
Abstract
Peptides are widely used for the diagnostics, prevention, and therapy of certain human diseases. How useful can they be for the disease caused by the SARS-CoV-2 coronavirus? In this review, we discuss the possibility of using synthetic and recombinant peptides and polypeptides for prevention of COVID-19 via blocking the interaction between the virus and its main receptor ACE2, as well as components of antiviral vaccines, in particular, against new emerging virus variants.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
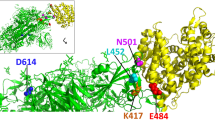
During the recent twenty years, β coronaviruses – SARS-CoV, MERS-CoV, and SARS-CoV-2 – have acquired the ability to infect human population, i.e., ceased to be exclusively animal pathogens. All these viruses cause respiratory diseases associated with acute respiratory distress syndrome and significant lung damage followed in some cases by the respiratory failure and death [1]. In December 2019, the first cases of pneumonia with an unknown etiology were detected in the city of Wuhan, Hubei province of China, which have attracted a worldwide attention. Metagenomic sequencing of RNA samples from the patients revealed a new variant of a virus from the Coronaviridae family, that was termed Wuhan-Hu-1 or WH-Human 1, also known as 2019-nCoV. The genome sequence of this virus was submitted to the GenBank under accession number MN908947. Coronaviruses are enveloped viruses with the genome represented by a ~27-32-kb single-stranded positive RNA (SARS-CoV-2 genome size is 29.9 kb), that encodes both structural and non-structural proteins. The genes in the SARS-CoV-2 genome are positioned in the following order: sixteen non-structural proteins (nsp1-16) are encoded by the 5′-end ORF1a/1b. The functions of some of these proteins are poorly understood, but it is known that most of them participate in the genome replication and in early transcription regulation. Structural proteins including outer membrane glycoprotein S, membrane glycoprotein E, matrix protein M, and nucleocapsid structural protein N, as well as several small non-structural auxiliary proteins (figure, a and b) are encoded at the 3′-end: the spike protein (S protein) is imbedded into the virion membrane and forms the “corona” on the surface of the viral particle [2]. This protein mediates virus attachment, fusion, and penetration into the cell, and comprises one of the main targets for the neutralizing antibodies produced in response to the infection, as a result inhibitors are being for suppression of the virus penetration into the cell, bioengineered antibodies, and potential therapeutic vaccines [3]. The homology between the SARS-CoV-2 genome and genome of the virus associated with the first outbreak of atypical pneumonia twenty years ago (SARS-CoV) is 80%, while the identity with the genome of bat coronavirus (CoV RaTG13) is 96.2% [4]. Although the S proteins of both viruses have very similar amino acid sequences, the SARS-CoV-2 S protein displays a higher affinity to the human angiotensin-converting enzyme 2 (ACE2) in comparison with the protein of the bat virus [5, 6]. The rate of genomic mutations for the SARS-CoV-2 virus was estimated as 1.25·10–6 nucleotides per replication cycle [7], which leads to a regular emergence of SARS-CoV-2 variants with a higher infectivity and ability to evade the immune defense of the host [8-10]. Some mutations that have emerged in the S protein sequence resulted in the increased affinity of SARS-CoV-2 to ACE2, which, likely, has enabled a very high rate of virus transmission [11-19] (figure, b). In order to determine the priorities in the monitoring and the analysis of these variants, the World Health Organization classified them into three categories, the highest attention given to the SARS-CoV-2 variants of concern (Table 1).

Schematic representation of SARS-CoV-2. a) Virion structure, b) S protein structure and mutations in virus variants. Black, red, and blue arrows indicate positions of peptide sequences selected for the development of vaccines inducing predominantly T cell-mediated immunity [30], CoVac-1 vaccine [31], and EpiVacCorona vaccine [32, 33], respectively.
Until recently the success in stopping the spread to SARS-CoV-2 virus has been very limited, and the damage caused by COVID-19 worldwide both in the healthcare sector and economics of developed countries has been significant [20]. Due to the lack of effective drugs against this particular virus, the main strategy in fighting the pandemics includes prophylactic measures, such as massive vaccination, distancing, and implementation of mask mandates. Vaccination is required for the production of neutralizing antibodies protecting an organism from the SARS-CoV-2 infection, as well as for the activation of cellular immune response, in which T lymphocytes play a central role.
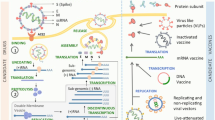
At present, there are five types of vaccines against SARS-CoV-2:
-
1)
Whole-virion vaccines that use chemically inactivated SARS-CoV-2 incapable of replication [21]. Although attenuation of live virus is a classic approach for the development of effective vaccines, no attenuated live vaccines have been reported for SARS-CoV-2. Live attenuated vaccines, as a rule, are associated with a risk of reversion, which makes this approach inapplicable for fighting highly pathogenic viruses. On the other hand, chemical inactivation could result in insufficient protective response, as in the case of Ebola virus, or cause harmful side effects, as was observed for the formaldehyde-inactivated respiratory syncytial virus in 1960s [22];
-
2)
Vaccines based on the modified mRNA packed into lipid particles have been developed for the efficient delivery of mRNA template for the S protein synthesis in cells, and they induce a strong immune response [23]. This type of vaccines was under evaluation for decades, mostly for tumor immunotherapy. Such vaccines could be adapted for a particular infectious agent and produced in large amounts much faster than in the case of other technologies;
-
3)
Vector vaccines based on the recombinant viruses for the delivery of S protein into the cell [24, 25]. Adenoviral vectors are the most studied ones; hence, they are most often used for the development of vaccines against various infectious diseases;
-
4)
Protein vaccines against SARS-CoV-2 that use recombinant S protein produced in different expression systems [26, 27]. Expressed full-size S protein can contain substitutions in the furin cleavage site [28];
-
5)
Peptide vaccines, which are to the topic of this review, are various synthetic peptides corresponding to the selected regions of viral proteins capable of inducing protective immune response [29]. They include vaccines inducing production of protective antibodies and those causing activation of T cell response [30-33].
Various platforms used for vaccine production could achieve different types of humoral and cell-mediated immune defense. It has become clear during the pandemics that the most valuable vaccine technologies should be easily adaptable for a particular virus and allow fast scaling up vaccine production.
MECHANISM OF SARS-CoV-2 INTERACTION WITH CELLS
The S proteins of SARS-CoV and SARS-CoV-2 are heavily glycosylated [34] 180 to 200-kDa transmembrane type 1 fusion proteins with the N-terminus located at the outer surface of the virus and short C-terminus buried in the inner membrane space [35]. The S protein uses ACE2 for the virus entry into the cell [36-38], while the transmembrane serine proteases (TMPRSS2) and cathepsin L initiate the rearrangements of the S protein structure [39, 40]. ACE2 is a type I transmembrane glycoprotein with the enzymatic (mono-carboxypeptidase) activity. Its main physiological function is conversion of biologically active vasoconstrictive and inflammatory peptide angiotensin-2 (8 amino acid residues) into angiotensin via cleavage of the C-terminal amino acid residues [41]. ACE2 is expressed in many organs and cells, but its highest expression was detected in the pulmonary alveolar epithelium and small intestine enterocytes [42], two major sites affected by the SARS-CoV-2. Moreover, the majority of pulmonary cells expressing ACE2 are type II cells of alveolar epithelium, which are more susceptible to the viral infection due to the high expression of genes associated with the virus replication [43]. The S protein is composed of two subunits, S1 and S2, that have two proteolytic sites: the polybasic furin cleavage site (S1/S2) and the site in the S2 subunit (S2′) [44, 45]. The cleavage at the S1/S2 site is required for the S protein activation and for the following hydrolysis of the S2′ site. Both these events are essential for the initiation of membrane fusion and virus entry into the cell.
