
Abstract
KIF11 gene mutations cause a rare autosomal dominant inheritable disease called microcephaly with or without chorioretinopathy, lymphedema, or mental retardation (MCLMR). Recently, such mutations were also found to be associated with familial exudative vitreoretinopathy (FEVR). Here, we report 7 novel KIF11 mutations identified by targeted gene capture in a cohort of 142 probands with FEVR who were diagnosed in our clinic between March 2015 and November 2015. These mutations were: p.L171V, c.790-2A>C, p.Q525*, p.Q842*, p.S936*, p.L983fs and p.R1025G. Phenotypic analysis revealed that all of the affected probands had advanced FEVR (stage 4 or above). Three had microcephaly and one had chorioretinopathy, which indicated a phenotypic overlap with MCLMR. Two mutations were also found in the families of the affected probands. One parent with a p.R1025G mutation had an avascular peripheral retina and abnormal looping vessels. However, one parent with p.L983fs had normal retina, which indicated incomplete penetration of the genotype. Our results further confirmed that KIF11 is causative of FEVR in an autosomal dominant manner. We also suggest the examination of MCLMR-like features, such as microcephaly, chorioretinopathy, for patients with FEVR and wide-field fundus photography for patients with MCLMR in future practice.
Similar content being viewed by others

Introduction
KIF11 (NM_004523, gene id: 3832) encodes a mitotic kinesin also known as Eg5. Eg5 has long been recognized as an important member of the kinesin-like protein family and is involved in the development of malignant cancer and angiogenesis1. Consequently, Eg5 is regarded as one of the most promising new targets for antimitotic drugs2. This protein has three distinctive functional domains: a microtubule-binding motor region, a stalk region and a tail region. In 2012, mutations in KIF11 were found to be associated with the development of a rare autosomal dominant inheritable disease called microcephaly with or without chorioretinopathy, lymphedema, or mental retardation (MCLMR) (OMIM 152950). The disease was first described by Jarmas et al. in 19813 and subsequently by Crowe and Dickerman in 19864. As the name indicates, patients with this disease often display a variable spectrum of central nervous system and ocular developmental anomalies. Thus far, 45 pathological mutations of KIF11 have been associated with MCLMR5,6,7,8,9,10.
Familial exudative vitreoretinopathy (FEVR) is an inheritable disorder of retinal blood vessel development that leads to the incomplete vascularization of the retina and poor vascular differentiation11. This condition was first described by Criswick and Schepens in 196912. The clinical manifestations of this disease are complicated and variable. Mild forms of the disease can be asymptomatic and only exhibit peripheral retinal vascular abnormalities, such as a peripheral avascular zone, venous telangiectasias and altered arterial tortuosity. Severe forms of FEVR are associated with retinal neovascularization, subretinal and intraretinal hemorrhages, exudates, retinal folds and tractional retinal detachment13. Thus far, approximately 50% of the clinically identified patients with FEVR have been found to be associated with the following four genes in the Wnt signaling pathway: NDP, FZD4, LRP5, and TSPAN1214,15. The critical roles of the Wnt pathway in ocular development and retinal vascular development have also been demonstrated in various tissue-specific Wnt pathway gene knockout mice16. Additionally, mutations in ZNF408 have also been found in FEVR patients17.
The association between KIF11 mutations and FEVR was first reported in November 201418. This study identified 5 KIF11 mutations in 72 screened FEVR probands and concluded that the mutations were inherited in an autosomal dominant manner. This study was followed by another recently published study that identified 4 novel mutations in a cohort of 48 FEVR patients19. These results indicate that KIF11 may be another important gene that is involved in the development of FEVR.
In this study, we report 7 novel KIF11 mutations in FEVR patients which were identified through targeted gene capture and we analyze the clinical phenotypes associated with these mutations.
Results
Cohort description and mutation detection rate
Between March 2015 and November 2015 we identified 142 FEVR probands based on clinical presentation from among the patients who came to our clinic. There were 87 males and 55 females. The medium age was 34 months and the range was 1.5 months to 53 years old. None of the patients had a history of premature birth.
