearing loss is slowed down [89], and senile changes in the salivary glands are less pronounced [90]. One would expect these animals to have a longer lifespan, but there is still no experimental evidence for this. On the contrary, according to the preliminary data, decrease of the level of KEAP1 expression leads to reduced survival in the cohort of two-year-old mice [91]. An additional problem in interpreting the effect of Nrf2 on longevity is the fact that KEAP1 interacts not only with Nrf2, but also with other cellular substrates [92, 93].
At the same time, there are indirect indications that the increased Nrf2 activity can indeed prolong life: it happens in mice deficient in glutathione transferase mGSTA4-4 (mammalian glutathione-S-transferase isoform A4-4) [94]. Since this enzyme provides detoxification of the final product of lipid oxidation, 4-hydroxynonenal (4-HNE), deletion of the corresponding gene was expected to contribute to oxidative damage accumulation and life shortening. It is likely that the compensatory activation of Nrf2 in these mice not only promoted 4-HNE detoxification but also slowed down aging.
An important model organism to study aging and role of mitochondria in this process are the so-called “mutator mice” with progeroid phenotype. These transgenic animals express mitochondrial DNA polymerase gamma lacking 3′→5′ exonuclease activity, which leads to accumulation of mtDNA mutations, respiratory chain dysfunction, oxidative stress, and premature aging [95, 96]. Lessening of the mitochondrial oxidative stress with the mitochondria-targeted antioxidant SkQ1 (plastoquinolyl-10(6′-decyltriphenyl)phosphonium) prolongs lifespan of these animals [97]. A rather unexpected involvement of Nrf2 in aging of the mutator mice was observed in the recent work by Lei et al. [98]: it turned out that mtDNA migrates from mitochondria to cytoplasm and activates the interferon response, which inhibits Nrf2, leading to the increased oxidative stress and accelerated aging.
The naked mole rat is widely known as a long-lived animal resistant to cancer and age-related diseases. The naked mole rats also have some neotenia traits, which may explain their phenomenal lifespan [99]. In comparison with other rodents, naked mole rats have elevated baseline Nrf2 levels, more Nrf2 in the nucleus, increased ability of Nrf2 to bind to ARE and, consequently, higher expression of Nrf2 targets [70, 100]. Researchers suggest that Nrf2 contributes significantly to high longevity of the naked mole rat [101].
One of the few reliable ways of prolonging lifespan of an animal is caloric restriction. Pearson et al. [102] found that caloric restriction also prolongs life of the Nrf2 knockout mice, hence, Nrf2 is not involved in the life extension caused by caloric restriction.
Little is known about the role of Nrf2 in human aging. It has been suggested that Nrf2 activity may be increased in centenarians due to specific diet containing its inducers, and that it is Nrf2 that could play a key role in such long lifespan [103]. However, there is still no experimental evidence that the long-livers actually have elevated Nrf2 activity.
Nrf2 AND AGE-RELATED CHANGES IN GENE EXPRESSION PROFILE
It is logical to assume that if Nrf2 is indeed one of the central regulators of aging, then the expression profile of its target genes in aging tissues should change accordingly. Numerous omics data on the age-related changes in various tissues and organs have been accumulated so far. Some of these changes do show signs of attenuation of Nrf2 transcriptional activity. For example, transcriptomic data indicate that the brains of Nrf2 knockout mice show such changes in signaling pathways that are typical for the brains of elderly people [104].
However, vast majority of the studies using an unbiased approach to analyze the patterns of senile changes do not find evidence of a marked change in the activity of Nrf2. Transcriptome analysis of the human skin samples (30-45 years old) demonstrates a central role of TNF (tumor necrosis factor), p53, and NF-κB in the age-related changes [105]. An independent meta-analysis of databases on 18 human tissues of different ages did not reveal Nrf2 among the transcription factors controlling transcription of the genes expression of which changes with age [106], which is consistent with the earlier work (see Stegeman and Weake for a review [107]). A full-scale epigenomic and transcriptomic landscape of four tissues in the aging mouse revealed induction of inflammatory response, but not Nrf2 activation [108]. Systemic analysis of the age-related changes in the human proteome also revealed no change in the amount of proteins, which are Nrf2 targets [109]. Discovery of the so-called “epigenetic clock,” which reflects biological age of humans and animals [110], unfortunately did not reveal transcription factors activity of which changes with aging [111].
It should be noted that omics technologies describing human and animal aging face a number of objective difficulties. Firstly, different genetic backgrounds and lifestyles contribute significantly to the detected changes. Secondly, the inevitable senile diseases and pathologies also alter gene expression profiles. Thirdly, cellular composition of the tissues changes significantly with age, thus perverting the data obtained. Unfortunately, most of the studies do not use preliminary cell sorting, which does not allow to draw unequivocal conclusions. Finally, Nrf2 activation could be transient, i.e., could increase and decrease rapidly. Various factors, such as diet and drug use, can influence this process. For example, consumption of broccoli shoots containing sulforaphane (an Nrf2 inducer) increases its expression in human blood [112], and many drugs are known to be Nrf2 activators [113].
