
Abstract
Comparative analysis of available literature data on the pathogenetic neuroendocrine mechanisms of depression and post-traumatic stress disorder (PTSD) is provided in this review to identify their common features and differences. We discuss the multidirectional modifications of the activity of cortical and subcortical structures of the brain, levels of neurotransmitters and their receptors, and functions of the hypothalamic-pituitary-adrenocortical axis in depression and PTSD. The analysis shows that these disorders are examples of opposite failures in the system of adaptive stress response of the body to stressful psychotraumatic events. On this basis, it is concluded that the currently widespread use of similar approaches to treat these disorders is not justified, despite the significant similarity of their anxiety-depressive symptoms; development of differential therapeutic strategies is required.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
INTRODUCTION
Depression, or major depressive disorder (MDD) is currently one of the most wide-spread diseases in the world, and not only among the psychiatric ones. A less common disease, post-traumatic stress disorder (PTSD), has also a post-stress nature. Despite the great role of genetic predisposition and belonging to different nosologic groups (F33.2 and F43.1 according to the International Classification of Diseases, ICD-10, respectively), the common feature of these disorders is their development as a result of intense or chronic exposure to stress (psychotraumatic nature), as well as a number of similar symptoms. Moreover, there is a high degree of comorbidity between PTSD and depression [1] that suggests the presence of common etiological factors leading to the development of these states. Yehuda and Antelman [2] have reported that duration and intensity of exposure to the traumatic agent is of great importance for formation of PTSD or depression-like state in animals. At the same time, a short-term exposure of high intensity leads to the development of PTSD more frequently, whereas depressive disorders develop as a result of prolonged exposure to stressful situations of mild intensity. However, despite a number of common symptoms and etiological factors, these two states have pronounced pathophysiological differences, and to some extent, even can be considered as opposites. This issue is discussed in the present review as an attempt to demonstrate that the considered post-stress conditions often exemplify differently directed disorders in the system of the organism’s adaptive response to psychotraumatic events. In our opinion, comprehensive comparative study of the pathogenetic mechanisms of these states could facilitate development of objective methods for their differential diagnosis, creation of selective therapeutical approaches, identification of the role of individual and hereditary predisposition and its targeted correction.
BRAIN STRUCTURES INVOLVED IN PATHOGENETIC CONTOURS IN PTSD AND DEPPRESSIVE DISORDER
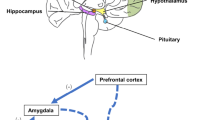
The cortico-limbic network and the default mode network (DMN) are the major neural networks investigated in the framework of MDD and PTSD pathogenesis. DMN includes ventromedial PFC (vmPFC) and dorsolateral PFC (dlPFC), lateral parietal cortex, and cingulate cortex with posterior cingulate gyrus and adjacent precuneus regions.
Connections between the brain structures in DMN, which combines orbitofrontal cortex with hypothalamus, amygdala, and hippocampus are changed in patients with MDD [3]. Pronounced microstructural changes are observed in the medial PFC and hippocampus of the patients with MDD, even in the remission state, as well as increase in the number of fibers connecting amygdala with other brain regions [4-6]. In addition to the decrease in the hippocampus volume, its activity was found to be asymmetric in the MDD patients, and increase in the blood flow in the region of amygdala nuclei was observed [7].
In the cortico-limbic networks the observed changes are associated not only with the grey matter, but also with the density of glial cells that is characteristic for MDD [8]. Increase in the number of connections between the amygdala, hippocampus, and basal nuclei could explain selective improvement of the memory about negative events in the patients with MDD in comparison with the healthy individuals not exhibiting such changes [9]. In patients with PTSD the described alterations specific for MDD were not observed in functioning of the cortico-limbic network and of DMN, however, a disorder was observed in the connections of the posterior cingulate cortex and precuneus with other DMN parts [10].
The PFC involvement in pathogenesis of the considered post-stress disorders was also confirmed by the data that the blood flow and glucose metabolism in patients with MDD were increased in the ventromedial PFC associated with regulation of emotions, whereas opposite changes were found in the dorsolateral PFC [11]. The importance of vmPFC in the MDD development was confirmed by the finding that the patients with damaged vmPFC got fewer points on the scale of the severity of depression symptoms than the patients with lesions in other parts of the brain [12]. In contrast to the patients with MDD, the patients with PTSD exhibited decreased blood flow in the vmPFC [13]. There are also data on the effect of dlPFC activity on manifestation of the PTSD symptoms: increased activity of dlPFC is associated with less pronounced symptoms of this disorder [14]. The results obtained in the experiments on animals demonstrated inhibitory effect of the prefrontal cortex neurons on the amygdala nuclei [15], which was in a good agreement with the data on changes in the activity of these structures PTSD.
Increase in the blood flow and glucose metabolism has been observed in the anterior cingulated cortex (ACC) of patients with MDD [16]. These data are in agreement with the studies on experimental animals. Thus, Barthas et al. [17] demonstrated involvement of ACC in the development of pathology in the model of chronic pain-induced depression [17]. In this case, chronic optogenetic stimulation of the ACC neurons led to formation of the depression-like behavior in intact animals. On the contrary, the blood flow intensity in the ACC was reduced in the patients with PTSD [18]. Thus, multidirectional changes were observed at the level of the cortex structures in these mental disorders: increase in the depressive disorders and decrease in PTSD.