Targeted gene capture followed by next-generation sequencing (NGS) was performed on 142 probands. Initial sequencing was performed using a custom retinal disease capture panel that included the following FEVR-related genes: NDP, FZD4, LRP5, TSPAN12, ZNF408, and KIF11. Pathological mutations, as judged by various analytical approaches described in the Methods, were further validated by Sanger sequencing in each proband and his/her parents and siblings (if any). We identified 52 probands with pathogenic mutations in the NDP, FZD4, LRP5, TSPAN12 and ZNF408 genes (36.62%) and 7 probands with novel mutations in KIF11 (4.93%). For the detection of the KIF11 mutations, the average sequencing depth was 389.94. The average coverage of the target region was 99.47%. Moreover, the average coverage of the targeted exons for >10X reads was 96.14% and that for >20X reads was 91.72%.
New KIF11 mutations and the associated clinical presentations
Seven novel KIF mutations were detected in 7 patients. The nature of the mutations, the predicted pathogenicities and the clinical presentations of the probands are listed in Table 1. With the exception of one patient (Patient No. 3), all were diagnosed with FEVR for the first time in our clinic.
We identified the following five de novo mutations: c.511C>G (p.L171V), c.790-2A>C, c.1573C>T (p.Q525*), c.2524C>T (p.Q842*) and c.2807C>G (p.S936*). All of these mutations were heterozygous for the respective sites. The affected probands all exhibited clinically advanced FEVR.
The carrier of the c.511C>G (p.L171V) mutation was a 4-month-old male. He was referred to our clinic for the inability to follow moving objects. A fundus examination revealed stage 4 FEVR with chorioretinopathy in both eyes. Inferotemporal dragging of the optic disc and macula by the fibrovascular mass was observed in the right eye. A retinal detachment involving the macula was observed in the left eye (Fig. 1a). At the age of 1 year, his head circumference was 39 cm, which was 6 standard deviations (SDs) below the mean.

Chromatograms of five de novo mutations and the corresponding fundus pictures of the affected probands.
The sequence variations are marked with red circles. OD and OS represent right and left eyes, respectively. (a) chromatograms and fundus pictures of a 4-month-old male with the c.511C>G (p.L171V) mutation. (b) chromatograms and fundus pictures of a 1-year-old female with the splicing mutation c.790-2A>C. (c) chromatograms and fundus pictures of a 7-year-old female with c.1573C>T (p.Q525*) mutation. (d) chromatograms and fundus pictures of a 10-month-old female with a c.2524C>T (p.Q842*) mutation. (e) chromatograms and fundus pictures of a 1-year-old male with a c.2807C>G (p.S936*) mutation. Notice the existence of chorioretinopathy in panel a (both eyes); the falciform retinal folds in panels a (OS), b (OD), c (OD), d (OS) and e (both eyes); the retrolenticular fibrotic masses in panels c (OS), d (OS) and e (both eyes); cataracts and microphthalmia in panel d (OD); dragged disc and macular in panels a (OD) and b (OS).
The splicing mutation c.790-2A>C affected the splicing of exons 7–8. The carrier was a 1-year-old female who exhibited a typical falciform retinal fold with a retrolenticular fibrotic mass in the right eye and an inferotemporal fibrotic proliferation with a dragged optic disc in the left eye (Fig. 1b). At the age of 2 years, her head circumference was 44 cm, which was 2 SDs below the mean.
The carrier of the c.1573C>T (p.Q525*) mutation was a 7-year-old girl. She had undergone panretinal photocoagulation therapy in her right eye prior to attending our clinic and presented with a retinal fold. The left eye exhibited total retinal detachment and microphthalmia (Fig. 1c). Her head circumference was normal.
The carrier of the c.2524C>T (p.Q842*) mutation was a 10-month-old female. She presented with total retinal detachment, cataracts and a shallow anterior chamber in the right eye and advanced FEVR features in the left eye that included retinal folds and a retrolenticular fibrotic mass (Fig. 1d). Her head circumference was 43 cm when she was 18 months old, which was 2 SDs below the mean. Additionally, she was diagnosed with mild mental retardation.
The carrier of the c.2807C>G (p.S936*) was a 1-year-old male. He exhibited bilateral falciform retinal detachment with a retrolenticular fibrotic mass (Fig. 1e). His head circumference was normal.
The frameshift mutation c.2949delG (p.L983fs) was detected in a 2-year-old boy who was heterozygous for the mutation and inherited it from his father. The boy exhibited bilateral FEVR with retinal folds and a fibrotic mass that obstructed the retinal imaging of the left eye (Fig. 2). However, we did not observe any retinal abnormality via fluorescein angiography in the father who also carried the mutation. The head circumference of this boy was normal.

Pedigree, chromatograms and fundus pictures of the frameshift mutation c.2949delG (p.L983fs).