CONCLUSION
Aging in organisms is accompanied by (i) accumulation of oxidative damage and (ii) development of chronic inflammation, “inflammaging”. Activation of the transcription factor Nrf2 may affect both of these factors, slowing down development of the senile changes. An indirect confirmation of this assumption is the fact that the long-lived animals, such as naked mole rat, have an increased level of Nrf2 activation. An important area of research should aim to obtain independent experimental data on the age-related dynamics of Nrf2 activity changes in animals and humans, since there is no data of this kind.
It is tempting to speculate that in order to successfully fight aging, humans must learn how to properly activate Nrf2, like naked mole rats do. However, we should consider that the long-lived organisms have evolved to adapt to high Nrf2 activity and fine-tuned complex systems of interactions of the signaling and metabolic pathways. Therefore, simple pharmacological activation of Nrf2 to prolong life does not seem to be the most promising approach, moreover, it could increase the risks of serious side effects. There is no reliable data in the literature that unequivocally prove that Nrf2 activation actually leads to increase of the lifespan of mammals.
Aging is accompanied by the changes in gene expression profile, which is tissue- and species-specific. These changes only to a small extent include the genes controlled by Nrf2. Thus, at the moment it cannot be concluded that Nrf2 is the “master regulator of the aging process”.
Abbreviations
- ARE:
-
antioxidant response element
- CS:
-
cellular senescence
- GSK3β:
-
glycogen synthase 3 beta
- KEAP1:
-
Kelch-like ECH(Nrf2)-associated protein-1
- Nrf2:
-
nuclear factor erythroid 2-related factor 2
- p62/SQSTM:
-
polyubiquitin-binding protein p62/sequestosome 1
- ROS:
-
reactive oxygen species
- SASP:
-
senescence-associated secretory phenotype
- β-TrCP:
-
beta-transducin repeat containing protein
References
Lewis, K. N., Mele, J., Hayes, J. D., and Buffenstein, R. (2010) Nrf2, a guardian of healthspan and gatekeeper of species longevity, Integr. Comp. Biol., 50, 829-843, https://doi.org/10.1093/icb/icq034.
Shilovsky, G. A. (2022) Lability of the Nrf2/Keap/ARE cell defense system in different models of cell aging and age-related pathologies, Biochemistry (Moscow), 87, 70-85, https://doi.org/10.1134/S0006297922010060.
Van der Rijt, S., Molenaars, M., McIntyre, R. L., Janssens, G. E., and Houtkooper, R. H. (2020) Integrating the hallmarks of aging throughout the tree of life: a focus on mitochondrial dysfunction, Front. Cell Dev. Biol., 8, 594416, https://doi.org/10.3389/fcell.2020.594416.
Moskalev, A. (2019) Biomarkers of Human Aging, Springer International Publishing, https://doi.org/10.1007/978-3-030-24970-0.
Harman, D. (1955) Aging: a theory based on free radical and radiation chemistry, J. Gerontol., 3, 298-300, https://doi.org/10.1093/geronj/11.3.298.
Gladyshev, V. N. (2014) The free radical theory of aging is dead. Long live the damage theory! Antioxid. Redox Signal., 20, 727-731, https://doi.org/10.1089/ars.2013.5228.
Skulachev, V. P., Anisimov, V. N., Antonenko, Y. N., Bakeeva, L. E., Chernyak, B. V., Erichev, V. P., et al. (2009) An attempt to prevent senescence: a mitochondrial approach, Biochim. Biophys. Acta, 1787, 437-461, https://doi.org/10.1016/j.bbabio.2008.12.008.
Franceschi, C., Capri, M., Monti, D., Giunta, S., Olivieri, F., Sevini, F., et al. (2007) Inflammaging and anti-inflammaging: a systemic perspective on aging and longevity emerged from studies in humans, Mech. Ageing Dev., 128, 92-105, https://doi.org/10.1016/j.mad.2006.11.016.
Da Costa, J. P., Vitorino, R., Silva, G. M., Vogel, C., Duarte, A. C., and Rocha-Santos, T. (2016) A synopsis on aging-theories, mechanisms and future prospects, Ageing Res. Rev., 29, 90-112, https://doi.org/10.1016/j.arr.2016.06.005.
Longo, V. D., Mitteldorf, J., and Skulachev, V. P. (2005) Programmed and altruistic ageing, Nat. Rev. Genet., 6, 866-872, https://doi.org/10.1038/nrg1706.
Skulachev, V. P. (1999) Phenoptosis: programmed death of an organism, Biochemistry (Moscow), 64, 1418-1426.
Goldsmith, T. C. (2016) Evolution of aging theories: why modern programmed aging concepts are transforming medical research, Biochemistry (Moscow), 81, 1406-1412, https://doi.org/10.1134/S0006297916120026.
Johnson, F. B., Sinclair, D. A., and Guarente, L. (1999) Molecular biology of aging, Cell, 96, 291-302, https://doi.org/10.1016/s0092-8674(00)80567-x.
Carlos Aledo, J., and Maria Blanco, J. (2015) Aging is neither a failure nor an achievement of natural selection, Curr. Aging Sci., 8, 4-10, https://doi.org/10.2174/1874609808666150421130033.
