
Abstract
The insulin-like growth factor (IGF) receptor (IGF-IR) is necessary for IGF signalling and has essential roles in cellular growth. In teleost fish, two distinct IGF-IR duplicates are conserved called IGF-IRa and IGF-IRb. However, while a salmonid-specific whole genome duplication (ssWGD) is known to have expanded several key genes within the IGF axis, its impact on the IGF-IR repertoire remains unresolved. Using bioinformatic and phylogenetic approaches, we establish that salmonids retain two IGF-IRa paralogues from ssWGD and a single IGF-IRb copy. We measured the tissue-specific and developmental transcriptional regulation of each IGF-IR gene, revealing tight co-expression between the IGF-IRa paralogues, but expression divergence comparing IGF-IRa and IGF-IRb genes. We also examined the regulation of each IGF-IR gene in fish challenged by bacterial and viral infections, adding to recent reports that the IGF axis has roles linking growth and immunity. While whole salmonid fry showed a small upregulation of IGF-IR expression during both types of infection, bacterial challenge caused striking downregulation of IGF-IRa1 and IGF-IRa2 in head kidney and spleen of adult fish, alongside genes coding IGF hormones, highlighting a strong repression of IGF-signalling in primary immune tissues. The reported immune-responsive regulation of IGF-IR genes adds to an emerging body of evidence that supports important cross-talk between master growth and immune pathways in vertebrates.
Similar content being viewed by others

Introduction
The transmembrane tyrosine kinase IGF-IR is a crucial component of the IGF axis, a signalling pathway with essential roles in the growth, migration, survival, proliferation and differentiation of vertebrate cells1,2,3. For example, the genetic ablation of IGF-IR in mice causes death during embryogenesis, in association with highly retarded growth phenotypes4. Both the IGF hormones (IGF-I and IGF-II) bind to IGF-IRs on target cell membranes, activating conserved signal transduction cascades within the cell, including canonical phosphoinositide 3-kinase/Akt and MAPK pathways3,5,6. Most understanding of the biology of IGF-IR comes from work with mammals, with relatively limited work done on IGF-IR from lower vertebrate lineages.
In teleost fish, core components of the IGF axis, including IGF-IR, have been expanded through gene duplication events, including whole genome duplication (WGD)7. The ancestor to teleosts experienced WGD ~350 Ma (the ‘teleost-specific WGD’: tsWGD) and ~12–20% of the created gene duplicates (paralogues) are retained in descendent lineages8,9,10. Teleosts retain paralogues of both IGF hormones11, multiple IGF binding proteins (IGFBPs)12,13, as well as IGF-IR14,15,16. The teleost IGF-IR paralogues IGF-IRa and IGF-IRb have overlapping yet distinct expression domains in embryonic and adult stages of zebrafish (Danio rerio) ontogeny14,15,16.
In salmonid fish, growth has major commercial relevance in relation to aquaculture, making the IGF axis a focal point of research interest. Interestingly, duplicated copies of several IGF axis components, including the IGF hormones17,18 and IGFBPs13,18 have been retained from an additional WGD event to tsWGD, which occurred in the salmonid ancestor ~95 Ma (the ‘salmonid-specific WGD’: ssWGD)19, and, from which ~50% of paralogues have been functionally retained10. In terms of IGF-IR, while two differentially expressed salmonid variants have been reported as partial sequences20,21,22, the evolutionary history of IGF-IRs in relation to tsWGD and ssWGD remains poorly resolved.
Therefore, the first aim of this study was to exploit recently developed genome-scale sequence data for salmonids10,23 and northern pike (Esox lucius), a closely related outgroup lineage to ssWGD24, to characterise the complete repertoire of full-length IGF-IR genes conserved in Atlantic salmon (Salmo salar) and rainbow trout (Oncorhynchus mykiss). The second aim was to quantify the expression of the complete salmonid IGF-IR gene repertoire in different adult tissues and stages of early development. The final aim was to characterize the expression responses of all salmonid IGF-IR genes during host defence responses to in vivo disease challenge, in light of an emerging role for the IGF axis in linking the growth and immune systems of salmonid fish25,26,27,28.