The entry of SARS-CoV-2 into the target cells occurs in several steps. The initial cleavage of the viral S protein at the polybasic S1/S2 site (RRAR) is catalyzed by furin. This process could be considered as an “initiation” resulting in the formation of non-covalently bound subunits S1 and S2 [46]. After the cleavage, the S1/S2 protomers form the mushroom-shaped trimers on the viral membrane. Each of the protomers (receptors-binding domains or RBDs) can assume two conformations – open/up or closed/down – to bind to the receptor [47]. The S1 subunit provides direct binding with ACE2, while the S2 subunit contains sequences required for the membrane fusion, which include the fusion domain (FD) and the α-helical hydrophobic heptad repeat 1 and 2 (HR1 and HR2) domains that combine in the process of receptor binding and form a coiled coil [48]. After binding of the S1 subunit RBD to ACE2 in the presence of TMPRSS2, the S2′ site is cleaved on the cell surface. The cleavage of the S2′ site exposes the fusion peptide (FP) [49], while dissociation of S1 from S2 causes significant conformational changes in the S2 subunit, especially in HR1, resulting in the translocation of FP closer to the membrane. The pair of heptad repeat domains creates an α-helical antiparallel complex between the HR1 and HR2, which brings closer the viral and cellular membranes [50, 51]. This conformation is very stable; it is believed that it helps to overcome the high energy barrier associated with the fusion of the membranes [52]. The fusion is initiated when the hydrophobic residues of the FP are incorporated into the host cell membrane [53]. Next, the HR1 and HR2 regions of the trimeric viral transmembrane protein interact with the formation of a bundle consisting of six helices, which brings closer the viral and cellular membranes resulting in the formation of fusion pore through which viral RNA is released into the target cell cytoplasm followed by the nucleocapsid removal and initiation of the replication cycle. If the target cell expresses insufficient amounts of TMPRSS2 or if the virus/ACE2 complex does not encounter TMPRSS2, then this complex is internalized into the cell via clathrin-mediated endocytosis and delivered to the lysosomes, where the S2′ site is cleaved by cathepsins that require acidic environment for their functioning. Next, the pore is formed via the abovementioned mechanism with the help of FP followed by the delivery of viral RNA into the cytoplasm and initiation of the virus replication cycle.
All these steps – cleavage of S1/S2 and S2′ sites, attachment to the cellular receptors, conformational changes in S1 and S2, exposure of FP – play a vital role in the process of SARS-CoV-2 infection and could be potential targets for inhibitors.
SARS-CoV and SARS-CoV-2 have almost identical structures, hence, it was assumed that they have a similar mechanism of penetration. However, molecular modeling revealed a stronger interaction of the SARS-CoV-2 RBD with the ACE2 receptor. In particular, the characteristic loop in the SARS-CoV-2 S protein contains flexible glycine residues, while the same region in the SARS-CoV protein has rigid proline residues, which makes the structures of the corresponding domains different. The major role in the interaction with the receptor likely belongs to phenylalanine residue F486 located in the flexible loop [54]. It is worth mentioning that the HR2 domains in SARS-CoV-2 and SARS-CoV are identical, while the HR1 domains display a certain variability [55]. This could probably affect the interaction between HR1 and HR2 and, hence, could explain the fact that SARS-CoV-2 exhibits higher capacity of fusion with the plasma membrane [56].
THE USE OF PEPTIDES FOR THE PROPHYLAXIS AND THERAPY OF VIRAL INFECTIONS
Identification of functional domains in the protein components of infectious agents provides an opportunity of using peptides as therapeutics, because these domains can be mimicked with short amino acid sequences. The choice of sequences for the synthesis is a complicated process that requires different types of analysis of experimental data. Traditionally, computational techniques and bioinformatics resources are used, based on numerous existing platforms. For example, the NHLBI-Ab Designer platform provides information required for the selection of optimal sequences for the production of peptide-targeted antibodies. The UniprotKB/Swiss-Prot, PAComplex, ORION, SPARKS-X, and PEPstrMOD databases containing information on the structure, functions, and properties of proteins help with selection of amino acid sequences for the synthesis. X-ray diffraction data are used for the identification of sites involved in the protein binding with the receptor, which together with the structural data could facilitate optimization of binding with the protein target (ex. see [57]). Another strategy for selecting the peptides with a high affinity to the target is phage display that uses combinatorial libraries of peptides displayed on the surface of filamentous phages [58, 59].
The technology of chemical synthesis and purification of peptides is well developed, but the cost of peptide production is much higher than in the case of using recombinant DNA. However, production of target polypeptides in bacterial or eukaryotic cell cultures presents certain risk of product contamination with cell metabolites. Peptide properties, such as high specificity, low toxicity, low immunogenicity, and long-term storage stability, make them valuable therapeutic agents [60].
One of the important factors affecting peptide functionality is their three-dimensional structure. The synthesis of short peptide sequences increases the risk of losing or distorting the 3D structure of the corresponding domain in the target protein, which could result in the loss of activity. Moreover, linear unmodified peptides exhibit low stability in the circulation, as they are cleaved fast by blood plasma proteases. To avoid proteolytic degradation, non-natural D-amino acids can be included in the peptide sequence, which are more resistant to the action of proteases. The development of therapeutic peptides with D-amino acids is a complicated task that could be solved using the mirror-image phage display technology employing the screening of D-amino acid versions of target proteins against the library of phage-displayed version of the L-amino acid protein. Despite the challenges of protein synthesis from D-amino acids, the platform for the screening of proteolysis-resistant peptides against large protein targets, such as viral glycoproteins, has been developed [61]. For this purpose, a viral particle decorated with the glycoprotein from Zaire strain of Ebola virus (rVSV–ZEBOV) was produced based on the vesicular stomatitis virus. The preservation of the glycoprotein 3D structure in this system significantly simplified the subsequent analysis. It was found that human cathelicidin antimicrobial peptide LL-37 (LLGDFFRKSKEKIGKEFKRIVQRIKDFLRNLVPRTES) suppressed infection with the recombinant virus by preventing its penetration into the cells. Another approach for increasing the activity of peptides is their conjugation with cholesterol, which enhances the antiviral properties of such peptides by targeting them to the lipid sites of interaction between the virus and the cells, where their fusion occurs [62]. Three peptides conjugated with cholesterol that prevented Ebola virus infection have been described: EBOV-5 [DWTKNIKDKIDKIIHDFVDKTLPDQS-C (PEG4-Chol)], EBOV-6 [DWTKNIKDKIDKIIHDFVDKTLPDQS-C (PEG12-Chol)], and EBOV-7 [IEPHDWTKNIKDKIDKIIHDFVDKTLPDQS-C (PEG12-Chol)]. Their efficiency was confirmed in the in vitro experiments and by testing with lethal doses of viral load in a mouse model. This type of peptides is considered as very promising for the development of specific therapy [63].
Peptides that inhibit fusion and penetration of human immunodeficiency virus (HIV) have been developed as well. The entry of HIV-1 and SARS-CoV-2 into host cells has some common features. The HIV-1 infection also occurs through the fusion of cell and viral membranes mediated by the virus envelope glycoproteins gp120 and gp41. First, the surface subunit of gp120 binds to the cellular receptor CD4 and co-receptors CCR5 or CXCR4, which initiates a cascade of conformational changes activating gp41 involvement in membrane fusion. The fusion peptide at the N-terminus of gp41 is incorporated into the cell membrane. Next, the membranes of the virus and the cell fuse with the pore formation [64]. The 36-amino acid synthetic peptide T20 (also known as Enfuvirtide or Fuzeon; YTSLIHSLIEESQNQQEKNEQELLELDKWASLWNWF) corresponding to the amino acid residues 638-673 of gp41 was approved for the HIV treatment [65] and is the first in class of antiretroviral agents used in the combined therapy. Clinical use of T20 for the treatment of HIV infection demonstrated that the antiviral peptide-based drugs could be a safe and effective alternative for the therapy of infectious diseases.
Currently existing methods of influenza prophylactics are based on impacting viral proteins M2 and neuraminidase N. Both these proteins are essential for the maintenance of normal life cycle of the virus. However, mutations of influenza virus and the properties of its multicomponent genome allow an emergence of variants resistant to the existing antiviral preparations. This implies the need for the development of new strategies to fight this viral infection. New and presumably more efficient strategies have been suggested based on the use of compounds targeting hemagglutinin glycoproteins (HAs). Infection with the influenza virus starts with the interaction of viral HA with sialic acids in the receptor complex on the surface of host cells. HA changes its spatial structure following the contact with the cell, that, in turn, facilitates virus penetration into the cell, where the virus replication cycle is initiated. Using phage display technology Matsubara et al. identified the HA-binding peptide (ARLPA) that interacted with the receptor-binding site of the viral HA and inhibited infection in the cell culture [66]. In order to make the synthesized peptides resistant to proteolytic enzymes, they can be modified with non-natural amino acids, such as β-alanines and diaminovaleric acid (ornithine). The cyclic structure of synthesized peptides, i.e., the absence of C- and N-ends common for the linear and branched polypeptides, also enhances peptide resistance to hydrolysis [67]. It should be noted that the majority of discussed peptides are considered as prophylactic agents, because they often disrupt the early stages of infection process [68].
PROPHYLACTICS AND THERAPY OF SARS-CoV-2 INFECTION. MOLECULAR TARGETS
The development of specific inhibitors of enzymes vital for the virus functions (e.g., RNA-dependent RNA polymerase) continues. The drugs developed for the treatment of infections caused by other RNA-containing viruses (influenza viruses, Ebola virus) have so far demonstrated only minor effects against SARS-CoV-2 infection. That is why the main attention in the context of the new coronavirus infection is being paid to such molecular targets as the RBD of the viral S protein, as well as ACE2 and TMPRSS2 protease, as main players during virus entry into the cell. There is a hope that the combination of inhibitors of two or more targets will be effective, as it has been shown for HIV-1.