Although the same neural networks and brain parts are associated with pathogenesis of MDD and PTSD, the observed changes in them are different in these pathologies, and, moreover, these changes are of opposite directions in some considered brain structures.
REARRANGEMENTS OF HYPOTHALAMIC-PITUITARY-ADRENOCORTICAL AXIS IN DEPRESSION AND PTSD, AND THEIR ASSOCIATION WITH NEUROPLASTICITY AND INFLAMMATION
Normally, glucocorticoid hormones bind to mineralocorticoid receptors (MR) at low concentrations, whereas at high concentrations (e.g., at stress response) they bind to glucocorticoid receptors (GR) through which the strength of the hypothalamic-pituitary-adrenocortical axis (HPA axis) reaction is regulated under stress conditions by the negative feedback mechanism. Glucocorticoids decrease secretion of corticotropin-releasing hormone (CRH) and adrenocorticotropic hormone (ACTH) and thus decrease their own level via the negative feedback on levels of pituitary gland, hypothalamus periventricular nucleus, and hippocampus [19].
Multilevel changes are observed in the functioning of HPA axis of the MDD and PTSD patients, which are specific and can be reproduced in animal models of these disorders. Furthermore, the patients with MDD display an increased level of cortisol in blood [20] and of CRH in hypothalamus [21], whereas the patients with PTSD can have both normal [22] and decreased basal levels of blood glucocorticoids [23]. Such changes can be caused by different sensitivities of the negative feedback mechanisms: they are considered sensitized in PTSD [24] and desensitized in MDD [25].
Changes in the density of glucocorticoid receptors in the brain structures involved in pathogenesis of MDD were found in the post-mortem study of the patients’ brains in the amygdala [26] and hippocampus [27]. Reduced level of glucocorticoid receptors in animal models of depression was observed in the hippocampus, PFC, and hypothalamus [28]. The observed decrease in the number of GRs on the background of increased level of glucocorticoids and CRH reflects the down-regulation phenomenon.
The chronically increased level of glucocorticoids in MDD can explain the previously mentioned decrease in the volume of hippocampus, where neurons are thought to be harmed by high concentrations of glucocorticoids [29]. It is likely that precisely the decrease of the inhibitory effect of hippocampus on CRH secretion in the hypothalamus paraventricular nucleus caused by this leads to the detected desensitization of the HPA axis. Development of the depression-like pathology observed in animals subjected to chronic injections of glucocorticoids speaks in favor of the pathogenetic role of hypercortisolemia in depression [30].
Glucocorticoids have a pronounced effect on neuroplasticity and neurogenesis; they decrease expression of the brain-derived neurotrophic factor (BDNF) [31], content of which decreases significantly in different brain section under the action of chronic stress and is reported for MDD and PTSD [32]. Such changes became a basis for the neuroplastic theory of depression development, and the antidepressive effect of serotoninergic preparations is now believed to be BDNF-mediated [32, 33].
In addition to the effect on the CNS functioning, such long-term high levels of glucocorticoids lead to the changes in immunity shifting the balance to chronic inflammation with high level of pro-inflammatory cytokines and neuroinflammation (which is especially important) accompanying the course of depressive disorder [34]. Introduction of proinflammatory cytokines leads to the development of depression-like symptoms [35], and exposure to lipopolysaccharides is used to induce depression in animal models [36]. Similarly to glucocorticoids, the cytokine IL-1β also decreases the BDNF expression [37]. As with other changes observed in depression, when it is difficult to find out whether they are the cause or consequence of the disease, this phenomenon became the basis for the inflammatory theory of depression development [38]. However, regardless of the secondary or primary nature of these changes, they undoubtedly have strong effect on the HPA axis functioning and the course of depression, respectively.
In patients with PTSD with decreased or unchanged basal level of cortisol in blood, the level of CRH in the cerebrospinal fluid is increased [39]. We also demonstrated hyperproduction of the hypothalamus-derived CRH in the rat PTSD model [40]. At the same time up-regulation of the number of glucocorticoid receptors was detected in the medial PFC [41] and hippocampus in PTSD models, while their number in the amygdala nuclei was unchanged [42].
Thus, it can be stated that modifications of the HPA axis functions in MDD and PTSD exhibit complex bidirectional interactions, that makes it difficult to separate them into conditionally primary (pathogenetic) and secondary due to subsequent rearrangements of the whole system via the feedback mechanisms. It seems that depression and to the lesser degree PTSD, as a disease with more heterogenous manifestations, also have heterogenous neuroendocrine disorders accompanying them.
MODIFICATIONS OF ACTIVITY OF THE BRAIN MONOAMINOERGIC SYSTEMS
The monoamine theory of MDD is the oldest among the existent theories of this disorder pathogenesis [43]. In the literature a great amount of data is accumulated on changes in the serotoninergic neuron functioning in MDD in humans [44], as well as in the models of depression-like conditions in rodents with a decreased level of serotonin found in the PFC and hippocampus [45]. This decrease was accompanied by a compensatory increase in the expression of serotonin receptors in these structures [46, 47]. Changes in the dopaminergic system in MDD include decrease in the levels of dopamine and its metabolites in the cerebrospinal fluid [48] and decrease in the density of dopamine receptors in the nucleus accumbens [49]. Concurrently, increase in the dopamine level in the ventral cover was shown in the rat model of chronic pain-induced depression [50]. The studies on the depression-like syndrome in experimental animals revealed increase in the expression of D2-receptors of dopamine in the prefrontal cortex [51] and decrease in the expression of D2-dopamine receptors in the nucleus accumbens [52]. Furthermore, the deficiency of D2-receptors observed in the limbic system structures of the models in the depression-like state is alleviated following long-term administration of the tricyclic antidepressant imipramine [53].