Results and Discussion
The complete salmonid IGF-IR repertoire
TBLASTn searches of the Atlantic salmon genome using northern pike IGF-IRa and IGF-IRb amino acid sequences as queries identified three putative genes coding IGF-IRs. Two genes were located on chromosome 10 and 16 (~81.0 and 29.6 Mb into 116.1 and 87.8 Mb total length of each chromosome, respectively) and encoded full-length IGF-IR sequences of 1,421 and 1,414 amino acids, respectively. The protein products of these genes shared 96% amino acid identity (and ~82/72% amino acid identity to zebrafish IGF-IRa/b) and are embedded within duplicated collinear segments of the genome retained from ssWGD10. Thus, our initial hypothesis was that these genes represented co-orthologues of IGF-IRa retained from ssWGD and they were named IGF-IRa1 and IGF-IRa2, in line with existing nomenclature14. BLASTn searches of a rainbow trout genome assembly using the Atlantic salmon IGF-IRa1 and IGF-IRa2 sequences as queries identified two distinct contigs (MMSRT019F_scaff_1700_1 and MMSRT039C_scaff_1472_3). Using the FGENESH+ algorithm29, putative IGF-IR genes were predicted from MMSRT019F_scaff_1700_1 and MMSRT039C_scaff_1472_3, coding 1,236 and 1,032 amino acids that shared ~96% identity to each other, but >99% respective identity to Atlantic salmon IGF-IRa1 and IGF-IRa2, suggesting two sets of 1:1 orthologues were identified when comparing the two salmonid species.
The third IGF-IR gene was located on the tip of Atlantic salmon chromosome 26 (~46.2 Mb into 47.8 Mb total chromosome length) and encoded a truncated 850 amino acids. The protein sequence for this gene shared 71/68% amino acid identity to zebrafish IGF-IRa/b, leaving uncertainty as to its relationship with other teleost IGF-IRs. The aforementioned location on chromosome 26 shares an extremely similar duplicated region at the start of chromosome 11 and has probably not yet completed the rediploidization process post-ssWGD (see ref. 10, around 10% of the duplicated genome is yet to undergo rediploidization). As the completion of rediploidization is necessary for ssWGD paralogues to diverge at the sequence level10, there is no expectation that IGF-IRb paralogues with distinct sequences should exist in the Atlantic salmon genome. This is clearly distinct to a common situation accounting for single copy salmonid genes, where two distinct paralogues originally existed, but one was lost during evolution10.
We also identified putative partial sequences for the third IGF-IR gene in rainbow trout, including a 2,550 bp transcript from within a published transcriptome13, that overlapped by 2,214 bp with the Atlantic salmon IGF-IR gene predicted on chromosome 26. The two salmonid IGF-IRb sequences shared 95% amino acid identity to each other in this region, but only 78% identity to the IGF-IRa paralogues, suggesting they were orthologues.
Evolution of salmonid IGF-IRs: phylogenetics and comparative genomics
Bayesian (Fig. 1) and maximum-likelihood (ML) (Supplementary Figure 1) phylogenetic analyses were performed using a high-confidence amino acid alignment including the novel IGF-IRs from salmonids, along with IGF-IR duplicates in diverse teleosts, a single IGF-IR from a ray-finned fish that never underwent tsWGD (i.e. spotted gar, Lepisosteus oculatus30) and single IGF-IRs from diverse lobe-finned fish lineages. We also included insulin receptor (INSR) sequences as an outgroup to IGF-IR, which was appropriate considering that INSR and IGF-IR have conserved extensive similarity since they duplicated from a single ancestral gene during early vertebrate evolution31,32,33.

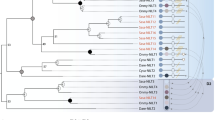
Bayesian phylogenetic analysis for salmonid genes encoding IGF-IR.
The tree was constructed using an amino acid alignment (1,177 amino acids length) including IGF-IR and INSR sequences from a broad range of vertebrates, employing the JTT+G model of amino acid substitution and a relaxed clock model. Branch lengths are scaled in relative time. Posterior probability support values are included for every node (white circles >0.99 posterior support). Nodes representing tsWGD and ssWGD are highlighted. (A,B) denote genes predicted from a genome assembly and published transcriptome, respectively (see results and discussion).
Our Bayesian analysis incorporated a relaxed molecular clock model, allowing probabilistic inference of the tree’s root34, which separated monophyletic IGF-IR and INSR clades with maximal posterior probability support (Fig. 1). Hence, we enforced the split of IGF-IR and INSR as the root in the ML analysis (Supplementary Figure 1). The topology of both trees was identical and congruent with known phylogenetic relationships of the included taxa (Fig. 1; Supplementary Figure 1). Thus, within the IGF-IR clade, a single divergence between ray- and lobe-finned fish was recovered with strong statistical support (Fig. 1; Supplementary Figure 1). Within the ray-finned fish IGF-IR clade, the spotted gar was the basal branch to two teleost-specific sister clades, each containing representatives of all major teleost lineages, including salmonids (Fig. 1; Supplementary Figure 1). This topology is consistent with duplication in the teleost ancestor followed by retention of IGF-IRa and IGF-IRb paralogues, in agreement with past work16. Within the IGF-IRa clade, a monophyletic salmonid clade branched as a sister group to northern pike, and split into two groups containing each included salmonid species (Fig. 1; Supplementary Figure 1). This topology matches to predictions considering the chromosomal location of these genes, in supporting the existence of gene duplicates retained from ssWGD that started diverging in the ancestor to Atlantic salmon and rainbow trout10. Within the IGF-IRb clade, the single salmonid copy of IGF-IRb branched as a sister group to northern pike (Fig. 1; Supplementary Figure 1).