S protein. The antiviral effect of monoclonal antibodies (both prophylactic and therapeutic) is based on blocking the interaction between the virus and ACE2 and prevention of virus fusion with the cell membrane. Antibodies targeting several epitopes of the S protein, especially its RBD, could block the virus interaction with ACE2 or prevent membrane fusion; hence, this mechanism underlies the action of neutralizing antibodies and monoclonal antibody therapy [69, 70]. There are several variants of human monoclonal antibodies that inhibit virus penetration via blocking its interaction with the receptor (e.g., anti-SARS-CoVAb – antibodies 80R and S230B) [71, 72]. It should be emphasized that the antibodies introduced artificially to an organism have a limited circulation time in the recipient, and such therapy/prophylactics does not lead to the formation of immunological memory.
ACE2. Blocking ACE2 can be another strategy for the inhibition of SARS-CoV-2 interaction with the cell. Indeed, high mutation rate and the emergence of new virus variants are the factors that complicate drug development, while the structure of ACE2 remains unchanged. On the other hand, ACE2 has multiple physiological functions in an organism and its binding to SARS-CoV-2 could reduce its activity by shifting the ACE/ACE2 balance towards the ACE activity. The genes encoding ACE and ACE2 have a similar structure, but the functions of these proteins are different. Both ACE and ACE2 are components of the renin-angiotensin system (RAS). Angiotensin synthesized in the liver is cleaved by renin into angiotensin-1 (Ang1), which is then converted into angiotensin-2 (Ang2) by ACE. Ang2 is a key participant of RAS; it binds to type I angiotensin receptor (AT1R). ACE2 acts as a negative regulator of the ACE–Ang2–AT1R complex activity. It hydrolyzes Ang2 into Ang(1-7), which causes Mas receptor-mediated vasodilation, decrease of blood pressure, and initiation of apoptosis. Moreover, ACE2 could interact with Ang1 and convert it into Ang(1-9), which then can be converted into Ang(1-7) by ACE. ACE1 and angiotensin receptor inhibitors can restore the balance in the ACE/ACE2 system, but this, in turn, can promote the binding of SARS-CoV-2 with ACE2 and stimulate disease progression. An increase in the ACE activity results in pulmonary vasoconstriction, as well as inflammatory and oxidative organ damage, which increase the risk of acute organ damage and systemic inflammation [73].
If the immune system is incapable of blocking SARS-CoV-2, the virus actively penetrates into the host cell, replicates there, and destroys it when exiting to the extracellular space. As a result, this may abolish inhibition of the angiotensin metabolic pathway, which promotes infection and inflammation. It was found that transgenic mice infected with SARS-CoV display the deficit of ACE; they have an increased level of Ang2 and develop severe respiratory disorders [74]. The absence of ACE2 with its protective activity results in the RAS dysfunction and development of acute pathological respiratory states. Interestingly, the protective function of ACE2 in acute lung damage has been observed not only in the course of coronavirus infection. Restoration of lung tissues upon administration of recombinant ACE2 was observed in laboratory mice with massive pulmonary edema, under severe hypoxia, and with inflammatory cellular infiltration [74].
The canonical concept of ligand–receptor interaction suggests specific ligand binding, usually with a high affinity, followed by transduction of the intracellular signal through the cytoplasmic part of the receptor. Many receptor classes include decoy receptors that serve as molecular traps for ligands and as inhibitors of receptor signaling components. The decoy receptor of type II interleukin-1 (IL-1RII) was the first example of such inhibition in the immune system [75, 76]. Later, decoy receptors for the members of the tumor necrosis factor family have been identified [77]. Obviously, soluble decoy receptors cannot activate signaling on their own, but they can limit the availability of ligands (in particular, proinflammatory cytokines) and the level of transmitted molecular signals.
Similar strategy could be used for the development of ACE2-based antiviral drugs. In particular, recombinant soluble ACE2 (sACE2) was used as a decoy that competed for the binding with the viral S protein but did not disturb the physiological functions of ACE2 [78]. Even considering mutations, the virus has only a limited repertoire for avoiding the sACE2-mediated neutralization without simultaneous decrease in its affinity for the membrane ACE2, which makes the virus less virulent. The drug based on sACE2 is currently undergoing phase II clinical trial [79]. In parallel, several research groups have designed their own variants of sACE2 to use for the development of high-affinity decoy receptors for SARS-CoV-2 [80-82]. Thus, a soluble form of ACE2 capable of efficient binding to the S protein was selected using saturating mutagenesis. The resulting soluble decoy receptor, ACE22.v2.4 (19-805 aa), contained 3 amino acid substitutions (T27Y, L79T, N330Y) and represented a highly soluble and stable monodisperse dimer. This protein was comparable in activity with the neutralizing antibodies and interacted with S proteins of both SARS-CoV-2 and SARS-CoV. Due to its high activity and ease of production, sACE22.v2.4 is considered as a good candidate for the pre-clinical and clinical trials. Despite the similarity to the natural ACE2, recombinant high-affinity decoy receptors could have additional variations at the interaction surface or in its vicinity, so that the virus spikes might be able to distinguish between the artificial and wild-type receptors [83].
What is the minimal length of the ACE2 fragment that can efficiently compete for the binding with the S protein? This important issue was addressed in a series of recent studies, that provided rather controversial results. One of the first peptides developed based on the structural analysis of ACE2 binding with the RBDs of SARS-CoV-2 and SARS-CoV, the 23-mer peptide SBP1 (IEEQAKTFLDKFNHEAEDLFYQS), was used in the study by Zhang et al. [84]. The peptide was designed based on the α1-helix in ACE2. Although the peptide was able to bind to the SARS-CoV-2 RBD, no data were presented on the inhibitsion of infection either in vitro or in vivo. The peptides based on the α1-helix of ACE2 were also used by the Kapoyan et al. for the development of non-modified peptide mimics with a high propensity for helix formation. However, an increase in the helical structure content resulted in the increase in hydrophobicity, which affected the solubility of the peptide, and, hence, lack of the neutralizing effect against the virus. The replacement of leucine by tyrosine caused a slight decrease in the helical content and significantly increased the neutralizing activity against SARS-CoV-2 (100% efficiency at a concentration of 1 µM in the infection of Calu-3 cells) [85].
Helical regions are among the main secondary structure elements in the protein–protein interactions. Very often, such elements are structurally unstable and sensitive to the proteolysis with peptidases. These limitations can be resolved by peptide optimization, such as incorporation of non-natural amino acid side chains or other chemical modifications. In order to stabilize the α-helical conformation in a peptide, non-natural amino acids should be located on the same side of the helix. These non-natural amino acids are then linked covalently with carbohydrate chains or disulfide bridges. These so-called “stapled” peptides demonstrate a significantly better pharmacological efficiency, increased affinity to the targets, and higher resistance to proteolytic cleavage. In comparison with low-molecular-weight drugs, which also often produce toxic metabolites, peptides show better binding due to their ability to interact with a large surface area of the protein molecule with a high selectivity and lesser toxicity [86, 87]. In particular, Morgan et al. used chemical methods to stabilize the peptides in order to maintain their helical structure [88]. The carbohydrate staples were introduced in the region of the kink in the α1 helix in ACE2, relative to the position of the histidine – H34. The staples were positioned at both sides of this histidine residue, as well as across the kink. Several stapled peptides have been produced, one of which (peptide 8: IEEQAKTFLDKFNHER8EDLFYQS5) demonstrated the highest helical content (72%). However, even this peptide did not block SARS-CoV-2 entry into the cells in in vitro experiments. The authors also used the cross-linked peptides, including the G-link peptide (IEEQAKTFLDKFNHEAEDLFYQSS-G-LGKGDFR) that had been previously found to neutralize SARS-CoV by another research group (see [88]), but observed no protective effect. Independently, Curreli et al. [89] developed a panel of longer 26-mer cross-linked peptides based on the sequence of ACE2 receptor. Three of these peptides demonstrated neutralizing activity in the in vitro system based on the pseudoviruses carrying the SARS-CoV-2 S protein and luciferase (marker) and ACE2-overexpressing cultured cells. These cross-linked peptides exhibited the blocking potential by acting as decoy sites for SARS-CoV-2 binding [89]. The structure of these peptides was stabilized by covalent cross-linking that provided additional stability to the α-helix, increased peptide resistance to proteolytic cleavage, and enhanced their blocking efficiency. In particular, peptides NYBSP-1 (TIEEQAKT-X-LDK-X-NHEAEDLFYQ-X-SLA-X-WN) and NYBSP-4 (TIEEQ-Z-KTFLDK-X-NHEAEDLFYQ-X-SLA-X-WN) neutralized 80-90% of the pseudovirus particles depending on concentration. It should be mentioned that the results of this work were confirmed in our studies.