Skulachev, M. V., and Skulachev, V. P. (2017) Programmed aging of mammals: proof of concept and prospects of biochemical approaches for anti-aging therapy, Biochemistry (Moscow), 82, 1403-1422, https://doi.org/10.1134/S000629791712001X.
Moi, P., Chan, K., Asunis, I., Cao, A., and Kan, Y. W. (1994) Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region, Proc. Natl. Acad. Sci. USA, 91, 9926-9930, https://doi.org/10.1073/pnas.91.21.9926.
Liu, T., Lv, Y.-F., Zhao, J.-L., You, Q.-D., and Jiang, Z.-Y. (2021) Regulation of Nrf2 by phosphorylation: consequences for biological function and therapeutic implications, Free Radic. Biol. Med., 168, 129-141, https://doi.org/10.1016/j.freeradbiomed.2021.03.034.
Ahmed, S. M. U., Luo, L., Namani, A., Wang, X. J., and Tang, X. (2017) Nrf2 signaling pathway: pivotal roles in inflammation, Biochim. Biophys. Acta. Mol. Bas. Dis., 1863, 585-597, https://doi.org/10.1016/j.bbadis.2016.11.005.
Chen, Q. M. (2021) Nrf2 for cardiac protection: pharmacological options against oxidative stress, Trends Pharmacol. Sci., 42, 729-744, https://doi.org/10.1016/j.tips.2021.06.005.
Kobayashi, E. H., Suzuki, T., Funayama, R., Nagashima, T., Hayashi, M., Sekine, H., et al. (2016) Nrf2 suppresses macrophage inflammatory response by blocking proinflammatory cytokine transcription, Nat. Commun., 7, 11624, https://doi.org/10.1038/ncomms11624.
Mills, E. L., Ryan, D. G., Prag, H. A., Dikovskaya, D., Menon, D., Zaslona, Z., et al. (2018) Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1, Nature, 556, 113-117, https://doi.org/10.1038/nature25986.
Kensler, T. W., Wakabayashi, N., and Biswal, S. (2007) Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway, Annu. Rev. Pharmacol. Toxicol., 47, 89-116, https://doi.org/10.1146/annurev.pharmtox.46.120604.141046.
Zinovkin, R. A., and Grebenchikov, O. A. (2020) Transcription factor Nrf2 as a potential therapeutic target for prevention of cytokine storm in COVID-19 patients, Biochemistry (Moscow), 85, 833-837, https://doi.org/10.1134/S0006297920070111.
Katoh, Y., Iida, K., Kang, M.-I., Kobayashi, A., Mizukami, M., Tong, K. I., et al. (2005) Evolutionary conserved N-terminal domain of Nrf2 is essential for the Keap1-mediated degradation of the protein by proteasome, Arch. Biochem. Biophys., 433, 342-350, https://doi.org/10.1016/j.abb.2004.10.012.
Motohashi, H., and Yamamoto, M. (2004) Nrf2-Keap1 defines a physiologically important stress response mechanism, Trends Mol. Med., 10, 549-557, https://doi.org/10.1016/j.molmed.2004.09.003.
Kwak, M.-K., Itoh, K., Yamamoto, M., and Kensler, T. W. (2002) Enhanced expression of the transcription factor Nrf2 by cancer chemopreventive agents: role of antioxidant response element-like sequences in the nrf2 promoter, Mol. Cell. Biol., 22, 2883-2892, https://doi.org/10.1128/mcb.22.9.2883-2892.2002.
Xie, L., Gu, Y., Wen, M., Zhao, S., Wang, W., Ma, Y., et al. (2016) Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation, Diabetes, 65, 3171-3184, https://doi.org/10.2337/db16-0020.
Jiang, T., Harder, B., Rojo de la Vega, M., Wong, P. K., Chapman, E., and Zhang, D. D. (2015) p62 links autophagy and Nrf2 signaling, Free Radic. Biol. Med., 88, 199-204, https://doi.org/10.1016/j.freeradbiomed.2015.06.014.
Hay, N., and Sonenberg, N. (2004) Upstream and downstream of mTOR, Genes Dev., 18, 1926-1945, https://doi.org/10.1101/gad.1212704.
Kim, D.-H., Sarbassov, D. D., Ali, S. M., King, J. E., Latek, R. R., Erdjument-Bromage, H., et al. (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery, Cell, 110, 163-175, https://doi.org/10.1016/s0092-8674(02)00808-5.
Ichimura, Y., Waguri, S., Sou, Y.-S., Kageyama, S., Hasegawa, J., Ishimura, R., et al. (2013) Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy, Mol. Cell, 51, 618-631, https://doi.org/10.1016/j.molcel.2013.08.003.
Saito, T., Ichimura, Y., Taguchi, K., Suzuki, T., Mizushima, T., Takagi, K., et al. (2016) p62/Sqstm1 promotes malignancy of HCV-positive hepatocellular carcinoma through Nrf2-dependent metabolic reprogramming, Nat. Commun., 7, 12030, https://doi.org/10.1038/ncomms12030.