The increased anxiety of rats in the PTSD model is accompanied by the increased level of dopamine in the PFC. It is known that dopaminergic fibers in this brain area are involved in realization of the fear-associated behavior [54], as well as in the emergence of emotionally colored memory [55], which is in agreement with the appearance of specific behavior symptoms in PTSD. Moreover, there are data on the anxiolytic effect of the dopamine D3-receptors antagonists in the rat PTSD models [56].
Changes in the serotoninergic synapses detected in the patients and in PTSD models are more heterogenous. In particular, increase in the density of serotonin 1A-type receptors in the hippocampus (5-HT1A) [57] and in the density of serotonin 2C-type receptors in the amygdala (5-HT2C) was shown [58]. At the same time, decrease in the expression of 5-HT1A receptors in the cingulate gyrus and suture nuclei was reported [59]. No changes were observed in the density of 5-HT1A receptors [57]. Despite some inconsistency of the available data, in general, excessive activation of serotoninergic synapses was observed in PTSD in the subcortical formations in response to stress conditions [60].
Decrease in the norepinephrine levels in the medial PFC and the nucleus accumbens was shown in the experimental animal model of depression, and substances facilitating increase in the norepinephrine level led to disappearance of the depression-like behavior [61]. Thus, the depressive disorder is characterized by the pronounced decrease in the activity of adrenergic neurons, whereas PTSD is characterized by activation of this system of the brain: this is associated with the increase in the norepinephrine level under conditions of chronic and acute stress in the PFC, hippocampus, and amygdala [60, 62]. Increased levels of norepinephrine and its metabolites in the amygdala were detected also in the rat PSTD models [63].
Based on the monoamine theory of depression several groups of antidepressants were developed, which affected the level of monoamines. These antidepressants are used up to now, however, therapeutic effect of these preparations is manifested several weeks after the beginning of the therapy, despite the immediate effect on the monoamine neurotransmission. Moreover, not all pharmaceuticals increasing the monoamine level in the synaptic cleft are antidepressants, such as stimulants [64]. Such findings suggest more complex and delayed mechanisms of the antidepressive effect based on the changes in neuroplasticity and reorganization of pathological neuronal bonds, but not on elimination of the neurotransmitters disbalance, as suggested in the monoamine theory of depression. Moreover, the observed changes in the functioning of the monoamine neurotransmission in MDD could be of the secondary nature.
CHANGES IN ACTIVITY OF THE BRAIN GLUTAMATERGIC SYSTEM
Enough data have been accumulated to date on changes in the glutamatergic system that emerge in the depressive and post-traumatic disorders, however, as in the case of other neurotransmitters systems, it is rather difficult to identify whether the observed changes are of primary or secondary nature.
Administration of ketamine, an NMDA receptor antagonist, rapidly leads to manifestation of antidepressive effect in humans with MDD [65], glutamate deficit was detected in the medial PFC [66], ACC [67], and the amygdala nuclei [68], and it was also accompanied by the increase in expression of the NMDA-receptors in the amygdala nuclei [69]. Decrease in the density of metabotropic receptors and of NMDA-receptors in the medial PFC has been reported recently for the patients with depression [70]. The antidepressive effect of ketamine could indicate involvement of the glutamate transmission disorder in the pathobiochemical mechanisms of MDD. Transient increase in the concentration of glutamate following ketamine injection could lead to the release of BDNF, which in its turn affects neuroplasticity disturbed in MDD [71].
Studies with PTSD patients revealed increase in the glutamate level in blood plasma [72], in the occipital-parietal areas of the cerebral cortex [73], and in the right hippocampus [74]. Decreased level of glutamate was found in the ventromedial PFC, which is characterized by the decreased activity in PTSD [75]. Increase in the density of the type-5 metabotropic receptors (mGluR5) was observed in the hyperactivated dorsolateral PFC [76].
Hence, in depression the glutamatergic system is suppressed in the prefrontal cortex, amygdala, and hippocampus, whereas the changes observed in the PTSD patients are more pronounced in the prefrontal cortex structures and characterized by sensitization of the neurons in this area to glutamate action.
PHARMACOTHERAPY OF MDD AND PTSD
The first choice of MDD treatment in the majority of cases are selective serotonin reuptake inhibitors (SSRIs) and/or of norepinephrine re-uptake. The second and third choices include non-selective inhibitors of monoamine reuptake and inhibitors of monoamine oxidase. However, the efficiency of antidepressants remains low and frequently is different from that of placebo only in the cases of severe depression [77]. To affect neuroplasticity more selectively and without side effects inherent for the existent monoamine antidepressants, a new class of therapeutics, “psychoplastogens”, is being developed [78].
To date the PTSD pharmacotherapy is mainly symptomatic, most often the SSRI-antidepressants are prescribed, but their efficiency remains questionable [79]. Some researchers believe that administration of SSRI in PTSD in the absence of a comorbid depressive disorder has not been justified [80]. Using of adrenoblockers in PTSD, even those with a preferentially peripheral action, such as prazosin [81] is reasonable for eliminating the biological positive feedback, which underlies development of panic attacks in the patients with anxiety disorders (table).