Bibilashvili et al. [90] constructed and evaluated chimeric peptides X1 and X2 (Table 2) based on the linear peptides h1 (IEEQAKTFLDKFNHEAEDLFYk) and h2 (DKWSAFLKEQSTIAQNleYPLQECI) corresponding to the fragments 21-42 and 64-88 of the α1- and α2-helices, respectively, of the extracellular ACE2 domain. The majority of amino acid residues participating in the interaction with the viral RBD were kept unchanged, but the X1 and X2 molecules differed in the location of disulfide bridges. The peptide X1 was a dimer, in which the terminal cysteine residues in the precursor h1 and h2 molecules were linked by a disulfide bond. The disulfide bridges in the X2 molecule were located in the middle of each precursor molecules. However, neither peptide noticeably neutralized the virus in the in vitro system based on Vero cells and SARS-CoV-2 virus [90]. Similar studies were conducted by our group using the 32-mer synthetic peptide P1 (STIEEQAKTFLDKFNHEAEDLFYQSSLASWNY) that included the entire α1-helix and the 80-mer recombinant peptide P2 (MSTIEEQAKTFLDKFNHEAEDLFYQSSLASWNYNTNITEENVQNMNNAGDKWSAFLKEQSTLAQMYPLQEIQNLTVKLQLQALQHHHHHH) that contained both α1- and α2-helices, as well as their derivatives based on the analysis of contact sites between the S protein RBD and ACE2 [91]. These peptides demonstrated good binding to the SARS-CoV-2 S protein in ELISA, but did not exhibit any neutralizing activity in the pseudovirus-based system and in the experiments with live SARS-CoV-2. They also did not prevent virus entry into HEK293T/ACE2 and Vero cells, which is in agreement with the conclusions of studies [88, 90], but contradicts the results of [89].
Finally, Larue et al. [92] synthesized and investigated short peptide inhibitors that could block the interaction of the S protein with ACE2. It was found that 17 out of 20 amino acid residues in the ACE2 molecule that formed contacts with the RBDs of SARS-CoV and SARS-CoV-2 were identical. Two peptides have been identified, SAP1 (27-TFLDKFNHEAEDLFYQ-42) and SAP6 (37-EDLFYQ-42), that inhibited virus entry into the cells in the in vitro experiments. The most interesting was the SAP6 peptide that contained the minimal consensus EDLFYQ including only 6 amino acids of the N-terminal α1-helix of ACE2 [92]. Considering the negative data on the infection blockade obtained by different research groups (excluding [89]), the effect of such short peptides seems doubtful. Short anti-ACE2 peptides have been also evaluated, dimerization of which increased their neutralizing activity against SARS-CoV-2. It was reported that many out of 26 analyzed peptides significantly affected the biological activity of the recombinant human ACE2. However, the peptide CPS4 (NNYLWWMTEYHD) and its dimer effectively blocked virus entry into the cell, but did not affect the functional activity of ACE2 [93]. It should be mentioned that no results on the in vivo activity have been reported for any of the discussed peptides (Table 2).
TMPRSS2. The fusion of the virus and cell membrane is mediated by serine proteinase TMPRSS2, which is a cell surface protein expressed in various human tissues and organs, including respiratory tract, prostate, and gastrointestinal tract. The protease activity of TMPRSS2 facilitates activation of viral glycoproteins, prostate cancer progression, and cleavage of endogenous substrates, including PAR2 receptor, which modulates inflammatory reactions, obesity, metabolism, and cancer. Inhibition of TMPRSS2 expression decreases the efficiency of virus entry into the cell, although it can also reduce the cleavage of endogenous substrates [94]. In order to block the virus entry into the cell, TMPRSS2 could be inhibited with peptides containing arginine [95] or hydrophobic amino acids (e.g., isoleucine) used to block the hydrophobic pocket at the S1 subunit of the S protein that binds the protease [96]. It is important to mention that the genetic knockout of TMPRSS2 in mice did not cause any noticeable phenotypic deviations, which makes this model suitable for pre-clinical studies. The strategy of TMPRSS2 inhibition for the prevention of infection with the influenza virus has been already tested in vitro using hydrophobic decanoylated peptide mimics. In particular, phosphorodiamidate morpholine oligomer conjugated with a peptide (PPMO) was synthesized that ensured expression of incomplete and inactive form of TMPRSS2 and prevented cell infection with the virus [97, 98].
PEPTIDE VACCINES
The ability of peptides to induce protective immunity in vivo in addition to the neutralization activity in vitro was first demonstrated in 1982 for the foot-and-mouth disease virus [99], although these results were not used in veterinary later. The first peptide vaccine was developed against the canine parvovirus from the Parvoviridae family, which is a small non-enveloped single-stranded DNA virus. The capsid of this virus consists of three different protein subunits (VPs) termed VP1, VP2, and VP3. A 37-amino acid sequence was identified at the N-terminus of VP2, that was identical in the samples derived from the infected animals of different species (cats, minks, and racoons). Based on this sequence, two peptides were selected for immunization: 1L15 (MSDGAVQPDGGQPAV) and 7L15 (QPDGGQPAVRNERAT). They were conjugated with KLH (keyhole limpet hemocyanin) and used successfully for dog immunization [100]. Following the success of this vaccine, the efforts have continued to produce a more immunogenic peptide based on the N-terminal sequence of VP2, which has opened the possibilities for using peptide vaccine in veterinary and medicine [101-103].
Abduljaleel et al. [30] attempted to develop a peptide vaccine against SARS-CoV-2 based on the antigenic epitopes of the virus. Twelve peptides 10-13 amino acids in length were selected: MDEFIERYKLEGY (ORF1ab), PYEDFQENWNTKH (ORF1a), LQDVVNQNAQALN (S protein), YDYCIPYNSVTSS (ORF3a), YVYSRVKNLNSSR (E protein), NGTITVEELKKLL (M protein), TENKYSQLDEEQP (ORF6), SPKLFIRQEEVQE (ORF7a), FSLELQDHNETCH (ORF7b), FYEDFLEYHDVRV (ORF8), DQELIRQGTDYKH (N protein), and SRNYIAQVDVVNF (ORF10). All peptides were linked via the GPGPG sequences to ensure spatial separation of the epitopes and to facilitate immunological processing of the antigens. The N-end of the construct was modified with a peptide adjuvant attached via the EAAAK linker to enhance the immune response. The final construct with a molecular mass of ~18.72 kDa consisted of ~156 amino acids.
Recently, the results of phase I clinical trial of a potential peptide vaccine against SARS-CoV-2 that induces the T-cell immunity have been published [31]. It is known that T lymphocytes exhibit partial protective effect even in the absence of antibodies and prevent the development of severe forms of COVID-19 [104]. Furthermore, unlike neutralizing antibodies, cytotoxic T lymphocytes could recognize epitopes of any SARS-CoV-2 antigen and not only of S protein antigens as a part of their effector function. In [31], healthy volunteers were immunized with a mixture of 6 linear 15-mer peptides that were fragments of the structural proteins S, N, M, and E and non-structural protein ORF8 (Table 3): P1_nuc (ASWFTALTQHGKEDL), P2_nuc (LLLLDRLNQLESKMS), P3_spi (ITRFQTLLALHRSYL), P4_env (FYVYSRVKNLNSSRV), P5_mem (LSYYKLGASQRVAGD), P6_ORF8 (SKWYIRVGARKSAPL). As one of the adjuvant components, the authors used a 9-mer lipopeptide, a strong agonist of the heteromeric innate immune receptor TLR1/TLR2 [105]. Based on the previous studies conducted by the same research group [106, 107], the peptides were selected in such way that the amino acid sequences presented by the MHC class II (DRB) molecules contained incorporated peptides presented by the frequent allele variants of the class I MHC. The T lymphocyte-mediated immune response was observed in all participants of the clinical study. Moreover, this response was maintained for the entire duration of the study (3 months) and was stronger than the response initiated as a result of infection.
It is known that conjugation of peptides with protein carriers and use of adjuvants help to initiate a strong immune response to peptides in animals or humans. The developers of EpiVacCorona peptide vaccine used this strategy with conserved sequences of the SARS-CoV-2 S protein to increase vaccine resistance to possible virus mutations (Table 3). Seven peptides 20-31 amino acid residues in length were designed and synthesized that carried presumable linear B-cell epitopes of the S protein. As a carrier protein, the authors used the chimeric recombinant protein MBP-6xHis-N_nCoV-2019 composed of the major part of the SARS-CoV-2 N protein and MBP protein from Escherichia coli expressed in a prokaryotic system; aluminum hydroxide was used as an adjuvant. Next, three most immunogenic peptides were selected for the conjugation: RLFRKSNLKPFERDISTEIYQAGS (corresponding to the receptor-binding motif of the SARS-CoV-2 S protein), KEIDRLNEVAKNLNESLIDLQE, and KNLNESLIDLQELGKYEQYIK (corresponding to the fragments of the S protein transmembrane domain). The conjugates of these peptide with the carrier protein were mixed at a 1 : 1 : 1 ratio, adsorbed on aluminum oxide, and used as components of the EpiVacCorona vaccine [32, 33]. It should be noted that locations of the B-cell epitopes determined in the independent studies [30] did not coincide with the positions of the selected peptides. The results of phase III clinical trials of this vaccine have not been published yet, but the efficacy of the EpiVacCorona has been questioned in the recent comparative study of Russian vaccines against the Delta variant of the virus [108].