Ma, K., Chen, G., Li, W., Kepp, O., Zhu, Y., and Chen, Q. (2020) Mitophagy, mitochondrial homeostasis, and cell fate, Front. Cell Dev. Biol., 8, 467, https://doi.org/10.3389/fcell.2020.00467.
Fan, Y., Xing, Y., Xiong, L., and Wang, J. (2020) Sestrin2 overexpression alleviates hydrogen peroxide-induced apoptosis and oxidative stress in retinal ganglion cells by enhancing Nrf2 activation via Keap1 downregulation, Chem. Biol. Interact., 324, 109086, https://doi.org/10.1016/j.cbi.2020.109086.
Kovaleva, I. E., Tokarchuk, A. V., Zheltukhin, A. O., Dalina, A. A., Safronov, G. G., Evstafieva, A. G., et al. (2020) Mitochondrial localization of SESN2, PLoS One, 15, e0226862, https://doi.org/10.1371/journal.pone.0226862.
Wu, T., Zhao, F., Gao, B., Tan, C., Yagishita, N., Nakajima, T., et al. (2014) Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis, Genes Dev., 28, 708-722, https://doi.org/10.1101/gad.238246.114.
Chowdhry, S., Zhang, Y., McMahon, M., Sutherland, C., Cuadrado, A., and Hayes, J. D. (2013) Nrf2 is controlled by two distinct β-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity, Oncogene, 32, 3765-3781, https://doi.org/10.1038/onc.2012.388.
Purdom-Dickinson, S. E., Sheveleva, E. V., Sun, H., and Chen, Q. M. (2007) Translational control of nrf2 protein in activation of antioxidant response by oxidants, Mol. Pharmacol., 72, 1074-1081, https://doi.org/10.1124/mol.107.035360.
Shang, G., Tang, X., Gao, P., Guo, F., Liu, H., Zhao, Z., et al. (2015) Sulforaphane attenuation of experimental diabetic nephropathy involves GSK-3 beta/Fyn/Nrf2 signaling pathway, J. Nutr. Biochem., 26, 596-606, https://doi.org/10.1016/j.jnutbio.2014.12.008.
Culbreth, M., Zhang, Z., and Aschner, M. (2017) Methylmercury augments Nrf2 activity by downregulation of the Src family kinase Fyn, Neurotoxicology, 62, 200-206, https://doi.org/10.1016/j.neuro.2017.07.028.
Li, W., Thakor, N., Xu, E. Y., Huang, Y., Chen, C., Yu, R., et al. (2010) An internal ribosomal entry site mediates redox-sensitive translation of Nrf2, Nucleic Acids Res., 38, 778-788, https://doi.org/10.1093/nar/gkp1048.
Lee, S. C., Zhang, J., Strom, J., Yang, D., Dinh, T. N., Kappeler, K., et al. (2017) G-quadruplex in the NRF2 mRNA 5′-untranslated region regulates de novo NRF2 protein translation under oxidative stress, Mol. Cell. Biol., 37, e00122-16, https://doi.org/10.1128/MCB.00122-16.
Kumari, R., and Jat, P. (2021) Mechanisms of cellular senescence: cell cycle arrest and senescence associated secretory phenotype, Front. Cell Dev. Biol., 9, 645593, https://doi.org/10.3389/fcell.2021.645593.
Rodier, F., Coppé, J.-P., Patil, C. K., Hoeijmakers, W. A. M., Muñoz, D. P., Raza, S. R., et al. (2009) Persistent DNA damage signalling triggers senescence-associated inflammatory cytokine secretion, Nat. Cell Biol., 11, 973-979, https://doi.org/10.1038/ncb1909.
Coppé, J.-P., Patil, C. K., Rodier, F., Sun, Y., Muñoz, D. P., Goldstein, J., et al. (2008) Senescence-associated secretory phenotypes reveal cell-nonautonomous functions of oncogenic Russian Academy of Sciences and the p53 tumor suppressor, PLoS Biol., 6, 2853-2868, https://doi.org/10.1371/journal.pbio.0060301.
Birch, J., and Gil, J. (2020) Senescence and the SASP: many therapeutic avenues, Genes Dev., 34, 1565-1576, https://doi.org/10.1101/gad.343129.120.
Cai, Y., Zhou, H., Zhu, Y., Sun, Q., Ji, Y., Xue, A., et al. (2020) Elimination of senescent cells by β-galactosidase-targeted prodrug attenuates inflammation and restores physical function in aged mice, Cell Res., 30, 574-589, https://doi.org/10.1038/s41422-020-0314-9.
Yuan, H., Xu, Y., Luo, Y., Wang, N.-X., and Xiao, J.-H. (2021) Role of Nrf2 in cell senescence regulation, Mol. Cell. Biochem., 476, 247-259, https://doi.org/10.1007/s11010-020-03901-9.
Kapeta, S., Chondrogianni, N., and Gonos, E. S. (2010) Nuclear erythroid factor 2-mediated proteasome activation delays senescence in human fibroblasts, J. Biol. Chem., 285, 8171-8184, https://doi.org/10.1074/jbc.M109.031575.