CONCLUSIONS
In the present review we attempted to generalize and provide comparative analysis of the accumulated data on mechanisms of MDD and PTSD pathogenesis. Wide variety of the involved neurotransmitters and neuroendocrine systems and of the brain contours along with abundance of different modality connections between them makes generalization of the pathological changes a really difficult problem. In total, it may be concluded that these mental disorders have many common features (from etiological factors to neurotransmitter and hormonal systems involved in pathogenesis of these conditions). However, it is obvious that based on the nature of the majority of pathogenetic changes considered in the review, these states can be opposed to each other, which makes application of similar pharmacotherapeutic treatment strategies of these different disorders unreasonable. Obviously, MDD and PTSD are examples of differently directed disorders in the stress-adaptation mechanisms and require development of different approaches to their treatment.
Abbreviations
- 5-HT1A:
-
1A-type serotonin receptors
- ACC:
-
anterior cingulate cortex
- BDNF:
-
brain-derived neurotrophic factor
- CRH:
-
corticotropin-releasing hormone
- DMN:
-
default mode network
- dlPFC:
-
dorsolateral prefrontal cortex
- GR:
-
glucocorticoid receptors
- HPA axis:
-
hypothalamic-pituitary-adrenocortical axis
- MDD:
-
major depressive disorder
- mPFC:
-
medial prefrontal cortex
- PFC:
-
prefrontal cortex
- PTSD:
-
post-traumatic stress disorder
- SSRI:
-
selective serotonin reuptake inhibitor
- vmPFC:
-
ventromedial prefrontal cortex
References
Kessler, R. C., Sonnega, A., Bromet, E., Hughes, M., and Nelson, C. B. (1995) Posttraumatic stress disorder in the National Comorbidity Survey, Arch. Gen. Psychiatry, 52, 1048-1060, https://doi.org/10.1001/archpsyc.1995.03950240066012.
Yehuda, R., and Antelman, S. M. (1993) Criteria for rationally evaluating animal models of posttraumatic stress disorder, Biol. Psychiatry, 33, 479-486, https://doi.org/10.1016/0006-3223(93)90001-t.
Li, B., Liu, L., Friston, K. J., Shen, H., Wang, L., et al. (2013) A treatment-resistant default mode subnetwork in major depression, Biol. Psychiatry, 74, 48-54, https://doi.org/10.1016/j.biopsych.2012.11.007.
Erk, S., Mikschl, A., Stier, S., Ciaramidaro, A., Gapp, V., et al. (2010) Acute and sustained effects of cognitive emotion regulation in major depression, J. Neurosci., 30, 15726-15734, https://doi.org/10.1523/JNEUROSCI.1856-10.2010.
Ma, N., Li, L., Shu, N., Liu, J., Gong, G., et al. (2007) White matter abnormalities in first-episode, treatment-naive young adults with major depressive disorder, Am. J. Psychiatry, 164, 823-826, https://doi.org/10.1176/ajp.2007.164.5.823.
Frodl, T., Jäger, M., Smajstrlova, I., Born, C., Bottlender, R., et al. (2008) Effect of hippocampal and amygdala volumes on clinical outcomes in major depression: a 3-year prospective magnetic resonance imaging study, J. Psychiatry Neurosci., 33, 423.
Ma, X., Liu, J., Liu, T., Ma, L., Wang, W., et al. (2019) Altered resting-state functional activity in medication-naive patients with first-episode major depression disorders. Healthy control: a quantitative meta-analysis, Front. Behav. Neurosci., 13, 89, https://doi.org/10.3389/fnbeh.2019.00089.
Bennett, M. R. (2011) The prefrontal–limbic network in depression: a core pathology of synapse regression, Prog. Neurobiol., 93, 457-467, https://doi.org/10.1016/j.pneurobio.2011.01.001.
Arnold, J. F., Zwiers, M. P., Fitzgerald, D. A., van Eijndhoven, P., Becker, E. S., et al. (2012) Fronto-limbic microstructure and structural connectivity in remission from major depression, Psychiatry Res. Neuroimaging, 204, 40-48, https://doi.org/10.1016/j.pscychresns.2012.07.010.
Bluhm, R. L., Williamson, P. C., Osuch, E. A., Frewen, P. A., Stevens, T. K., et al. (2009) Alterations in default network connectivity in posttraumatic stress disorder related to early-life trauma, J. Psychiatry Neurosci., 34, 187.
Goldman, S., Mendlewicz, J., Luxen, A., Delvenne, V., Lotstra, F., et al. (1994) Frontal and parietal metabolic disturbances in unipolar depression, Biol. Psychiatry, 36, 381-388, https://doi.org/10.1016/0006-3223(94)91213-0.
Koenigs, M., Huey, E. D., Calamia, M., Raymont, V., Tranel, D., and Grafman, J. (2008) Distinct regions of prefrontal cortex mediate resistance and vulnerability to depression, J. Neurosci., 28, 12341-12348, https://doi.org/10.1523/JNEUROSCI.2324-08.2008.
Bremner, J. D., Staib, L. H., Kaloupek, D., Southwick, S. M., Soufer, R., and Charney, D. S. (1999) Neural correlates of exposure to traumatic pictures and sound in Vietnam combat veterans with and without posttraumatic stress disorder: a positron emission tomography study, Biol. Psychiatry, 45, 806-816, https://doi.org/10.1016/s0006-3223(98)00297-2.