In addition to phylogenetic analysis, we established the intron-exon structure of each salmonid IGF-IR gene, along with the domain organization of encoded IGF-IR proteins in comparison to northern pike and zebrafish (Fig. 2). The complete coding sequence of Atlantic salmon IGF-IRa1 and IGF-IRa2, in common with IGF-IRa orthologues from northern pike and zebrafish, is organized into 21 exons of conserved length, separated by introns of variable size (Fig. 2A). Both genes encode proteins with the full repertoire of conserved protein domains expected for IGF-1R1,2,3 (Fig. 2A).

Inferred intron-exon structures and encoded protein domain organizations of IGF-IRa (A) and IGF-IRb (B) genes in Atlantic salmon, northern pike and zebrafish. Quantitative information on the lengths of exons (boxes, to scale), introns (lines, not to scale) and protein domains (boxes, to scale) are presented. Conserved protein domain abbreviations: ‘FU’ is Furin-like repeats; ‘FN’ is Fibronectin type III; ‘TM’ is the transmembrane helix region; ‘TyrKc’ is Tyrosine kinase. Diagonal lines represent missing information; dashed lines represent missing data. (C) Genomic arrangement of IGF-IRb gene models within the Atlantic salmon genome assembly.
However, the Atlantic salmon IGF-IRb gene (on chromosome 26) is comprised of four separate gene models, which together account for 18 (out of 21) exons (Fig. 2B). These gene fragments are arranged in an unusual pattern, being located on both DNA strands partly interrupted by other genes (Fig. 2C). Assuming this genomic arrangement is correct, such gene fragments cannot be translated to generate a functional IGF-IR protein. An important alternate possibility is an artefact in the genome assembly. In this respect, we identified an Atlantic salmon transcript within a transcriptome assembly (NCBI accession: GEGX01316105.1) that spans the first two IGF-IRb fragments in the genome assembly (i.e. those located on different strands interrupted by a distinct gene) and furthermore, includes bases missing from the genome (Fig. 2B). This transcript encodes the first 1,044 amino acids of IGF-IRb, has the expected conserved protein domains and matches directly to exons 1–16 of the zebrafish IGF-IRb gene (Fig. 2B). This transcript should not exist if the IGF-IRb genomic arrangement predicted within the Atlantic salmon genome is correct. Thus, the genomic arrangement of IGF-IRb in the Atlantic salmon genome is likely an assembly artefact.
In summary, these analyses confirm that the IGF-IR gene repertoire of salmonids was shaped by both the tsWGD and ssWGD duplication events.
Tissue-specific and developmental transcriptional regulation of salmonid IGF-IR repertoire
Our next goal was to quantify the expression of each identified salmonid IGF-IR gene in different adult tissues and during different stages of early development. We used qPCR to quantify transcripts for the three IGF-IR genes in eleven tissues of adult Atlantic salmon13 (Fig. 3A). IGF-IRa1 and IGF-IRa2 were expressed in several tissues, including brain, eye, heart, fast muscle and gill and also had highly similar expression patterns according to Spearman’s rank correlation test (Fig. 3A,B, P < 0.0001), suggesting strong conservation of ancestral regulation across tissues. While IGF-IRb was expressed in many of the same tissues as IGF-IRa1 and IGF-IRa2, there were some notable quantitative differences, including higher expression in brain, head kidney, spleen and stomach (Fig. 3A). The liver is the predominant source of endocrine IGFs and IGFBPs13,35 but exhibited negligible expression of all three IGF-IRs (Fig. 3A), as reported for other teleosts14,36. This suggests that the liver does not require IGF signalling in healthy, satiated fish. Such a lack of IGF-IR expression may also ensure that IGFs are efficiently released from liver into the circulation without competition from IGF-IR.

Tissue-specific (A,B) and developmental (C,D) transcriptional regulation of salmonid IGF-IR genes. Data are presented as mean transcript levels (n = 4 to 5) +SD for each gene. Different letters indicate significant differences (P < 0.05) between the three stages of development for each gene (one-way ANOVAs testing the effect of developmental stage provided in Table 1). Scatterplots of ranked data illustrate co-expression of IGF-IRa1 and IGF-IRa2 transcript levels across the examined tissues and developmental stages.
Next, the transcript expression of each salmonid IGF-IR gene was quantified in whole rainbow trout fry at three stages of development: hatching (H), first feeding (FF) and 3-weeks post first feeding (3wFF)26,37 (Fig. 3C; Table 1). While IGF-IRb expression was unchanged during this period, IGF-IRa1 and IGF-IRa2 were significantly co-upregulated between H and 3wFF (Fig. 3C,D; Table 1). While this period coincides with the onset of complex growth regulation, reflected by upregulation of IGF-I and IGF-II hormones and diverse IGFBP subtypes26, further interpretation of these data is limited by the fact that the samples used were whole fry, leading to a lack of tissue-specific context.