Other peptides associated with immunogenic T-cell epitopes have been described in [106, 109, 110]. In particular, several dozens of epitopes that induced immune response after disease or vaccination in the majority of carriers of a particular allele variant of MHC have been revealed in the recent systematic study that evaluated a panel of peptides for their immunogenicity [111]. Similar peptide panel could be used for evaluating the T cell-mediated immune response in individuals after infection or vaccination and for estimating its duration. It should be mentioned that these T-cell epitopes are only slightly affected by mutations in the most common variants of the virus; hence, the development of new vaccines based on such epitopes seems promising.
CONCLUSIONS
In this review, we discussed possible applications of synthetic peptides in the fight against COVID-19 pandemics. The majority of discussed strategies is related to diagnostics and prophylactics of SARS-CoV-2 infection. First, these are strategies used to block virus entry into the cell. Other strategies involve the use of peptides for the development of prophylactic vaccines. Many authors believe that although the use of synthetic peptides to block the virus entry is very appealing (e.g., in the composition of prophylactic formulation for inhalation), such preparations are unlikely to become competitive in comparison with monoclonal antibodies and high-affinity decoy receptors. The synthesis of stapled peptides (Table 2) capable of virus entry blockade is extremely expensive. Hence, it seems highly unlikely that the COVID-19 pandemics could be stopped with the help of such peptides. With regard to peptide vaccines, the chances for the success of T-cell activating peptide vaccines, which are now at the early stages of clinical trials, are rather high. Although there is no convincing information so far on the efficiency of B cell-activating vaccines, this area of research remains also very promising.
References
Shereen, M. A., Khan, S., Kazmi, A., Bashir, N., and Siddique, R. (2020) COVID-19 infection: Origin, transmission, and characteristics of human coronaviruses, J. Adv. Res., 24, 91-98, https://doi.org/10.1016/j.jare.2020.03.005.
Wang, N., Shang, J., Jiang, S., and Du, L. (2020) Subunit vaccines against emerging pathogenic human coronaviruses, Front. Microbiol., 11, 298, https://doi.org/10.3389/fmicb.2020.00298.
Du, L., He, Y., Zhou, Y., Liu, S., Zheng, B. J., and Jiang, S. (2009) The spike protein of SARS-CoV – a target for vaccine and therapeutic development, Nat. Rev. Microbiol., 7, 226-236, https://doi.org/10.1038/nrmicro2090.
Zhou, P., Yang, X. L., Wang, X. G., Hu, B., Zhang, L., et al. (2020) A pneumonia outbreak associated with a new coronavirus of probable bat origin, Nature, 579, 270-273, https://doi.org/10.1038/s41586-020-2012-7.
Wrapp, D., Wang, N., Corbett, K. S., Goldsmith, J. A., Hsieh, C. L., et al. (2020) Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation, Science, 367, 1260-1263, https://doi.org/10.1126/science.abb2507.
Ortega, J. T., Serrano, M. L., Pujol, F. H., and Rangel, H. R. (2020) Role of changes in SARS-CoV-2 spike protein in the interaction with the human ACE2 receptor: An in silico analysis, EXCLI J., 19, 410-417, https://doi.org/10.17179/excli2020-1167.
Amicone, M., Borges, V., Alves, M. J., Isidro, J., Zé-Zé, L., et al. (2021) Mutation rate of SARS-CoV-2 and emergence of mutators during experimental evolution, bioRxiv, https://doi.org/10.1101/2021.05.19.444774.
Volz, E., Mishra, S., Chand, M., Barrett, J. C., Johnson, R., et al. (2021) Assessing transmissibility of SARS-CoV-2 lineage B.1.1.7 in England, Nature, 593, 266-269, https://doi.org/10.1038/s41586-021-03470-x.
Wibmer, C. K., Ayres, F., Hermanus, T., Madzivhandila, M., Kgagudi, P., et al. (2021) SARS-CoV-2 501Y. V2 escapes neutralization by South African COVID-19 donor plasma, Nat. Med., 27, 622-625, https://doi.org/10.1038/s41591-021-01285-x.
Tegally, H., Wilkinson, E., Giovanetti, M., Iranzadeh, A., Fonseca, V., et al. (2021) Detection of a SARS-CoV-2 variant of concern in South Africa, Nature, 592, 438-443, https://doi.org/10.1038/s41586-021-03402-9.
Muik, A., Wallisch, A. K., Sänger, B., Swanson, K. A., Mühl, J., et al. (2021) Neutralization of SARS-CoV-2 lineage B.1.1.7 pseudovirus by BNT162b2 vaccine-elicited human sera, Science, 371, 1152-1153, https://doi.org/10.1126/science.abg6105.
Davies, N. G., Abbott, S., Barnard, R. C., Jarvis, C. I., Kucharski, A. J., et al. (2021) Estimated transmissibility and impact of SARS-CoV-2 lineage B.1.1.7 in England, Science, 372, eabg3055, https://doi.org/10.1126/science.abg3055.
Yan, R., Zhang, Y., Li, Y., Xia, L., Guo, Y., and Zhou, Q. (2020) Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2, Science, 367, 1444-1448, https://doi.org/10.1126/science.abb2762.
Belouzard, S., Chu, V. C., and Whittaker, G. R. (2009) Activation of the SARS coronavirus spike protein via sequential proteolytic cleavage at two distinct sites, Proc. Natl. Acad. Sci. USA, 106, 5871-5876, https://doi.org/10.1073/pnas.0809524106.
Barrett, C. T., Neal, H. E., Edmonds, K., Moncman, C. L., Thompson, R., et al. (2021) Effect of clinical isolate or cleavage site mutations in the SARS-CoV-2 spike protein on protein stability, cleavage, and cell-cell fusion, J. Biol. Chem., 297, 100902, https://doi.org/10.1016/j.jbc.2021.100902.
Singh, J., Rahman, S. A., Ehtesham, N. Z., Hira, S., and Hasnain, S. E. (2021) SARS-CoV-2 variants of concern are emerging in India, Nat. Med., 27, 1131-1133, https://doi.org/10.1038/s41591-021-01397-4.
Borisova, N. I., Kotov, I. A., Kolesnikov, A. A., Kaptelova, V. V., Speranskaya, A. S., et al. (2021) Monitoring the spread of the SARS-CoV-2 (Coronaviridae: Coronavirinae: Betacoronavirus; Sarbecovirus) variants in the Moscow region using targeted high-throughput sequencing [in Russia], Vopr. Virusol., 66, 269-278, https://doi.org/10.36233/0507-4088-72.
Kumar, S., Thambiraja, T. S., Karuppanan, K., and Subramaniam, G. (2021) Omicron and Delta variant of SARS-CoV-2: A comparative computational study of spike protein, J. Med. Virol., 94, 1641-1649, https://doi.org/10.1002/jmv.27526.
Gong, S. Y., Chatterjee, D., Richard, J., Prévost, J., Tauzin, A., et al. (2021) Contribution of single mutations to selected SARS-CoV-2 emerging variants spike antigenicity, Virology, 563, 134-145, https://doi.org/10.1016/j.virol.2021.09.001.
Jhun, H., Park, H. Y., Hisham, Y., Song, C. S., and Kim, S. (2021) SARS-CoV-2 Delta (B. 1.617.2) Variant: A unique T478K mutation in receptor binding motif (RBM) of spike gene, Immune Netw., 21, e32, https://doi.org/10.4110/in.2021.21.e32.
Gao, Q., Bao, L., Mao, H., Wang, L., Xu, K., et al. (2020) Development of an inactivated vaccine candidate for SARS-CoV-2, Science, 369, 77-81, https://doi.org/10.1126/science.abc1932.
Rauch, S., Jasny, E., Schmidt, K. E., and Petsch, B. (2018) New vaccine technologies to combat outbreak situations, Front. Immunol., 9, 1963, https://doi.org/10.3389/fimmu.2018.01963.
Corbett, K. S., Edwards, D. K., Leist, S. R., Abiona, O. M., Boyoglu-Barnum, S., et al. (2020) SARS-CoV-2 mRNA vaccine design enabled by prototype pathogen preparedness, Nature, 586, 567-571, https://doi.org/10.1038/s41586-020-2622-0.