In the pedigree, M sign represents a variant; +, a normal allele. In the chromatograms, the variation is marked with red cycle. OD and OS represent right and left eyes, respectively. This mutation was detected in a 2-year-old boy and it was inherited from his father. Fundus examination showed bilateral FEVR with retinal folds. The microphthalmic left eye had cataracts and fibrotic mass which obstructed retinal imaging.
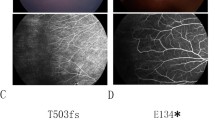
Another missense mutation, i.e., c.3073A>G (p.R1025G), was detected in a 1-year-old boy. This mutation was also detected in his father. The boy exhibited peripheral fibrovascular proliferation with exudates in the right eye and microphthalmia with cataracts in the left eye. His head circumference was normal. Fundus fluorescein angiography of his father revealed an avascular peripheral retina and vascular looping in the right eye (Fig. 3).

Pedigree, chromatograms and fundus pictures of the missense mutation c.3073A>G (p.R1025G).
In the pedigree, M sign represents a variant; +, a normal allele. In the chromatograms, the variation is marked with red cycle. OD and OS represent right and left eyes, respectively. This mutation was detected in a 1-year-old boy and it was inherited from his father. Fundus examination showed a peripheral fibrovascular proliferation with exudates in the right eye and microphthalmia with cataracts in the left eye. Fundus fluorescein angiography of his father (bottom) revealed an avascular peripheral retina (asterisk) and vascular looping in the right eye (arrows).
Discussion
Despite the extensive research on the protein product of KIF11, mutations of this gene and their associations with hereditary diseases were only recently discovered. A literature search identified 6 studies that reported approximately 45 mutations involved in MCLMR5,6,7,8,9,10 and 2 studies that reported 9 KIF11 mutations associated with FEVR18,19. One mutation was found in both MCLMR and FEVR patients. These two studies on FEVR patients reported the KIF11 mutation detection rates of 6.94% (5 out of 72 screened) and 8.33% (4 out of 48 screened), respectively18,19. Here, we reported the identification of 7 additional novel heterozygous mutations in KIF11 in 142 screened FEVR probands, which represents a mutation detection rate of 4.93% among all of the FEVR patients. The present study is by far the largest cohort study that has screened for KIF11 mutations in FEVR patients. The distribution of KIF11 mutations found thus far in the FEVR patients is presented in Fig. 4. A similar summary of the mutations found in MCLMR patients is also available10. In both diseases, approximately 40% of the mutations are located in the motor region, 20% in the stalk region and 40% in the tail region.

Unlike in MCLMR, the hallmarks of which are both central nervous system and ocular developmental defects, the primary defect of FEVR is limited to abnormal retinal vasculature. The shared clinical features of advanced FEVR and MCLMR include retinal folds, retinal detachment, cataracts and microphthalmia. Chorioretinopathy and optic nerve atrophy are not typical presentations of FEVR20. In the seven FEVR probands who carried KIF11 mutations, we observed a phenotypic overlap between these two diseases. We identified 3 probands with microcephaly, 1 with chorioretinopathy, 1 with mild mental retardation and 3 with microphthalmia (Table 1). A similar phenotypic overlap has also been reported previously by two groups18,19. At the protein level, these findings are consistent with the function of the KIF11 protein. In vivo studies in zebra fish and chicken embryos have revealed that the KIF11 protein is predominantly expressed in lymphoblasts and endothelial cells. Defective KIF11 protein causes developmental and vascular defects21. An interesting question to pursue is why some KIF11 mutations cause broader defects that manifest as MCLMR while others cause relatively focused diseases such as FEVR.
Incomplete penetrance and variable expressivity of the KIF11 mutations were also observed in the affected FEVR patients. Among the two inherited heterozygous mutations (p.L983fs and p.R1025G) found in this study, one parent (with a p.R1025G mutation) exhibited a mild clinical phenotype, whereas the other parent (with a p.L983fs mutation) appeared to be normal. Variable expressivity of the KIF11 mutations was demonstrated by c.790-2A>C. A similar mutation (c.790-1G>T), which also affects the splicing of exons 7–8, has been previously reported by two separate groups. This mutation caused FEVR in one patient and MCLMR in the other patient8,18. Both mutations were de novo. All three of the probands had microcephaly. However, the ocular abnormalities in the MCLMR patient were different than those in the two FEVR patients. Between the two FEVR patients, it appeared that the patient in our study had a milder presentation than the one reported by Robitaille et al.18. However, because our patient was younger and it was her first diagnosis of the disease, it is possible that the condition may progress to greater severity.