Regulation of salmonid IGF-IR repertoire during disease challenge
We recently demonstrated that key genes from the IGF-signalling axis, notably IGFBP-1A1 and IGFBP-6A2, were strongly induced during in vivo disease challenges, suggesting roles linking growth to the host defence response26. However, this past study lacked any data on the IGF-IR genes characterized here. Therefore, the expression of the novel salmonid IGF-IR repertoire was examined in whole rainbow trout fry challenged separately with Aeromonas salmonicida (AS), the causative bacterial agent of furunculosis, and viral hemorrhagic septicemia virus (VHSv), during H, FF and 3wFF stages26,37 (Fig. 4; Table 2). There was a statistically significant effect of AS on transcript levels of IGF-IRa1 and IGF-IRa2, but not IGF-IRb, and for IGF-IRa1 a significant treatment*stage interaction (Fig. 4A; Table 2). In addition, we observed a statistically significant effect of VHSv on all three IGF-IR genes, and a significant treatment*stage interaction for IGF-IRa1 and IGF-IRa2 (Fig. 4C; Table 2). The effect of AS and VHSv on IGF-IR transcript levels was associated with a small level of induction (Fig. 4A,C). We also observed a highly significant (P < 0.0001) co-expression of IGF-IRa1 and IGF-IRa2 (Fig. 4B,D), as observed in different tissues. However, the magnitude of induction varied depending on the nature of the pathogen and developmental stage. The highest induction of IGF-IRa1 and IGF-IRa2 in response to AS occurred at H, whereas the largest effect of VHSv on both genes was observed at FF (Fig. 4A,C; Table 2).

Transcriptional response of IGF-IR genes to AS (A,B) and VHSv (C,D) challenge at three stages of rainbow trout development. Data are presented as fold change (treatment/control) values for each gene, along with asterisks denoting a significant (P < 0.05) effect of treatment at each stage of development (two-way ANOVAs testing the effect of AS and VHSv challenge provided in Table 2). For AS, n = 4 for treatments and n = 5 for PBS controls. For VHSv, n = 3 to 5 for treatments and n = 5 for PBS controls. Scatterplots of ranked data illustrate co-expression of IGF-IRa1 and IGF-IRa2 transcript levels in control and AS/VHSv infected fish.
When interpreting these findings, we were initially surprised that IGF-IRa1 and IGF-IRa2 were induced rather than repressed by infection, as this is counterintuitive to past suggestions that IGF-signalling is restricted during disease to facilitate energetic reallocation towards immunity25,26,38. However, as our samples come from whole animals, the tissue-specific context of this finding remains unknown and it possible that downregulation in some tissues is being counteracted by strong upregulation elsewhere, leading to a small average upregulation observed in the global mRNA message. Under this scenario, it is important to note our previous observation that an IGFBP-1A subtype was strongly upregulated in response to AS and VHSv, which is expected to restrict IGF hormones in the circulation26. Thus, an upregulation of IGF-IRs within certain tissues may act to maintain IGF-signalling locally, despite endocrine restriction of IGF signalling.
To add an immune tissue-specific context to our study, we measured the expression of the salmonid IGF-IR genes, along with genes coding IGF-I and IGF-II hormones, in spleen and head kidney from rainbow trout challenged in vivo by Yersinia ruckeri (YR), the causative bacterial agent of enteric redmouth disease39 (Fig. 5; Table 3). While this is a distinct pathogen challenge to that performed in whole fry, we previously observed consistent immune-responsive regulation of IGF-axis gene components to both YR and AS, suggesting underlying control by immune pathways that are activated in response to different bacteria species. Interestingly, IGF-IRa1 and IGF-IRa2 were each highly repressed in concert with IGF-I in both tissues, whereas IGF-IRb was lowly expressed in both tissues (Fig. 5A–C,E–G; Table 3). Downregulation of IGF-I expression in response to YR challenge has been previously reported in liver and head kidney of juvenile rainbow trout27. However, in response to YR, IGF-II did not change significantly in head kidney, but was modestly repressed in spleen (Fig. 5D,H; Table 3). Interestingly, in our past work, we also observed a striking upregulation of IGFBP-1A1 and IGFBP-6A2 subtypes in response to YR infection in the same primary immune tissues, which was likely governed through conserved immune cytokine networks26. Previously, we suggested the strong induction of these IGFBP subtypes served to either restrict availability of IGF-hormones to IGF-IR or stimulate the development of immune cells through mechanisms that are IGF-independent26. The results presented here are consistent with this hypothesis, indicating an overall strong repression of IGF-signalling in immune tissues during infection.