Aupperle, R. L., Allard, C. B., Grimes, E. M., Simmons, A. N., Flagan, T., et al. (2012) Dorsolateral prefrontal cortex activation during emotional anticipation and neuropsychological performance in posttraumatic stress disorder, Arch. Gen. Psychiatry., 69, 360-371, https://doi.org/10.1001/archgenpsychiatry.2011.1539.
Rosenkranz, J. A., and Grace, A. A. (2002) Cellular mechanisms of infralimbic and prelimbic prefrontal cortical inhibition and dopaminergic modulation of basolateral amygdala neurons in vivo, J. Neurosci., 22, 324-337, https://doi.org/10.1523/JNEUROSCI.22-01-00324.2002.
Sundermann, B., Beverborg, M. O., and Pfleiderer, B. (2014) Toward literature-based feature selection for diagnostic classification: a meta-analysis of resting-state fMRI in depression, Front. Hum. Neurosci., 8, 692, https://doi.org/10.3389/fnhum.2014.00692.
Barthas, F., Sellmeijer, J., Hugel, S., Waltisperger, E., Barrot, M., and Yalcin, I. (2015) The anterior cingulate cortex is a critical hub for pain-induced depression, Biol. Psychiatry, 77, 236-245, https://doi.org/10.1016/j.biopsych.2014.08.004.
Britton, J. C., Phan, K. L., Taylor, S. F., Fig, L. M., and Liberzon, I. (2005) Corticolimbic blood flow in posttraumatic stress disorder during script-driven imagery, Biol. Psychiatry, 57, 832-840, https://doi.org/10.1016/j.biopsych.2004.12.025.
De Kloet, E. R., Vreugdenhil, E., Oitzl, M. S., and Joëls, M. (1998) Brain corticosteroid receptor balance in health and disease, Endocr. Rev., 19, 269-301, https://doi.org/10.1210/edrv.19.3.0331.
Murri, M. B., Pariante, C., Mondelli, V., Masotti, M., Atti, A. R., et al. (2014) HPA axis and aging in depression: systematic review and meta-analysis, Psychoneuroendocrinology, 41, 46-62, https://doi.org/10.1016/j.psyneuen.2013.12.004.
Nemeroff, C. B., and Owens, M. J. (2002) Treatment of mood disorders, Nat. Neurosci., 5, 1068-1070, https://doi.org/10.1038/nn943.
Meewisse, M. L., Reitsma, J. B., de Vries, G. J., Gersons, B. P. R., and Olff, M. (2007) Cortisol and post-traumatic stress disorder in adults: systematic review and meta-analysis, Br. J. Psychiatry, 191, 387-392, https://doi.org/10.1192/bjp.bp.106.024877.
Pan, X., Wang, Z., Wu, X., Wen, S. W., and Liu, A. (2018) Salivary cortisol in post-traumatic stress disorder: a systematic review and meta-analysis, BMC Psychiatry, 18, 324, https://doi.org/10.1186/s12888-018-1910-9.
Franco, A. J., Chen, C., Scullen, T., Zsombok, A., Salahudeen, A. A., et al. (2016) Sensitization of the hypothalamic-pituitary-adrenal axis in a male rat chronic stress model, Endocrinology, 157, 2346-2355, https://doi.org/10.1210/en.2015-1641.
Yehuda, R., Teicher, M. H., Trestman, R. L., Levengood, R. A., and Siever, L. J. (1996) Cortisol regulation in posttraumatic stress disorder and major depression: a chronobiological analysis, Biol. Psychiatry, 40, 79-88, https://doi.org/10.1016/0006-3223(95)00451-3.
Wang, Q., Verweij, E. W. E., Krugers, H. J., Joels, M., Swaab, D. F., and Lucassen, P. J. (2013) Distribution of the glucocorticoid receptor in the human amygdala; changes in mood disorder patients, Brain Struct. Funct., 219, 1615-1626, https://doi.org/10.1007/s00429-013-0589-4.
Wang, Q., Joels, M., Swaab, D. F., and Lucassen, P. J. (2012) Hippocampal GR expression is increased in elderly depressed females, Neuropharmacology, 62, 527-533, https://doi.org/10.1016/j.neuropharm.2011.09.014.
Chen, J., Wang, Z., Zuo, W., Zhang, S., Chu, S., and Chen, N. (2016) Effects of chronic mild stress on behavioral and neurobiological parameters – role of glucocorticoid, Horm. Behav., 78, 150-159, https://doi.org/10.1016/j.yhbeh.2015.11.006.
Sapolsky, R. M. (1986) Glucocorticoid toxicity in the hippocampus: reversal by supplementation with brain fuels, J. Neurosci., 6, 2240-2244, https://doi.org/10.1523/JNEUROSCI.06-08-02240.1986.
Gourley, S. L., Wu, F. J., Kiraly, D. D., Ploski, J. E., Kedves, A. T., et al. (2008) Regionally specific regulation of ERK MAP kinase in a model of antidepressant – sensitive chronic depression, Biol. Psychiatry, 63, 353-359, https://doi.org/10.1016/j.biopsych.2007.07.016.
Numakawa, T., Adachi, N., Richards, M., Chiba, S., and Kunugi, H. (2013) Brain-derived neurotrophic factor and glucocorticoids: reciprocal influence on the central nervous system, Neuroscience, 239, 157-172, https://doi.org/10.1016/j.neuroscience.2012.09.073.