Volonte, D., Liu, Z., Musille, P. M., Stoppani, E., Wakabayashi, N., Di, Y.-P., et al. (2013) Inhibition of nuclear factor-erythroid 2-related factor (Nrf2) by caveolin-1 promotes stress-induced premature senescence, Mol. Biol. Cell, 24, 1852-1862, https://doi.org/10.1091/mbc.E12-09-0666.
Hiebert, P., Wietecha, M. S., Cangkrama, M., Haertel, E., Mavrogonatou, E., Stumpe, M., et al. (2018) Nrf2-mediated fibroblast reprogramming drives cellular senescence by targeting the matrisome, Dev. Cell, 46, 145-161.e10, https://doi.org/10.1016/j.devcel.2018.06.012.
Wu, S., Lu, H., and Bai, Y. (2019) Nrf2 in cancers: a double-edged sword, Cancer Med., 8, 2252-2267, https://doi.org/10.1002/cam4.2101.
Zhou, T., Zhang, M., Zhao, L., Li, A., and Qin, X. (2016) Activation of Nrf2 contributes to the protective effect of Exendin-4 against angiotensin II-induced vascular smooth muscle cell senescence, Am. J. Physiol. Cell Physiol., 311, C572-C582, https://doi.org/10.1152/ajpcell.00093.2016.
Romero, A., San Hipólito-Luengo, Á., Villalobos, L. A., Vallejo, S., Valencia, I., Michalska, P., et al. (2019) The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation, Aging Cell, 18, e12913, https://doi.org/10.1111/acel.12913.
Wang, Z., Chen, Z., Jiang, Z., Luo, P., Liu, L., Huang, Y., et al. (2019) Cordycepin prevents radiation ulcer by inhibiting cell senescence via NRF2 and AMPK in rodents, Nat. Commun., 10, 2538, https://doi.org/10.1038/s41467-019-10386-8.
Chen, L., Yang, R., Qiao, W., Zhang, W., Chen, J., Mao, L., et al. (2019) 1,25-Dihydroxyvitamin D exerts an antiaging role by activation of Nrf2-antioxidant signaling and inactivation of p16/p53-senescence signaling, Aging Cell, 18, e12951, https://doi.org/10.1111/acel.12951.
Lu, D., Le, Y., Ding, J., Dou, X., Mao, W., and Zhu, J. (2021) CLIC1 Inhibition protects against cellular senescence and endothelial dysfunction via the Nrf2/HO-1 pathway, Cell Biochem. Biophys., 79, 239-252, https://doi.org/10.1007/s12013-020-00959-6.
Kovac, S., Angelova, P. R., Holmström, K. M., Zhang, Y., Dinkova-Kostova, A. T., and Abramov, A. Y. (2015) Nrf2 regulates ROS production by mitochondria and NADPH oxidase, Biochim. Biophys. Acta, 1850, 794-801, https://doi.org/10.1016/j.bbagen.2014.11.021.
Suh, J. H., Shenvi, S. V., Dixon, B. M., Liu, H., Jaiswal, A. K., Liu, R.-M., et al. (2004) Decline in transcriptional activity of Nrf2 causes age-related loss of glutathione synthesis, which is reversible with lipoic acid, Proc. Natl. Acad. Sci. USA, 101, 3381-3386, https://doi.org/10.1073/pnas.0400282101.
Matsumaru, D., and Motohashi, H. (2021) The KEAP1-NRF2 system in healthy aging and longevity, Antioxidants (Basel), 10, 1929, https://doi.org/10.3390/antiox10121929.
Safdar, A., deBeer, J., and Tarnopolsky, M. A. (2010) Dysfunctional Nrf2-Keap1 redox signaling in skeletal muscle of the sedentary old, Free Radic. Biol. Med., 49, 1487-1493, https://doi.org/10.1016/j.freeradbiomed.2010.08.010.
Gounder, S. S., Kannan, S., Devadoss, D., Miller, C. J., Whitehead, K. J., Odelberg, S. J., et al. (2012) Impaired transcriptional activity of Nrf2 in age-related myocardial oxidative stress is reversible by moderate exercise training, PLoS One, 7, e45697, https://doi.org/10.1371/journal.pone.0045697.
Duan, W., Zhang, R., Guo, Y., Jiang, Y., Huang, Y., Jiang, H., et al. (2009) Nrf2 activity is lost in the spinal cord and its astrocytes of aged mice, In vitro Cell Dev. Biol. Anim., 45, 388-397, https://doi.org/10.1007/s11626-009-9194-5.
Ungvari, Z., Bailey-Downs, L., Sosnowska, D., Gautam, T., Koncz, P., Losonczy, G., et al. (2011) Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response, Am. J. Physiol. Heart Circ. Physiol., 301, H363-H372, https://doi.org/10.1152/ajpheart.01134.2010.
Baek, M.-K., Lee, H., Kim, K.-O., Kwon, H.-J., Chung, M.-H., Park, H.-M., et al. (2017) Age-related changes in nuclear factor erythroid 2-related factor 2 and reactive oxygen species and mitochondrial structure in the tongues of Fischer 344 rats, Clin. Exp. Otorhinolaryngol., 10, 357-362, https://doi.org/10.21053/ceo.2016.01095.