Transcriptional response of genes coding IGF-IRs and IGF hormones to YR challenge in head kidney (A–D) and spleen (E–H) of rainbow trout. Data are presented as mean transcript levels (n = 4) +SD for each gene, along with asterisks denoting significant (P < 0.05) effect of treatment (one-way ANOVA testing the effect of YR challenge provided in Table 3).
Materials and Methods
Sequence retrieval
Annotated amino acid sequences for genes coding IGF-IR and INSR were retrieved from GenBank (http://ncbi.nlm.nih.gov/) or Ensembl databases (http://www.ensembl.org/index.html). This included the following genome assemblies: northern pike (E. lucius, NCBI accession: GCA_000721915.2), Japanese puffer (Takifugu rubripes, Ensembl database: FUGU 4.0), Nile tilapia (Oreochromis niloticus, Ensembl database: Orenil1.0), zebrafish (D. rerio, Ensembl database: GRCz10), cave fish (Astyanax mexicanus, Ensembl database: AstMex102), spotted gar (L. oculatus, Ensembl database: LepOcu1), coelacanth (Latimeria chalumnae, Ensembl database: LatCha1), chicken (Gallus gallus, Ensembl database: Galgal4), anole lizard (Anolis carolinensis, Ensembl database: AnoCar2.0), human (Homo sapiens, Ensembl database: GRCh38.p5), cow (Bos taurus, Ensembl database: UMD3.1) and western clawed frog (Xenopus tropicalis, Ensembl database: JGI 4.2). Salmonid IGF-IR genes were acquired using northern pike IGF-IRa and IGF-IRb amino acid sequences as queries in tBLASTn searches40 of Atlantic salmon and rainbow trout genome assemblies through SalmoBase (http://salmobase.org/index.php) and the National Animal Genome Research Program (http://www.animalgenome.org/), respectively. Rainbow trout coding sequences were predicted from the corresponding genomic DNA using the FGENESH+ algorithm29 via a webserver (http://www.softberry.com/berry.phtml). Gene predictions were trained on zebrafish gene-finding parameters, but with guidance from the more similar northern pike IGF-IRa and IGF-IRb amino acid sequences. Accession numbers for all studied sequences are provided within figures.
Phylogenetic and comparative genomic analyses
Phylogenetic analysis was performed on IGF-IR and INSR amino acid sequences sampled from diverse vertebrate lineages. Sequence alignment was performed using MAFFT v741 via the GUIDANCE webserver42. The original alignment length was 1,628 amino acids with an overall GUIDANCE alignment score of 0.95. The GUIDANCE algorithm43 was used to identify and discard aligned sites below a statistical confidence cut-off score of 0.93. A final resulting alignment of 1,177 amino acids length (provided in Data file S1) was uploaded to MEGA v.644 to estimate the best-fitting amino acid substitution model using ML. Bayesian phylogenetic analysis was performed using BEAST v1.7.544, employing the best-fitting substitution model (JTT+G), an uncorrelated lognormal relaxed clock model34, a Yule speciation prior46 and a UPGMA starting tree. Two runs of BEAST were performed, each with a Markov chain Monte Carlo (MCMC) chain length of 10,000,000 generations, sampled every 1,000 generations. The MCMC traces were analysed for mixing and convergence properties via Tracer v1.6 (http://beast.bio.ed.ac.uk/Tracer). For all estimated parameters, effective sample size values exceeded 100. The two runs were combined in LogCombiner v1.7.5 (http://beast.bio.ed.ac.uk/LogCombiner), discarding 10% of tress as burn-in. A maximum clade credibility tree was generated using TreeAnnotator v1.7.545. The ML phylogenetic analysis was performed on the same data under the PROTGAMMAJTT amino acid substitution model in RAxML v8.1.2147. The ML tree was generated via raxmlGUI v1.5b148 with 100 rapid bootstrap replicates49.
The intron-exon structures of IGF-IR genes were determined by aligning IGF-IR mRNAs with the corresponding genomic DNA using Spidey (http://www.ncbi.nlm.nih.gov/spidey/). The protein domain organization of IGF-IR genes were predicted using SMART v7.0 (http://smart.embl-heidelberg.de/) and transmembrane helix regions were predicted via the TMHMM Server v2.0 (http://www.cbs.dtu.dk/services/TMHMM/).