Folegatti, P. M., Ewer, K. J., Aley, P. K., Angus, B., Becker, S., et al. (2020) Safety and immunogenicity of the ChAdOx1 nCoV-19 vaccine against SARS-CoV-2: a preliminary report of a phase 1/2, single-blind, randomised controlled trial, Lancet, 396, 467-478, https://doi.org/10.1016/s0140-6736(20)31604-4.
Logunov, D. Y., Dolzhikova, I. V., Shcheblyakov, D. V., Tukhvatulin, A. I., Zubkova, O. V., et al. (2021) Safety and efficacy of an rAd26 and rAd5 vector-based heterologous prime-boost COVID-19 vaccine: an interim analysis of a randomised controlled phase 3 trial in Russia, Lancet, 397, 671-681, https://doi.org/10.1016/s0140-6736(21)00234-8.
Yang, J., Wang, W., Chen, Z., Lu, S., Yang, F., et al. (2020) A vaccine targeting the RBD of the S protein of SARS-CoV-2 induces protective immunity, Nature, 586, 572-577, https://doi.org/10.1038/s41586-020-2599-8.
Wang, J. (2021) New strategy for COVID-19 vaccination: targeting the receptor-binding domain of the SARS-CoV-2 spike protein, Cell Mol. Immunol., 18, 243-244, https://doi.org/10.1038/s41423-020-00584-6.
Tian, J. H., Patel, N., Haupt, R., Zhou, H., Weston, S., et al. (2021) SARS-CoV-2 spike glycoprotein vaccine candidate NVX-CoV2373 immunogenicity in baboons and protection in mice, Nat. Commun., 12, 372, https://doi.org/10.1038/s41467-020-20653-8.
Di Natale, C., La Manna, S., De Benedictis, I., Brandi, P., and Marasco, D. (2020) Perspectives in peptide-based vaccination strategies for syndrome coronavirus 2 pandemic, Front. Pharmacol., 11, 578382, https://doi.org/10.3389/fphar.2020.578382.
Abduljaleel, Z., Al-Allaf, F. A., and Aziz, S. A. (2021) Peptides-based vaccine against SARS-nCoV-2 antigenic fragmented synthetic epitopes recognized by T cell and β-cell initiation of specific antibodies to fight the infection, Biodes. Manuf., 4, 490-505, https://doi.org/10.1007/s42242-020-00114-3.
Heitmann, J. S., Bilich, T., Tandler, C., Nelde, A., Maringer, Y., et al. (2021) A COVID-19 peptide vaccine for the induction of SARS-CoV-2 T cell immunity, Nature, 601, 617-622, https://doi.org/10.1038/s41586-021-04232-5.
Ryzhikov, A. B., Ryzhikov, E. A., Bogryantseva, M. P., Danilenko, E. D., Imatdinov, I. R., et al. (2021) Immunogenicity and protectivity of the Peptide Vaccine against SARS-CoV-2, Ann. Russ. Acad. Med. Sci., 76, 5-19, https://doi.org/10.15690/vramn1528.
Ryzhikov, A. B., Ryzhikov, E. A., Bogryantseva, M. P., Usova, S. V., Danilenko, E. D., et al. (2021) A single blind, placebo-controlled randomized study of the safety, reactogenicity and immunogenicity of the “EpiVacCorona” Vaccine for the prevention of COVID-19, in volunteers aged 18-60 years (phase I-II), Russ. J. Infect. Immun., 11, 283-296, https://doi.org/10.15789/2220-7619-ASB-1699.
Watanabe, M., Omahdi, Z., and Yamasaki, S. (2020) Direct binding analysis between C-type lectins and glycans using immunoglobulin receptor fusion proteins, Methods Mol. Biol., 2132, 119-128, https://doi.org/10.1007/978-1-0716-0430-4_12.
Walls, A. C., Park, Y. J., Tortorici, M. A., Wall, A., McGuire, A. T., et al. (2020) Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein, Cell, 181, 281-292.e286, https://doi.org/10.1016/j.cell.2020.02.058.
Tripet, B., Howard, M. W., Jobling, M., Holmes, R. K., Holmes, K. V., et al. (2004) Structural characterization of the SARS-coronavirus spike S fusion protein core, J. Biol. Chem., 279, 20836-20849, https://doi.org/10.1074/jbc.M400759200.
Li, W., Moore, M. J., Vasilieva, N., Sui, J., Wong, S. K., et al. (2003) Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus, Nature, 426, 450-454, https://doi.org/10.1038/nature02145.
Shang, J., Wan, Y., Luo, C., Ye, G., Geng, Q., et al. (2020) Cell entry mechanisms of SARS-CoV-2, Proc. Natl. Acad. Sci. USA, 117, 11727-11734, https://doi.org/10.1073/pnas.2003138117.
Hussain, M., Jabeen, N., Amanullah, A., Baig, A. A., Aziz, B., et al. (2020) Structural basis of SARS-CoV-2 spike protein priming by TMPRSS2, bioRxiv, https://doi.org/10.1101/2020.04.21.052639.
Hoffmann, M., Kleine-Weber, H., Schroeder, S., Krüger, N., Herrler, T., et al. (2020) SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor, Cell, 181, 271-280.e278, https://doi.org/10.1016/j.cell.2020.02.052.
VanPatten, S., He, M., Altiti, A., Cheng, K. F., Ghanem, M. H., et al. (2020) Evidence supporting the use of peptides and peptidomimetics as potential SARS-CoV-2 (COVID-19) therapeutics, Future Med. Chem., 12, 1647-1656, https://doi.org/10.4155/fmc-2020-0180.
Hamming, I., Timens, W., Bulthuis, M. L., Lely, A. T., Navis, G., et al. (2004) Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis, J. Pathol., 203, 631-637, https://doi.org/10.1002/path.1570.
Zhao, Y., Zhao, Z., Wang, Y., Zhou, Y., Ma, Y., et al. (2020) Single-cell RNA expression profiling of ACE2, the receptor of SARS-CoV-2, Am. J. Respir. Crit. Care Med., 202, 756-759, https://doi.org/10.1164/rccm.202001-0179LE.
Millet, J. K., and Whittaker, G. R. (2015) Host cell proteases: critical determinants of coronavirus tropism and pathogenesis, Virus Res., 202, 120-134, https://doi.org/10.1016/j.virusres.2014.11.021.
Shulla, A., Heald-Sargent, T., Subramanya, G., Zhao, J., Perlman, S., et al. (2011) A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry, J. Virol., 85, 873-882, https://doi.org/10.1128/jvi.02062-10.
Chambers, J. P., Yu, J., Valdes, J. J., and Arulanandam, B. P. (2020) SARS-CoV-2, early entry events, J. Pathog., 2020, 9238696, https://doi.org/10.1155/2020/9238696.
Gur, M., Taka, E., Yilmaz, S. Z., Kilinc, C., Aktas, U., et al. (2020) Conformational transition of SARS-CoV-2 spike glycoprotein between its closed and open states, J. Chem. Phys., 153, 075101, https://doi.org/10.1063/5.0011141.
Xu, C., Wang, Y., Liu, C., Zhang, C., Han, W., et al. (2021) Conformational dynamics of SARS-CoV-2 trimeric spike glycoprotein in complex with receptor ACE2 revealed by cryo-EM, Sci. Adv., 7, eabe5575, https://doi.org/10.1126/sciadv.abe5575.
Jackson, C. B., Farzan, M., Chen, B., and Choe, H. (2022) Mechanisms of SARS-CoV-2 entry into cells, Nat. Rev. Mol. Cell Biol., 23, 3-20, https://doi.org/10.1038/s41580-021-00418-x.
Tang, T., Bidon, M., Jaimes, J. A., Whittaker, G. R., and Daniel, S. (2020) Coronavirus membrane fusion mechanism offers a potential target for antiviral development, Antiviral. Res., 178, 104792, https://doi.org/10.1016/j.antiviral.2020.104792.
Bosch, B. J., van der Zee, R., de Haan, C. A., and Rottier, P. J. (2003) The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex, J. Virol., 77, 8801-8811, https://doi.org/10.1128/jvi.77.16.8801-8811.2003.
White, J. M., Delos, S. E., Brecher, M., and Schornberg, K. (2008) Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme, Crit. Rev. Biochem. Mol. Biol., 43, 189-219, https://doi.org/10.1080/10409230802058320.
Millet, J. K., and Whittaker, G. R. (2018) Physiological and molecular triggers for SARS-CoV membrane fusion and entry into host cells, Virology, 517, 3-8, https://doi.org/10.1016/j.virol.2017.12.015.
Chen, Y., Guo, Y., Pan, Y., and Zhao, Z. J. (2020) Structure analysis of the receptor binding of 2019-nCoV, Biochem. Biophys. Res. Commun., 525, 135-140, https://doi.org/10.1016/j.bbrc.2020.02.071.
Xia, S., Zhu, Y., Liu, M., Lan, Q., Xu, W., et al. (2020) Fusion mechanism of 2019-nCoV and fusion inhibitors targeting HR1 domain in spike protein, Cell. Mol. Immunol., 17, 765-767, https://doi.org/10.1038/s41423-020-0374-2.