Shih, P.-H., and Yen, G.-C. (2007) Differential expressions of antioxidant status in aging rats: the role of transcriptional factor Nrf2 and MAPK signaling pathway, Biogerontology, 8, 71-80, https://doi.org/10.1007/s10522-006-9033-y.
Zhou, L., Zhang, H., Davies, K. J. A., and Forman, H. J. (2018) Aging-related decline in the induction of Nrf2-regulated antioxidant genes in human bronchial epithelial cells, Redox Biol., 14, 35-40, https://doi.org/10.1016/j.redox.2017.08.014.
Ungvari, Z., Bailey-Downs, L., Gautam, T., Sosnowska, D., Wang, M., Monticone, R. E., et al. (2011) Age-associated vascular oxidative stress, Nrf2 dysfunction, and NF-κB activation in the nonhuman primate Macaca mulatta, J. Gerontol. A Biol. Sci. Med. Sci., 66, 866-875, https://doi.org/10.1093/gerona/glr092.
Li, M., Liu, R.-M., Timblin, C. R., Meyer, S. G., Mossman, B. T., and Fukagawa, N. K. (2006) Age affects ERK1/2 and NRF2 signaling in the regulation of GCLC expression, J. Cell Physiol., 206, 518-525, https://doi.org/10.1002/jcp.20496.
Lewis, K. N., Wason, E., Edrey, Y. H., Kristan, D. M., Nevo, E., and Buffenstein, R. (2015) Regulation of Nrf2 signaling and longevity in naturally long-lived rodents, Proc. Natl. Acad. Sci. USA, 112, 3722-3727, https://doi.org/10.1073/pnas.1417566112.
Castiglione, G. M., Xu, Z., Zhou, L., and Duh, E. J. (2020) Adaptation of the master antioxidant response connects metabolism, lifespan and feather development pathways in birds, Nat. Commun., 11, 2476, https://doi.org/10.1038/s41467-020-16129-4.
Jones, O. R., Scheuerlein, A., Salguero-Gómez, R., Camarda, C. G., Schaible, R., Casper, B. B., et al. (2014) Diversity of ageing across the tree of life, Nature, 505, 169-173, https://doi.org/10.1038/nature12789.
Charmantier, A., Perrins, C., McCleery, R. H., and Sheldon, B. C. (2006) Quantitative genetics of age at reproduction in wild swans: support for antagonistic pleiotropy models of senescence, Proc. Natl. Acad. Sci. USA, 103, 6587-6592, https://doi.org/10.1073/pnas.0511123103.
Fulop, G. A., Kiss, T., Tarantini, S., Balasubramanian, P., Yabluchanskiy, A., Farkas, E., et al. (2018) Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation, Geroscience, 40, 513-521, https://doi.org/10.1007/s11357-018-0047-6.
Ahn, B., Pharaoh, G., Premkumar, P., Huseman, K., Ranjit, R., Kinter, M., et al. (2018) Nrf2 deficiency exacerbates age-related contractile dysfunction and loss of skeletal muscle mass, Redox Biol., 17, 47-58, https://doi.org/10.1016/j.redox.2018.04.004.
Kitaoka, Y., Tamura, Y., Takahashi, K., Takeda, K., Takemasa, T., and Hatta, H. (2019) Effects of Nrf2 deficiency on mitochondrial oxidative stress in aged skeletal muscle, Physiol. Rep., 7, e13998, https://doi.org/10.14814/phy2.13998.
Zhao, Z., Chen, Y., Wang, J., Sternberg, P., Freeman, M. L., Grossniklaus, H. E., et al. (2011) Age-related retinopathy in NRF2-deficient mice, PLoS One, 6, e19456, https://doi.org/10.1371/journal.pone.0019456.
Hoshino, T., Tabuchi, K., Nishimura, B., Tanaka, S., Nakayama, M., Ishii, T., et al. (2011) Protective role of Nrf2 in age-related hearing loss and gentamicin ototoxicity, Biochem. Biophys. Res. Commun., 415, 94-98, https://doi.org/10.1016/j.bbrc.2011.10.019.
Yoh, K., Itoh, K., Enomoto, A., Hirayama, A., Yamaguchi, N., Kobayashi, M., et al. (2001) Nrf2-deficient female mice develop lupus-like autoimmune nephritis, Kidney Int., 60, 1343-1353, https://doi.org/10.1046/j.1523-1755.2001.00939.x.
Han, K., Jin, X., Guo, X., Cao, G., Tian, S., Song, Y., et al. (2021) Nrf2 knockout altered brain iron deposition and mitigated age-related motor dysfunction in aging mice, Free Radic. Biol. Med., 162, 592-602, https://doi.org/10.1016/j.freeradbiomed.2020.11.019.
He, F., Antonucci, L., Yamachika, S., Zhang, Z., Taniguchi, K., Umemura, A., et al. (2020) NRF2 activates growth factor genes and downstream AKT signaling to induce mouse and human hepatomegaly, J. Hepatol., 72, 1182-1195, https://doi.org/10.1016/j.jhep.2020.01.023.