Quantitative gene expression analyses
The transcript levels of salmonid IGF-IR genes were determined using quantitative polymerase chain reaction (qPCR) under four different experimental contexts. In each case, the cDNA templates employed have been described elsewhere13,26,37,39. Firstly, to assess tissue-specific expression, we employed cDNA samples synthesised from total RNA extracted from eleven different tissues sampled before from juvenile Atlantic salmon (n = 4)13. Secondly, to establish regulation during early salmonid development, we employed cDNA samples synthesised from total RNA extracted from whole rainbow trout fry at three stages of development: hatching, first-feeding, and 3 weeks post-first feeding (i.e. H, FF and 3wFF; n = 5)26,37. Thirdly, to examine regulation during disease challenge, we employed cDNA samples synthesised from total RNA extracted from whole rainbow trout fry challenged by AS or VHSv at H, FF and 3wFF (n = 3 to 5)26,37. Finally, to determine expression in primary immune tissues during disease challenge, we employed cDNA samples synthesised from total RNA extracted from head kidney and spleen tissues of rainbow trout challenged by Y. ruckeri (n = 4)26,39. New qPCR primers pairs were designed for each salmonid IGF-IR gene in order to bind regions that distinguish duplicated IGF-IR genes retained from the tsWGD and ssWGD. In addition, primers were predicted to produce no self- or cross-dimers via NetPrimer (PREMIER Biosoft) and to be positioned in different exons. Details of all primers used in the study are provided in Table 4. Finally, qPCR, including technical execution, efficiency calculation, normalization and relative quantification, was performed as described in ref. 26, using an Mx3005P qPCR System with Brilliant III Ultra-Fast SYBR Green (Agilent Technologies), along with the software LinRegPCR50 (to calculate efficiency) and GenEx (MultiD Analyses AB) for data analysis. Regarding the normalization strategy, we tested the combined stability of five reference genes (namely: RpL4, RpS13, RpS29, ACTB and EF1A, see Table 4) using NormFinder51 within GenEx (done separately for each different experimental context). Based on the results, we employed the following combinations of reference genes for normalization: RpS29 and EF1A for the Atlantic salmon tissue-specific expression study; RpL4 and EF1A for expression analyses involving rainbow trout fry, including AS/VHSv challenges; RpL4 and RpS29 to assess immune tissue-specific expression responses to Y. ruckeri challenge.
A statement confirming that all animal experiments (from which samples used within this study were derived) were done in accordance with appropriate guidelines and regulations is provided elsewhere34.
Statistical analyses
All statistical analyses were performed in Minitab v.17 (Minitab Inc.). The effect of developmental stage on transcript levels of IGF-IR genes was established using one-way analysis of variance (ANOVA) with H, FF and 3wFF as fixed factors - Tukey’s post hoc test was applied to identify significant differences between the three developmental stages for each gene. The effect of AS and VHSv challenge on transcript levels of IGF-IR genes at the different stages of development was established using two-way ANOVA, with treatment, developmental stage and treatment*stage as model variables. One-way ANOVA was used to identify the effect of treatment at each stage of development for each gene, with PBS and AS/VHSv as fixed factors. The effect of YR challenge on transcript levels of genes coding IGF-IRs and IGF hormones in head kidney and spleen was tested using one-way ANOVA, with PBS and YR as fixed factors. When model residuals did not conform to the assumptions of normality (Anderson-Darling test) and/or homoscedasticity (Levene’s test) using raw data, we employed natural-log transformations. When data failed these assumptions after transformation, a nonparametric Kruskal-Wallis test was used. In addition, Spearman’s correlation was performed to evaluate co-expression of salmonid-specific gene duplicates (i.e. IGF-IRa1 and IGF-IRa2) under different experimental contexts.
Additional Information
How to cite this article: Alzaid, A. et al. The complete salmonid IGF-IR gene repertoire and its transcriptional response to disease. Sci. Rep. 6, 34806; doi: 10.1038/srep34806 (2016).
References
LeRoith, D., Werner, H., Beitner-Johnson, D. & Roberts, C. H. Jr. Molecular and cellular aspects of the insulin-like growth factor I receptor. Endocr. Rev. 16, 143–163 (1995).
Baserga, R., Hongo, A., Rubini, M., Prisco, M. & Valentinis, B. The IGF-I receptor in cell growth, transformation and apoptosis. Biochim. Biophys. Acta 1332, F105–F126 (1997).
Adams, T. E., Epa, V. C., Garrett, T. P. J. & Ward, C. W. Structure and function of the type 1 insulin-like growth factor receptor. Cell. Mol. Life Sci. 57, 1050–1093 (2000).
Liu, J. P., Baker, J., Perkins, A. S., Robertson, E. J. & Efstratiadis, A. Mice carrying null mutations of the genes encoding insulin-like growth factor I (Igf-1) and type 1 IGF receptor (Igf1r). Cell 75, 59–72 (1993).
Siddle, K. Signalling by insulin and IGF receptors: supporting acts and new players. J. Mol. Endocrinol. 47, R1–R10 (2011).
Siddle, K. Molecular basis of signaling specificity of insulin and IGF receptors: neglected corners and recent advances. Front. Endocrinol. 3, 1–24 (2012).
Johnston, I. A., Bower, N. I. & Macqueen, D. J. Growth and the regulation of myotomal muscle mass in teleost fish. J. Exp. Biol. 214, 1617–1628 (2011).
Jaillon, O. et al. Genome duplication in the teleost fish Tetraodon nigroviridis reveals the early vertebrate proto-karyotype. Nature 431, 946–957 (2004).