Xia, S., Liu, M., Wang, C., Xu, W., Lan, Q., et al. (2020) Inhibition of SARS-CoV-2 (previously 2019-nCoV) infection by a highly potent pan-coronavirus fusion inhibitor targeting its spike protein that harbors a high capacity to mediate membrane fusion, Cell Res., 30, 343-355, https://doi.org/10.1038/s41422-020-0305-x.
Smyth, M. S., and Martin, J. H. (2000) X Ray crystallography, Mol. Pathol., 53, 8-14, https://doi.org/10.1136/mp.53.1.8.
Smith, G. P. (1985) Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface, Science, 228, 1315-1317, https://doi.org/10.1126/science.4001944.
Trier, N. H., Hansen, P. R., and Houen, G. (2012) Production and characterization of peptide antibodies, Methods, 56, 136-144, https://doi.org/10.1016/j.ymeth.2011.12.001.
Lee, A. C., Harris, J. L., Khanna, K. K., and Hong, J. H. (2019) A comprehensive review on current advances in peptide drug development and design, Int. J. Mol. Sci., 20, 2383, https://doi.org/10.3390/ijms20102383.
Rabinowitz, J. A., Lainson, J. C., Johnston, S. A., and Diehnelt, C. W. (2018) Non-natural amino acid peptide microarrays to discover Ebola virus glycoprotein ligands, Chem. Commun., 54, 1417-1420, https://doi.org/10.1039/c7cc08242h.
Ingallinella, P., Bianchi, E., Ladwa, N. A., Wang, Y. J., Hrin, R., et al. (2009) Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency, Proc. Natl. Acad. Sci. USA, 106, 5801-5806, https://doi.org/10.1073/pnas.0901007106.
Pessi, A., Bixler, S. L., Soloveva, V., Radoshitzky, S., Retterer, C., et al. (2019) Cholesterol-conjugated stapled peptides inhibit Ebola and Marburg viruses in vitro and in vivo, Antiviral Res., 171, 104592, https://doi.org/10.1016/j.antiviral.2019.104592.
Chen, G., Cook, J. D., Ye, W., Lee, J. E., and Sidhu, S. S. (2019) Optimization of peptidic HIV-1 fusion inhibitor T20 by phage display, Protein Sci., 28, 1501-1512, https://doi.org/10.1002/pro.3669.
Ding, X., Zhang, X., Chong, H., Zhu, Y., Wei, H., Wu, X., et al. (2017) Enfuvirtide (T20)-based lipopeptide is a potent HIV-1 cell fusion inhibitor: Implications for viral entry and inhibition, J. Virol., 91, https://doi.org/10.1128/jvi.00831-17.
Matsubara, T., Onishi, A., Saito, T., Shimada, A., Inoue, H., et al. (2010) Sialic acid-mimic peptides as hemagglutinin inhibitors for anti-influenza therapy, J. Med. Chem., 53, 4441-4449, https://doi.org/10.1021/jm1002183.
Kadam, R. U., Juraszek, J., Brandenburg, B., Buyck, C., Schepens, W. B. G., et al. (2017) Potent peptidic fusion inhibitors of influenza virus, Science, 358, 496-502, https://doi.org/10.1126/science.aan0516.
Shilovskiy, I. P., Andreev, S. M., Kozhikhova, K. V., Nikolskii, A. A., and Khaitov, M. R. (2019) Prospects for the use of peptides against respiratory syncytial virus [in Russian], Mol. Biol. (Mosk), 53, 541-560, https://doi.org/10.1134/s002689841904013x.
Brouwer, P. J. M., Caniels, T. G., van der Straten, K., Snitselaar, J. L., Aldon, Y., et al. (2020) Potent neutralizing antibodies from COVID-19 patients define multiple targets of vulnerability, Science, 369, 643-650, https://doi.org/10.1126/science.abc5902.
Wang, C., Li, W., Drabek, D., Okba, N. M. A., van Haperen, R., Osterhaus, A., et al. (2020) A human monoclonal antibody blocking SARS-CoV-2 infection, Nat. Commun., 11, 2251, https://doi.org/10.1038/s41467-020-16256-y.
Sui, J., Li, W., Murakami, A., Tamin, A., Matthews, L. J., Wong, S. K., et al. (2004) Potent neutralization of severe acute respiratory syndrome (SARS) coronavirus by a human mAb to S1 protein that blocks receptor association, Proc. Natl. Acad. Sci. USA, 101, 2536-2541, https://doi.org/10.1073/pnas.0307140101.
Walls, A. C., Xiong, X., Park, Y. J., Tortorici, M. A., Snijder, J., et al. (2019) Unexpected receptor functional mimicry elucidates activation of coronavirus fusion, Cell, 176, 1026-1039.e1015, https://doi.org/10.1016/j.cell.2018.12.028.
Khavinson, V., Linkova, N., Dyatlova, A., Kuznik, B., and Umnov, R. (2020) Peptides: prospects for use in the treatment of COVID-19, Molecules, 25, 4389, https://doi.org/10.3390/molecules25194389.
Kuba, K., Imai, Y., Rao, S., Gao, H., Guo, F., et al. (2005) A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury, Nat. Med., 11, 875-879, https://doi.org/10.1038/nm1267.
Mantovani, A., Locati, M., Vecchi, A., Sozzani, S., and Allavena, P. (2001) Decoy receptors: a strategy to regulate inflammatory cytokines and chemokines, Trends Immunol., 22, 328-336, https://doi.org/10.1016/s1471-4906(01)01941-x.
Dinarello, C. A. (1997) Interleukin-1, Cytokine Growth Factor Rev., 8, 253-265, https://doi.org/10.1016/s1359-6101(97)00023-3.
Idriss, H. T., and Naismith, J. H. (2000) TNF alpha and the TNF receptor superfamily: structure-function relationship(s), Microsc Res. Tech., 50, 184-195, https://doi.org/10.1002/1097-0029(20000801)50:3<184::Aid-jemt2>3.0.Co;2-h.
Pinto, D., Park, Y. J., Beltramello, M., Walls, A. C., Tortorici, M. A., et al. (2020) Cross-neutralization of SARS-CoV-2 by a human monoclonal SARS-CoV antibody, Nature, 583, 290-295, https://doi.org/10.1038/s41586-020-2349-y.
Zoufaly, A., Poglitsch, M., Aberle, J. H., Hoepler, W., Seitz, T., et al. (2020) Human recombinant soluble ACE2 in severe COVID-19, Lancet Respir. Med., 8, 1154-1158, https://doi.org/10.1016/s2213-2600(20)30418-5.
Chan, K. K., Dorosky, D., Sharma, P., Abbasi, S. A., Dye, J. M., et al. (2020) Engineering human ACE2 to optimize binding to the spike protein of SARS coronavirus 2, Science, 369, 1261-1265, https://doi.org/10.1126/science.abc0870.
Lu, J., and Sun, P. D. (2020) High affinity binding of SARS-CoV-2 spike protein enhances ACE2 carboxypeptidase activity, bioRxiv, https://doi.org/10.1101/2020.07.01.182659.
Glasgow, A., Glasgow, J., Limonta, D., Solomon, P., Lui, I., et al. (2020) Engineered ACE2 receptor traps potently neutralize SARS-CoV-2, bioRxiv, https://doi.org/10.1101/2020.07.31.231746.
Chan, K. K., Tan, T. J. C., Narayanan, K. K., and Procko, E. (2020) An engineered decoy receptor for SARS-CoV-2 broadly binds protein S sequence variants, bioRxiv, https://doi.org/10.1101/2020.10.18.344622.
Zhang, G., Pomplun, S., Loftis, A. R., Tan, X., Loas, A., et al. (2020), Investigation of ACE2 N-terminal fragments binding to SARS-CoV-2 Spike RBD, bioRxiv, https://doi.org/10.1101/2020.03.19.999318.
Karoyan, P., Vieillard, V., Gómez-Morales, L., Odile, E., Guihot, A., et al. (2021) Human ACE2 peptide-mimics block SARS-CoV-2 pulmonary cells infection, Commun. Biol., 4, 197, https://doi.org/10.1038/s42003-021-01736-8.
Rezaei Araghi, R., and Keating, A. E. (2016) Designing helical peptide inhibitors of protein-protein interactions, Curr. Opin. Struct. Biol., 39, 27-38, https://doi.org/10.1016/j.sbi.2016.04.001.
Ali, A. M., Atmaj, J., Van Oosterwijk, N., Groves, M. R., and Dömling, A. (2019) Stapled peptides inhibitors: A new window for target drug discovery, Comput. Struct. Biotechnol. J., 17, 263-281, https://doi.org/10.1016/j.csbj.2019.01.012.