Strong, R., Miller, R. A., Antebi, A., Astle, C. M., Bogue, M., Denzel, M. S., et al. (2016) Longer lifespan in male mice treated with a weakly estrogenic agonist, an antioxidant, an α-glucosidase inhibitor or a Nrf2-inducer, Aging Cell, 15, 872-884, https://doi.org/10.1111/acel.12496.
Selvarani, R., Mohammed, S., and Richardson, A. (2021) Effect of rapamycin on aging and age-related diseases – past and future, GeroScience, 43, 1135-1158, https://doi.org/10.1007/s11357-020-00274-1.
Wang, R., Yu, Z., Sunchu, B., Shoaf, J., Dang, I., Zhao, S., et al. (2017) Rapamycin inhibits the secretory phenotype of senescent cells by a Nrf2-independent mechanism, Aging Cell, 16, 564-574, https://doi.org/10.1111/acel.12587.
Mohammed, I., Hollenberg, M. D., Ding, H., and Triggle, C. R. (2021) A critical review of the evidence that metformin is a putative anti-aging drug that enhances healthspan and extends lifespan, Front. Endocrinol., 12, 718942, https://doi.org/10.3389/fendo.2021.718942.
Onken, B., and Driscoll, M. (2010) Metformin induces a dietary restriction-like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN-1, PLoS One, 5, e8758, https://doi.org/10.1371/journal.pone.0008758.
Allard, J. S., Perez, E. J., Fukui, K., Carpenter, P., Ingram, D. K., and de Cabo, R. (2016) Prolonged metformin treatment leads to reduced transcription of Nrf2 and neurotrophic factors without cognitive impairment in older C57BL/6J mice, Behav. Brain Res., 301, 1-9, https://doi.org/10.1016/j.bbr.2015.12.012.
Wakabayashi, N., Itoh, K., Wakabayashi, J., Motohashi, H., Noda, S., Takahashi, S., et al. (2003) Keap1-null mutation leads to postnatal lethality due to constitutive Nrf2 activation, Nat. Genet., 35, 238-245, https://doi.org/10.1038/ng1248.
Oishi, T., Matsumaru, D., Ota, N., Kitamura, H., Zhang, T., Honkura, Y., et al. (2020) Activation of the NRF2 pathway in Keap1-knockdown mice attenuates progression of age-related hearing loss, NPJ Aging Mech. Dis., 6, 14, https://doi.org/10.1038/s41514-020-00053-4.
Wati, S. M., Matsumaru, D., and Motohashi, H. (2020) NRF2 pathway activation by KEAP1 inhibition attenuates the manifestation of aging phenotypes in salivary glands, Redox Biol., 36, 101603, https://doi.org/10.1016/j.redox.2020.101603.
Taguchi, K., Maher, J. M., Suzuki, T., Kawatani, Y., Motohashi, H., and Yamamoto, M. (2010) Genetic analysis of cytoprotective functions supported by graded expression of Keap1, Mol. Cell. Biol., 30, 3016-3026, https://doi.org/10.1128/MCB.01591-09.
Lee, D.-F., Kuo, H.-P., Liu, M., Chou, C.-K., Xia, W., Du, Y., et al. (2009) KEAP1 E3 ligase-mediated downregulation of NF-κB signaling by targeting IKKβ, Mol. Cell, 36, 131-140, https://doi.org/10.1016/j.molcel.2009.07.025.
Mulvaney, K. M., Matson, J. P., Siesser, P. F., Tamir, T. Y., Goldfarb, D., Jacobs, T. M., et al. (2016) Identification and characterization of MCM3 as a Kelch-like ECH-associated protein 1 (KEAP1) substrate, J. Biol. Chem., 291, 23719-23733, https://doi.org/10.1074/jbc.M116.729418.
Singh, S. P., Niemczyk, M., Saini, D., Sadovov, V., Zimniak, L., and Zimniak, P. (2010) Disruption of the mGsta4 gene increases life span of C57BL mice, J. Gerontol. A Biol. Sci. Med. Sci., 65, 14-23, https://doi.org/10.1093/gerona/glp165.
Trifunovic, A., Wredenberg, A., Falkenberg, M., Spelbrink, J. N., Rovio, A. T., Bruder, C. E., et al. (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase, Nature, 429, 417-423, https://doi.org/10.1038/nature02517.
Kujoth, G. C., Hiona, A., Pugh, T. D., Someya, S., Panzer, K., Wohlgemuth, S. E., et al. (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging, Science, 309, 481-484, https://doi.org/10.1126/science.1112125.
Shabalina, I. G., Vyssokikh, M. Y., Gibanova, N., Csikasz, R. I., Edgar, D., Hallden-Waldemarson, A., et al. (2017) Improved health-span and lifespan in mtDNA mutator mice treated with the mitochondrially targeted antioxidant SkQ1, Aging, 9, 315-339, https://doi.org/10.18632/aging.101174.
Lei, Y., Guerra Martinez, C., Torres-Odio, S., Bell, S. L., Birdwell, C. E., Bryant, J. D., et al. (2021) Elevated type I interferon responses potentiate metabolic dysfunction, inflammation, and accelerated aging in mtDNA mutator mice, Sci. Adv., 7, https://doi.org/10.1126/sciadv.abe7548.