Lee, B. H., and Kim, Y. K. (2010) The roles of BDNF in the pathophysiology of major depression and in antidepressant treatment, Psychiatry Invest., 7, 231, https://doi.org/10.4306/pi.2010.7.4.231.
Duman, R. S., and Monteggia, L. M. (2006) A neurotrophic model for stress-related mood disorders, Biol. Psychiatry, 59, 1116-1127, https://doi.org/10.1016/j.biopsych.2006.02.013.
Loftis, J. M., Huckans, M., and Morasco, B. J. (2010) Neuroimmune mechanisms of cytokine-induced depression: current theories and novel treatment strategies, Neurobiol. Dis., 37, 519-533, https://doi.org/10.1016/j.nbd.2009.11.015.
Capuron, L., and Dantzer, R. (2003) Cytokines and depression: the need for a new paradigm, Brain Behav. Immun., 17, 119-124, https://doi.org/10.1016/S0889-1591(02)00078-8.
Kubera, M., Curzytek, K., Duda, W., Leskiewicz, M., Basta-Kaim, A., et al. (2013) A new animal model of (chronic) depression induced by repeated and intermittent lipopolysaccharide administration for 4 months, Brain Behav. Immun., 31, 96-104, https://doi.org/10.1016/j.bbi.2013.01.001.
Barrientos, R. M., Sprunger, D. B., Campeau, S., Higgins, E. A., Watkins, L. R., et al. (2003) Brain-derived neurotrophic factor mRNA downregulation produced by social isolation is blocked by intrahippocampal interleukin-1 receptor antagonist, Neuroscience, 121, 847-853, https://doi.org/10.1016/S0306-4522(03)00564-5.
Smith, R. S. (1991) The macrophage theory of depression, Med. Hypotheses, 35, 298-306, https://doi.org/10.1016/0306-9877(91)90272-Z.
Baker, D. G., West, S. A., Nicholson, W. E., Kasckow, J. W., Hill, K. K., et al. (1999) Corticotropin-releasing hormone levels and adrenocortical activity in combat veterans with posttraumatic stress disorder, Am. J. Psychiatry, 156, 585-588, https://doi.org/10.1176/ajp.156.4.585.
Mironova, V., Rybnikova, E., and Pivina, S. (2013) Effect of inescapable stress in rodent models of depression and posttraumatic stress disorder on CRH and vasopressin immunoreactivity in the hypothalamic paraventricular nucleus, Acta Physiol. Hung., 100, 395-410, https://doi.org/10.1556/APhysiol.100.2013.4.4.
Wen, L., Han, F., and Shi, Y. (2014) Changes in the glucocorticoid receptor and Ca2+/calreticulin-dependent signalling pathway in the medial prefrontal cortex of rats with post-traumatic stress disorder, J. Mol. Neurosci., 56, 24-34, https://doi.org/10.1007/s12031-014-0464-7.
Eagle, A. L., Knox, D., Roberts, M. M., Mulo, K., Liberzon, I., et al. (2013) Single prolonged stress enhances hippocampal glucocorticoid receptor and phosphorylated protein kinase B levels, Neurosci. Res., 75, 130-137, https://doi.org/10.1016/j.neures.2012.11.001.
Mulinari, S. (2012) Monoamine theories of depression: historical impact on biomedical research, J. Hist. Neurosci., 21, 366-392, https://doi.org/10.1080/0964704X.2011.623917.
Andrews, P. W., Bharwani, A., Lee, K. R., Fox, M., and Thomson, J. A. Jr. (2015) Is serotonin an upper or a downer? The evolution of the serotonergic system and its role in depression and the antidepressant response, Neurosci. Biobehav. Rev., 51, 164-188, https://doi.org/10.1016/j.neubiorev.2015.01.018.
Lu, Q., Mouri, A., Yang, Y., Kunisawa, K., Teshigawara, T., et al. (2019) Chronic unpredictable mild stress-induced behavioral changes are coupled with dopaminergic hyperfunction and serotonergic hypofunction in mouse models of depression, Behav. Brain Res., 372, 112053, https://doi.org/10.1016/j.bbr.2019.112053.
Ossowska, G., Nowa, G., Kata, R., Klenk-Majewska, B., Danilczuk, Z., and Zebrowska-Lupina, I. (2001) Brain monoamine receptors in a chronic unpredictable stress model in rats, J. Neural Transm., 108, 311-319, https://doi.org/10.1007/s007020170077.
Garcia-Garcia, A. L., Meng, Q., Canetta, S., Gardier, A. M., Guiard, B. P., et al. (2017) Serotonin signaling through prefrontal cortex 5-HT1A receptors during adolescence can determine baseline mood-related behaviors, Cell Rep., 18, 1144-1156, https://doi.org/10.1016/j.celrep.2017.01.021.
Roy, A., de Jong, J., and Linnoila, M. (1989) Cerebrospinal fluid monoamine metabolites and suicidal behavior in depressed patients: a 5-year follow-up study, Arch. Gen. Psychiatry, 46, 609-612, https://doi.org/10.1001/archpsyc.1989.01810070035005.
Kram, M. L., Kramer, G. L., Ronan, P. J., Steciuk, M., and Petty, F. (2002) Dopamine receptors and learned helplessness in the rat: an autoradiographic study, Prog. Neuropsychopharmacol. Biol. Psychiatry, 26, 639-645, https://doi.org/10.1016/s0278-5846(01)00222-6.