Braasch, I. & Postlethwait, J. H. Polyploidy in fish and the teleost genome duplication In Polyploidy genome evolution (eds Soltis, P. S. & Soltis, D. E. ) Ch. 17, 341–384 (Springer, 2012).
Lien, S. et al. The Atlantic salmon genome provides insights into rediploidization. Nature 533, 200–205 (2016).
Zou, S., Kamei, H., Modi, Z. & Duan, C. Zebrafish IGF genes: gene duplication, conservation and divergence, and novel roles in midline and notochord development. PLoS One 4, e7026 (2009).
Daza, D. O., Sundström, G., Bergqvist, C. A., Duan, C. & Larhammar, D. Evolution of the insulin-like growth factor binding protein (IGFBP) family. Endocrinology 152, 2278–2289 (2011).
Macqueen, D. J., Garcia De La Serrana, D. & Johnston, I. A. Evolution of ancient functions in the vertebrate insulin-like growth factor system uncovered by study of duplicated salmonid fish genomes. Mol. Biol. Evol. 30, 1060–1076 (2013).
Maures, T. et al. Structural, biochemical, and expression analysis of two distinct insulin-like growth factor I receptors and their ligands in zebrafish. Endocrinology 143, 1858–1871 (2002).
Nakao, N., Tanaka, M., Higashimoto, Y. & Nakashima, K. Molecular cloning, identification and characterization of four distinct receptor subtypes for insulin and IGF-I in Japanese flounder, Paralichthys olivaceus. J. Endocrinol. 173, 365–75 (2002).
Schlueter, P. et al. Gene duplication and functional divergence of the zebrafish insulin-like growth factor 1 receptors. FASEB J. 20, E462–E471 (2006).
Wallis, A. & Devlin, R. H. Duplicate insulin-like growth factor-I genes in salmon display alternative splicing pathways. Mol. Endocrinol. 7, 409–422 (1993).
Lappin, F. M., Shaw, R. L. & Macqueen, D. J. Targeted sequencing for high-resolution evolutionary analyses following genome duplication in salmonid fish: proof of concept for key components of the insulin-like growth factor axis. Mar. Genomics (in press).
Macqueen, D. J. & Johnston, I. A. A well-constrained estimate for the timing of the salmonid whole genome duplication reveals major decoupling from species diversification. Proc. Biol. Sci. 281, 20132881 (2014).
Chan, S. J., Plisetskaya, E. M., Urbinati, E., Jin, Y. & Steiner, D. F. Expression of multiple insulin and insulin-like growth factor receptor genes in salmon gill cartilage. Proc. Natl. Acad. Sci. USA 94, 12446–12451 (1997).
Greene, M. W. & Chen, T. T. Characterization of teleost insulin receptor family members. II. Developmental expression of insulin receptor messenger RNAs in rainbow trout. Gen. Comp. Endocrinol. 115, 270–281 (1999).
Gabillard, J. C. et al. Differential expression of the two GH genes during embryonic development of rainbow trout Oncorhynchus mykiss in relation with the IGFs system. Mol. Reprod. Dev. 64, 32–40 (2003).
Berthelot, C. et al. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat. Commun. 5, 3657 (2014).
Rondeau, E. B. et al. The genome and linkage map of the northern pike (Esox lucius): conserved synteny revealed between the salmonid sister group and the neoteleostei. PLoS One 9, e102089 (2014).
Pooley, N. J., Tacchi, L., Secombes, C. J. & Martin, S. A. M. Inflammatory responses in primary muscle cell cultures in Atlantic salmon (Salmo salar). BMC Genomics 14, 747 (2013).
Alzaid, A. et al. Cross-talk between growth and immunity: coupling of the IGF axis to conserved cytokine pathways in rainbow trout. Endocrinology 157, 1942–1955 (2016).
Wenger, M. et al. Developmental oestrogen exposure differentially modulates IGF-I and TNF-α expression levels in immune organs of Yersinia ruckeri-challenged young adult rainbow trout (Oncorhynchus mykiss). Gen. Comp. Endocrinol. 205, 168–175 (2014).
Franz, A. et al. Endocrine and Local IGF-I in the Bony Fish Immune System. Biology (Basel). 5, E9 (2016).
Solovyev, V. Statistical approaches in eukaryotic gene prediction In Handbook of statistical genetics 3ed edn Vol. 1 (eds Balding, D. J., Bishop, M. & Cannings, C. ) Ch. 4, 97–159 (Wiley, 2007).
Braasch, I. et al. The spotted gar genome illuminates vertebrate evolution and facilitates human-teleost comparisons. Nat. Genet. 48, 427–437 (2016).
Pashmforoush, M., Chan, S. J. & Steiner, D. F. Structure and expression of the insulin-like peptide receptor from amphioxus. Mol. Endocrinol. 10, 857–866 (1996).