Morgan, D. C., Morris, C., Mahindra, A., Blair, C. M., Tejeda, G., et al. (2021) Stapled ACE2 peptidomimetics designed to target the SARS-CoV-2 spike protein do not prevent virus internalization, Pept. Sci. (Hoboken), 113, e24217, https://doi.org/10.1002/pep2.24217.
Curreli, F., Victor, S. M. B., Ahmed, S., Drelich, A., Tong, X., et al. (2020) Stapled Peptides based on human angiotensin-converting enzyme 2 (ACE2) potently inhibit SARS-CoV-2 infection in vitro, mBio, 11, e02451-20, https://doi.org/10.1128/mBio.02451-20.
Bibilashvili, R. S., Sidorova, M. V., Dudkina, U. S., Palkeeva, M. E., Molokoedov, A. S., et al. (2021) Peptide inhibitors of the interaction of the SARS-CoV-2 receptor-binding domain with the ACE2 cell receptor, Biochem. Mosc. Suppl. B Biomed. Chem., 15, 274-280, https://doi.org/10.1134/s199075082104003x.
Krut, V. G., Astrakhantseva, I. V., Chuvpilo, S. A., Efimov, G. A., Ambaryan, S. G., et al. (2021) Antibodies to the N-terminal domain of angiotensin-converting enzyme (ACE2) that block its interaction with SARS-CoV-2 S protein, Dokl. Biochem. Biophys., 4, 1-4, https://doi.org/10.1134/S160767292201001X.
Larue, R. C., Xing, E., Kenney, A. D., Zhang, Y., Tuazon, J. A., et al. (2021) Rationally designed ACE2-derived peptides inhibit SARS-CoV-2, Bioconjug. Chem., 32, 215-223, https://doi.org/10.1021/acs.bioconjchem.0c00664.
Adhikary, P., Kandel, S., Mamani, U. F., Mustafa, B., Hao, S., et al. (2021) Discovery of small anti-ACE2 peptides to inhibit SARS-CoV-2 infectivity, Adv. Ther. (Weinh), 4, 2100087, https://doi.org/10.1002/adtp.202100087.
Wettstein, L., Kirchhoff, F., and Münch, J. (2022) The transmembrane protease TMPRSS2 as a therapeutic target for COVID-19 treatment, Int. J. Mol. Sci., 23, 1351, https://doi.org/10.3390/ijms23031351.
Iwata-Yoshikawa, N., Okamura, T., Shimizu, Y., Hasegawa, H., Takeda, M., et al. (2019) TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after Coronavirus infection, J. Virol., 93, e01815-18, https://doi.org/10.1128/jvi.01815-18.
Lucas, J. M., Heinlein, C., Kim, T., Hernandez, S. A., Malik, M. S., et al. (2014) The androgen-regulated protease TMPRSS2 activates a proteolytic cascade involving components of the tumor microenvironment and promotes prostate cancer metastasis, Cancer Discov., 4, 1310-1325, https://doi.org/10.1158/2159-8290.Cd-13-1010.
Bertram, S., Glowacka, I., Blazejewska, P., Soilleux, E., Allen, P., et al. (2010) TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells, J. Virol., 84, 10016-10025, https://doi.org/10.1128/jvi.00239-10.
Böttcher-Friebertshäuser, E., Freuer, C., Sielaff, F., Schmidt, S., et al. (2010) Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors, J. Virol., 84, 5605-5614, https://doi.org/10.1128/jvi.00140-10.
Bittle, J. L., Houghten, R. A., Alexander, H., Shinnick, T. M., Sutcliffe, J. G., et al. (1982) Protection against foot-and-mouth disease by immunization with a chemically synthesized peptide predicted from the viral nucleotide sequence, Nature, 298, 30-33, https://doi.org/10.1038/298030a0.
Langeveld, J. P., Casal, J. I., Osterhaus, A. D., Cortés, E., de Swart, R., et al. (1994) First peptide vaccine providing protection against viral infection in the target animal: studies of canine parvovirus in dogs, J. Virol., 68, 4506-4513, https://doi.org/10.1128/JVI.68.7.4506-4513.1994.
Casal, J. I., Langeveld, J. P., Cortés, E., Schaaper, W. W., van Dijk, E., et al. (1995) Peptide vaccine against canine parvovirus: Identification of two neutralization subsites in the N terminus of VP2 and optimization of the amino acid sequence, J. Virol., 69, 7274-7277, https://doi.org/10.1128/JVI.69.11.7274-7277.1995.
DiMarchi, R., Brooke, G., Gale, C., Cracknell, V., Doel, T., et al. (1986) Protection of cattle against foot-and-mouth disease by a synthetic peptide, Science, 232, 639-641, https://doi.org/10.1126/science.3008333.
Broekhuijsen, M. P., van Rijn, J. M., Blom, A. J., Pouwels, P. H., Enger-Valk, B. E., et al. (1987) Fusion proteins with multiple copies of the major antigenic determinant of foot-and-mouth disease virus protect both the natural host and laboratory animals, J. Gen. Virol., 68, 3137-3143, https://doi.org/10.1099/0022-1317-68-12-3137.
Rydyznski Moderbacher, C., Ramirez, S. I., Dan, J. M., Grifoni, A., Hastie, K. M., et al. (2020) Antigen-specific adaptive immunity to SARS-CoV-2 in acute COVID-19 and associations with age and disease severity, Cell, 183, 996-1012.e1019, https://doi.org/10.1016/j.cell.2020.09.038.
Rammensee, H. G., Wiesmüller, K. H., Chandran, P. A., Zelba, H., Rusch, E., et al. (2019) A new synthetic toll-like receptor 1/2 ligand is an efficient adjuvant for peptide vaccination in a human volunteer, J. Immunother Cancer, 7, 307, https://doi.org/10.1186/s40425-019-0796-5.102.
Nelde, A., Bilich, T., Heitmann, J. S., Maringer, Y., Salih, H. R., et al. (2021) SARS-CoV-2-derived peptides define heterologous and COVID-19-induced T cell recognition, Nat. Immunol., 22, 74-85, https://doi.org/10.1038/s41590-020-00808-x.
Bilich, T., Nelde, A., Heitmann, J. S., Maringer, Y., Roerden, M., Bauer, J., et al. (2021) T cell and antibody kinetics delineate SARS-CoV-2 peptides mediating long-term immune responses in COVID-19 convalescent individuals, Sci. Transl. Med., 13, https://doi.org/10.1126/scitranslmed.abf7517.
Barchuk, A., Cherkashin, M., Bulina, A., Berezina, N., Rakova, T., et al. (2021) Vaccine effectiveness against referral to hospital and severe lung injury associated with COVID-19: a population-based case-control study in St. Petersburg, Russia, medRxiv, https://doi.org/10.1101/2021.08.18.21262065.
Shomuradova, A. S., Vagida, M. S., Sheetikov, S. A., Zornikova, K. V., Kiryukhin, D., et al. (2020) SARS-CoV-2 epitopes are recognized by a public and diverse repertoire of human T cell receptors, Immunity, 53, 1245-1257.e1245, https://doi.org/10.1016/j.immuni.2020.11.004.
Ferretti, A. P., Kula, T., Wang, Y., Nguyen, D. M. V., Weinheimer, A., et al. (2020) Unbiased screens show CD8+ T cells of COVID-19 patients recognize shared epitopes in SARS-CoV-2 that largely reside outside the spike protein, Immunity, 53, 1095-1107.e1093, https://doi.org/10.1016/j.immuni.2020.10.006.
Titov, A., Shaykhutdinova, R., Shcherbakova, O. V., Serdyuk, Y. V., Sheetikov, S. A., et al. (2022) Immunogenic epitope panel for accurate detection of non-cross-reactive T cell response to SARS-CoV-2, JCI Insight, 7, e157699, https://doi.org/10.1172/jci.insight.157699.
Acknowledgments
The authors express their gratitude to A. A. Ishmukhametov for his support and interest in this study, and to R. A. Abagyan, I. V. Yampolsky, and G. A. Maleev for helpful discussions.
Funding
This work was supported by Sirius University of Science and Technology (project IMB-NIR-2103), Russian Foundation for Basic Research (project 20-04-60338), and Ministry of Science and Higher Education of the Russian Federation (agreement 075-03-2021-448/3, topic no. 121122300151-5).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
L.I.K. is working at the Chumakov Federal Scientific Center for Research and Development of Immunobiological Products, Russian Academy of Sciences, which is the developer and producer of the CoviVac vaccine against coronavirus infection. This article does not contain data on human participants or animals performed by any of the authors.
Rights and permissions
About this article

Cite this article
Krut’, V.G., Chuvpilo, S.A., Astrakhantseva, I.V. et al. Will Peptides Help to Stop COVID-19?. Biochemistry Moscow 87, 590–604 (2022). https://doi.org/10.1134/S0006297922070021
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297922070021