Ji, N. N., Kang, J., Hua, R., and Zhang, Y. M. (2018) Involvement of dopamine system in the regulation of the brain corticotropin-releasing hormone in paraventricular nucleus in a rat model of chronic visceral pain, Neurol. Res., 40, 650-657, https://doi.org/10.1080/01616412.2018.1460702.
Chen, C., Yang, J. M., Hu, T. T., Xu, T. J., Xu, W. P., and Wei, W. (2013) Elevated dopamine D2 receptor in prefrontal cortex of CUMS rats is associated with downregulated cAMP-independent signaling pathway, Can. J. Physiol. Pharmacol., 91, 750-758, https://doi.org/10.1139/cjpp-2012-0399.
Qiao, H., Yang, S., Xu, C., Ma, X. M., and An, S. C. (2020) Involvement of D2 receptor in the NAc in chronic unpredictable stress-induced depression-like behaviors, Stress, 23, 318-327, https://doi.org/10.1080/10253890.2019.1673361.
Papp, M., Klimek, V., and Willner, P. (1994) Parallel changes in dopamine D2 receptor binding in limbic forebrain associated with chronic mild stress-induced anhedonia and its reversal by imipramine, Psychopharmacology, 115, 441-446, https://doi.org/10.1007/BF02245566.
Shah, A. A., Sjovold, T., and Treit, D. (2004) Selective antagonism of medial prefrontal cortex D4 receptors decreases fear-related behaviour in rats, Eur. J. Neurosci., 19, 3393-3397, https://doi.org/10.1111/j.0953-816X.2004.03447.x.
Laviolette, S. R., Lipski, W. J., and Grace, A. A. (2005) A subpopulation of neurons in the medial prefrontal cortex encodes emotional learning with burst and frequency codes through a dopamine D4 receptor-dependent basolateral amygdala input, J. Neurosci., 25, 6066-6075, https://doi.org/10.1523/JNEUROSCI.1168-05.2005.
Horvitz, J. C., Stewart, T., and Jacobs, B. L. (1997) Burst activity of ventral tegmental dopamine neurons is elicited by sensory stimuli in the awake cat, Brain Res., 759, 251-258, https://doi.org/10.1016/s0006-8993(97)00265-5.
Harvey, B. H., Naciti, C., Brand, L., and Stein, D. J. (2003) Endocrine, cognitive and hippocampal/cortical 5HT1A/2A receptor changes evoked by a time-dependent sensitization (TDS) stress model in rats, Brain Res., 983, 97-107, https://doi.org/10.1016/s0006-8993(03)03033-6.
Harada, K., Yamaji, T., and Matsuoka, N. (2008) Activation of the serotonin 5-HT2C receptor is involved in the enhanced anxiety in rats after single-prolonged stress, Pharmacol. Biochem. Behav., 89, 11-16, https://doi.org/10.1016/j.pbb.2007.10.016.
Neumeister, A., Bain, E., Nugent, A. C., Carson, R. E., Bonne, O., et al. (2004) Reduced serotonin type 1A receptor binding in panic disorder, J. Neurosci., 24, 589-591, https://doi.org/10.1523/JNEUROSCI.4921-03.2004.
Krystal, J. H., and Neumeister, A. (2009) Noradrenergic and serotonergic mechanisms in the neurobiology of posttraumatic stress disorder and resilience, Brain Res., 1293, 13-23, https://doi.org/10.1016/j.brainres.2009.03.044.
Shen, M., Yang, Y., Wu, Y., Zhang, B., Wu, H., et al. (2018) L-theanine ameliorate depressive-like behavior in a chronic unpredictable mild stress rat model via modulating the monoamine levels in limbic-cortical-striatal-pallidal-thalamic-circuit related brain regions, Phytother. Res., 33, 412-421, https://doi.org/10.1002/ptr.6237.
Bremner, J. D., Krystal, J. H., Southwick, S. M., and Charney, D. S. (1996) Noradrenergic mechanisms in stress and anxiety. I. Preclinical studies, Synapse, 23, 28-38, https://doi.org/10.1002/(SICI)1098-2396(199605)23:1<28::AID-SYN4>3.0.CO;2-J.
Ronzoni, G., Del Arco, A., Mora, F., and Segovia, G. (2016) Enhanced noradrenergic activity in the amygdala contributes to hyperarousal in an animal model of PTSD, Psychoneuroendocrinology, 70, 1-9, https://doi.org/10.1016/j.psyneuen.2016.04.018.
Bear, M., Connors, B., and Paradiso, M. A. (2020) Neuroscience: Exploring the Brain, Jones & Bartlett Learning, Burlington, USA.
Berman, R. M., Cappiello, A., Anand, A., Oren, D. A., Heninger, G. R., et al. (2000) Antidepressant effects of ketamine in depressed patients, Biol. Psychiatry, 47, 351-354, https://doi.org/10.1016/S0006-3223(99)00230-9.
Arnone, D., Mumuni, A. N., Jauhar, S., Condon, B., and Cavanagh, J. (2015) Indirect evidence of selective glial involvement in glutamate-based mechanisms of mood regulation in depression: meta-analysis of absolute prefrontal neuro-metabolic concentrations, Eur. Neuropsychopharmacol., 25, 1109-1117, https://doi.org/10.1016/j.euroneuro.2015.04.016.