Hernández-Sánchez, C., Mansilla, A., de Pablo, F. & Zardoya, R. Evolution of the insulin receptor family and receptor isoform expression in vertebrates. Mol. Biol. Evol. 25, 1043–1053 (2008).
Reinecke, M. & Collet, C. The Phylogeny of the Insulin-like Growth Factors. Int. Rev. Cytol. 183, 1–94 (1998).
Drummond, A. J., Ho, S. Y. W., Phillips, M. J. & Rambaut, A. Relaxed phylogenetics and dating with confidence. PLoS Biol. 4, e88 (2006).
Ohlsson, C. et al. The role of liver-derived insulin-like growth factor-I. Endocr. Rev. 30, 494–535 (2009).
Zhang, J., Shi, Z., Cheng, Q. & Chen, X. Expression of insulin-like growth factor I receptors at mRNA and protein levels during metamorphosis of Japanese flounder (Paralichthys olivaceus). Gen. Comp. Endocrinol. 173, 78–85 (2011).
Castro, R. et al. Disparate developmental patterns of immune responses to bacterial and viral infections in fish. Sci. Rep. 5, 15458 (2015).
Benbassat, C. A. et al. Interleukin-1α (IL-1α) and tumor necrosis factor α (TNFα) regulate insulin-like growth factor binding protein-1 (IGFBP-1) levels and mRNA abundance in vivo and in vitro. Horm. Metab. Res. 31, 209–215 (1999).
Harun, N. O., Wang, T. & Secombes, C. J. Gene expression profiling in naïve and vaccinated rainbow trout after Yersinia ruckeri infection: insights into the mechanisms of protection seen in vaccinated fish. Vaccine 29, 4388–4399 (2011).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Katoh, K. & Standley, D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780 (2013).
Penn, O. et al. GUIDANCE: a web server for assessing alignment confidence scores. Nucleic Acids Res. 38, W23–W28 (2010).
Penn, O., Privman, E., Landan, G., Graur, D. & Pupko, T. An alignment confidence score capturing robustness to guide tree uncertainty. Mol. Biol. Evol. 27, 1759–1767 (2010).
Tamura, K., Stecher, G., Peterson, D., Filipski, A. & Kumar, S. MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729 (2013).
Drummond, A. J., Suchard, M. A., Xie, D. & Rambaut, A. Bayesian phylogenetics with BEAUti and the BEAST 1.7. Mol. Biol. Evol. 29, 1969–1973 (2012).
Gernhard, T. The conditioned reconstructed process. J. Theor. Biol. 253, 769–778 (2008).
Stamatakis, A. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313 (2014).
Silvestro, D. & Michalak, I. raxmlGUI: a graphical front-end for RAxML. Org. Divers. Evol. 12, 335–337 (2012).
Stamatakis, A., Hoover, P. & Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771 (2008).
Ruijter, J. M. et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res. 37, e45 (2009).
Andersen, C. L., Jensen, J. L. & Orntoft, T. F. Normalization of real-time quantitative reverse transcription-PCR data: a model-based variance estimation approach to identify genes suited for normalization, applied to bladder and colon cancer data sets. Cancer Res 64, 5245–5250 (2004).
Macqueen, D. J., Kristjánsson, B. K. & Johnston, I. A. Salmonid genomes have a remarkably expanded akirin family, coexpressed with genes from conserved pathways governing skeletal muscle growth and catabolism. Physiol. Genomics 42, 134–148 (2010).
Bower, N. I., Li, X., Taylor, R. & Johnston, I. A. Switching to fast growth: the insulin-like growth factor (IGF) system in skeletal muscle of Atlantic salmon. J. Exp. Biol. 211, 3859–3870 (2008).
Tacchi, L. et al. Ubiquitin E3 ligase atrogin-1 (Fbox-32) in Atlantic salmon (Salmo salar): sequence analysis, genomic structure and modulation of expression. Comp. Biochem. Physiol. - B Biochem. Mol. Biol. 157, 364–373 (2010).
Acknowledgements
This work was supported financially by the European Community’s Seventh Framework Program (FP7/2007-13) under Grant 442 Agreements 222719 (LIFECYCLE). AA was supported by a PhD studentship from Kuwait University. The authors thank Mr Anthony K. Redmond (University of Aberdeen) for assisting with gene prediction and phylogenetic analysis.
Author information
Authors and Affiliations
Contributions
Designed research: D.J.M. and S.A.M.M. Performed phylogenetic and comparative genomic analyses, molecular biology and qPCR: A.A. analysed qPCR data: A.A. and D.J.M. Performed statistics: A.A. and D.J.M. Interpreted results and wrote the paper: all authors.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Electronic supplementary material
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Alzaid, A., Martin, S. & Macqueen, D. The complete salmonid IGF-IR gene repertoire and its transcriptional response to disease. Sci Rep 6, 34806 (2016). https://doi.org/10.1038/srep34806
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep34806
- Springer Nature Limited