Skulachev, V. P., Holtze, S., Vyssokikh, M. Y., Bakeeva, L. E., Skulachev, M. V., Markov, A. V., et al. (2017) Neoteny, prolongation of youth: from naked mole rats to “naked apes” (humans), Physiol. Rev., 97, 699-720, https://doi.org/10.1152/physrev.00040.2015.
Lewis, K. N., Mele, J., Hornsby, P. J., and Buffenstein, R. (2012) Stress resistance in the naked mole-rat: the bare essentials – a mini-review, Gerontology, 58, 453-462, https://doi.org/10.1159/000335966.
Bruns, D. R., Drake, J. C., Biela, L. M., Peelor, F. F., Miller, B. F., and Hamilton, K. L. (2015) Nrf2 signaling and the slowed aging phenotype: evidence from long-lived models, Oxid. Med. Cell. Longev., 2015, 732596, https://doi.org/10.1155/2015/732596.
Pearson, K. J., Lewis, K. N., Price, N. L., Chang, J. W., Perez, E., Cascajo, M. V., et al. (2008) Nrf2 mediates cancer protection but not prolongevity induced by caloric restriction, Proc. Natl. Acad. Sci. USA, 105, 2325-2330, https://doi.org/10.1073/pnas.0712162105.
Davinelli, S., Willcox, D. C., and Scapagnini, G. (2012) Extending healthy ageing: nutrient sensitive pathway and centenarian population, Immun. Ageing, 9, 9, https://doi.org/10.1186/1742-4933-9-9.
Rojo, A. I., Pajares, M., Rada, P., Nuñez, A., Nevado-Holgado, A. J., Killik, R., et al. (2017) NRF2 deficiency replicates transcriptomic changes in Alzheimer’s patients and worsens APP and TAU pathology, Redox Biol., 13, 444-451, https://doi.org/10.1016/j.redox.2017.07.006.
Haustead, D. J., Stevenson, A., Saxena, V., Marriage, F., Firth, M., Silla, R., et al. (2016) Transcriptome analysis of human ageing in male skin shows mid-life period of variability and central role of NF-κB, Sci. Rep., 6, 26846, https://doi.org/10.1038/srep26846.
Alfego, D., Rodeck, U., and Kriete, A. (2018) Global mapping of transcription factor motifs in human aging, PLoS One, 13, e0190457, https://doi.org/10.1371/journal.pone.0190457.
Stegeman, R., and Weake, V. M. (2017) Transcriptional signatures of aging, J. Mol. Biol., 429, 2427-2437, https://doi.org/10.1016/j.jmb.2017.06.019.
Benayoun, B. A., Pollina, E. A., Singh, P. P., Mahmoudi, S., Harel, I., Casey, K. M., et al. (2019) Remodeling of epigenome and transcriptome landscapes with aging in mice reveals widespread induction of inflammatory responses, Genome Res., 29, 697-709, https://doi.org/10.1101/gr.240093.118.
Johnson, A. A., Shokhirev, M. N., Wyss-Coray, T., and Lehallier, B. (2020) Systematic review and analysis of human proteomics aging studies unveils a novel proteomic aging clock and identifies key processes that change with age, Ageing Res. Rev., 60, 101070, https://doi.org/10.1016/j.arr.2020.101070.
Horvath, S. (2013) DNA methylation age of human tissues and cell types, Genome Biol., 14, 3156, https://doi.org/10.1186/gb-2013-14-10-r115.
Bell, C. G., Lowe, R., Adams, P. D., Baccarelli, A. A., Beck, S., Bell, J. T., et al. (2019) DNA methylation aging clocks: challenges and recommendations, Genome Biol., 20, 249, https://doi.org/10.1186/s13059-019-1824-y.
Dinkova-Kostova, A. T., Fahey, J. W., Kostov, R. V., and Kensler, T. W. (2017) KEAP1 and done? Targeting the NRF2 pathway with sulforaphane, Trends Food Sci. Technol., 69, 257-269, https://doi.org/10.1016/j.tifs.2017.02.002.
Taguchi, K., and Kensler, T. W. (2020) Nrf2 in liver toxicology, Arch. Pharm. Res., 43, 337-349, https://doi.org/10.1007/s12272-019-01192-3.
Acknowledgments
The authors are grateful to Mr. E. S. Egorov for help in editing the article.
Funding
The work was supported by the Lomonosov Moscow State University Interdisciplinary Research and Education School “Molecular Technology of Living Systems and Synthetic Biology.”
Author information
Authors and Affiliations
Contributions
R. A. Zinovkin – review concept; L. A. Zinovkina – figures preparation; R. A. Zinovkin, L. A. Zinovkina, and N. D. Kondratenko – writing and editing the text.
Corresponding author
Ethics declarations
The authors declare no conflict of interest. This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zinovkin, R.A., Kondratenko, N.D. & Zinovkina, L.A. Does Nrf2 Play a Role of a Master Regulator of Mammalian Aging?. Biochemistry Moscow 87, 1465–1476 (2022). https://doi.org/10.1134/S0006297922120045
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297922120045