Luykx, J. J., Laban, K. G., van den Heuvel, M. P., Boks, M. P., Mandl, R. C., et al. (2012) Region and state specific glutamate downregulation in major depressive disorder: a meta-analysis of (1)H–MRS findings, Neurosci. Biobehav. Rev., 36, 198-205, https://doi.org/10.1016/j.neubiorev.2011.05.014.
Knox, D., Perrine, S. A., George, S. A., Galloway, M. P., and Liberzon, I. (2010) Single prolonged stress decreases glutamate, glutamine, and creatine concentrations in the rat medial prefrontal cortex, Neurosci. Lett., 480, 16-20, https://doi.org/10.1016/j.neulet.2010.05.052.
Karolewicz, B., Szebeni, K., Gilmore, T., Maciag, D., Stockmeier, C. A., and Ordway, G. A. (2009) Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression, Int. J. Neuropsychopharmacol., 12, 143-153, https://doi.org/10.1017/S1461145708008985.
Gulyaeva, N. V. (2017) Interplay between brain BDNF and glutamatergic systems: a brief state of the evidence and association with the pathogenesis of depression, Biochemistry (Moscow), 82, 301-307, https://doi.org/10.1134/S0006297917030087.
Deschwanden, A., Karolewicz, B., Feyissa, A. M., Treyer, V., Ametamey, S. M., et al. (2011) Reduced metabotropic glutamate receptor 5 density in major depression determined by [(11)C]ABP688 PET and postmortem study, Am. J. Psychiatry, 168, 727-734, https://doi.org/10.1176/appi.ajp.2011.09111607.
Nishi, D., Hashimoto, K., Noguchi, H., Hamazaki, K., Hamazaki, T., and Matsuoka, Y. (2015) Glutamatergic system abnormalities in posttraumatic stress disorder, Psychopharmacology, 232, 4261-4268, https://doi.org/10.1007/s00213-015-4052-5.
Meyerhoff, D. J., Mon, A., Metzler, T., and Neylan, T. C. (2014) Cortical gamma-aminobutyric acid and glutamate in posttraumatic stress disorder and their relationships to self-reported sleep quality, Sleep, 37, 893-900, https://doi.org/10.5665/sleep.3654.
Rosso, I. M., Crowley, D. J., Silveri, M. M., Rauch, S. L., and Jensen, J. E. (2017) Hippocampus glutamate and n-acetyl aspartate markers of excitotoxic neuronal compromise in posttraumatic stress disorder, Neuropsychopharmacology, 42, 1698-1705, https://doi.org/10.1038/npp.2017.32.
Ousdal, O. T., Milde, A. M., Craven, A. R., Ersland, L., Endestad, T., et al. (2019) Prefrontal glutamate levels predict altered amygdala–prefrontal connectivity in traumatized youths, Psychol. Med., 49, 1822-1830, https://doi.org/10.1017/S0033291718002519.
Holmes, S. E., Girgenti, M. J., Davis, M. T., Pietrzak, R. H., DellaGioia, N., et al. (2017) Altered metabotropic glutamate receptor 5 markers in PTSD: in vivo and postmortem evidence, Proc. Natl. Acad. Sci. USA, 114, 8390-8395, https://doi.org/10.1073/pnas.1701749114.
Kirsch, I., Deacon, B. J., Huedo-Medina, T. B., Scoboria, A., Moore, T. J., and Johnson, B. T. (2008) Initial severity and antidepressant benefits: a meta-analysis of data submitted to the Food and Drug Administration, PLoS Med., 5, e45, https://doi.org/10.1371/journal.pmed.0050045.
Cameron, L. P., Tombari, R. J., Lu, J., Pell, A. J., Hurley, Z. Q., et al. (2021) A non-hallucinogenic psychedelic analogue with therapeutic potential, Nature, 589, 474-479, https://doi.org/10.1038/s41586-020-3008-z.
Ravindran, L. N., and Stein, M. B. (2009) Pharmacotherapy of PTSD: premises, principles, and priorities, Brain Res., 1293, 24-39, https://doi.org/10.1016/j.brainres.2009.03.037.
Torrens, M., Fonseca, F., Mateu, G., and Farré, M. (2005) Efficacy of antidepressants in substance use disorders with and without comorbid depression: a systematic review and meta-analysis, Drug Alcohol Dependence, 78, 1-22, https://doi.org/10.1016/j.drugalcdep.2004.09.004.
Raskind, M. A., Thompson, C., Petrie, E. C., Dobie, D. J., Rein, R. J., et al. (2002) Prazosin reduces nightmares in combat veterans with posttraumatic stress disorder, J. Clin. Psychiatry, 63, 565-568, https://doi.org/10.4088/jcp.v63n0705.
Funding
The work was financially supported by the Russian Foundation for Basic Research (project no. 19-015-00336).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors declare no conflicts of interest in financial or any other sphere. This article does not contain any studies with human participants or animals performed by any of the authors.
Rights and permissions
Open access. This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution, and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit https://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stupin, K.N., Zenko, M.Y. & Rybnikova, E.A. Comparative Analysis of Pathobiochemical Changes in Major Depression and Post-Traumatic Stress Disorder. Biochemistry Moscow 86, 729–736 (2021). https://doi.org/10.1134/S0006297921060109
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0006297921060109