Overall, we noticed that the 7 probands carrying the KIF11 mutations identified in this study all had advanced FEVR; three had stage 4 and four had stage 5 FEVR. This association between severe FEVR phenotypes and KIF11 mutations has also previously been observed by another group18. This association also seems to differ from the clinical presentations associated with NDP, FZD4, LRP5 and TSPAN12 gene mutations, which display broader phenotypic spectra and primarily vary from stage 2 to stage 5. However, additional studies are needed to conclude whether mutations of KIF11 have more profound pathogenic effects in FEVR.
In conclusion, we identified 7 novel KIF11 mutations during the screening of 142 FEVR patients. FEVR due to KIF11 mutations represents a mechanism that leads to incomplete development of the retinal vasculature that differs from the well-known mechanisms related to the Wnt signaling pathway. Combined with the findings from other groups, the existing evidence suggests that KIF11 mutations in FEVR patients are often associated with severe clinical manifestations, although incomplete phenotype penetration and variation exist. We suggest that FEVR patients in the clinic undergo examinations for typical MCLMR features, such as microcephaly, chorioretinopathy, lymphedema and mental retardation and that MCLMR patients undergo wide-field fundus photography. We also suggest the inclusion of the KIF11 gene in future genetic screens for FEVR.
Methods
Study subjects and clinical data collection
This study was approved by the Institutional Review Board of Xin Hua Hospital affiliated to the Shanghai Jiao Tong University School of Medicine. All work was performed in accordance with the approved study protocol. Informed written consent was obtained from the parents or guardians of each participant because they were all under-aged children.
The FEVR probands were identified from among the patients who attended our clinic between March 2015 and November 2015 based on the presence of at least one of the following retinal vascular developmental anomalies as previously described22: a lack of peripheral retinal vascular development with or without variable degrees of nonperfusion, vitreoretinal traction, subretinal exudation, retinal neovascularization occurring at any age or total retinal detachment with a fibrotic mass behind the lens. All participants were born full-term. The disease severities of all probands were further classified according to the staging system described by Ranchod et al.20. Each proband was subjected to the following ocular examinations: wide-field fundus photography using either a RetCam (Clarity Medical Systems, Pleasanton, CA, USA) or an Optos 200Tx (Optos, Inc., Marlborough, MA, USA) and indirect ophthalmoscopy with a 28D lens and scleral depression when needed. Additionally, their direct family members, primarily the parents, were subjected to fundus fluorescein angiography using Spectralis HRA2 (Heidelberg Engineering GmbH, Germany).
Targeted gene capture and next-generation sequencing (NGS)
Targeted gene capture and sequencing were performed by MyGenostics (MyGenostics, MD, USA). Briefly, peripheral blood was drawn from each proband and their direct family members and the genomic DNA was extracted and fragmented. Illumina adapters were added to the fragments and the samples were size-selected for the 350–400 bp products. This pool of DNA fragments was amplified using PCR and allowed to hybridize with DNA capture probes that were specifically designed for the targeted genes. The captured DNA fragments were eluted, amplified again and subjected to NGS using an Illumina HiSeq 2000 (Illumina, Inc., San Diego, CA, USA). A custom Genetic Pediatric Retinal Diseases Panel based on targeted exome capture technology was used and covered the following twenty-one genes: ABCB6, GDF6, LRP5, RS1, SOX2, TENM3, VSX2, FZD4, IKBKG, NDP, SALL2, STRA6, TSPAN12, YAP1, GDF3, KIF11, PAX6, SHH, TBX1, TUBA8, and ZNF408.
Data analysis
The sequenced reads were mapped to the UCSC hg19 (http://genome.ucsc.edu) human reference genome using the Burrows Wheeler Aligner (BWA) (http://bio-bwa.sourceforge.net/bwa.shtml). Variants were detected with GATK and further annotated using the 1000 Genomes database, ESP6500, dbSNP and the company’s own in-house database of 800 samples. The pathogenicity of each variant was assessed with the following databases: PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/), Sorting Intolerant From Tolerant (http://sift.jcvi.org/www/SIFT_enst_submit.html), Mutation Taster (http://www.mutationtaster.org/) and GERP++ (http://mendel.stanford.edu/SidowLab/downloads/gerp/index.html). For the autosomal dominant mutations, a reported minor allele frequency of more than 0.01 according to the Exome Aggregation Consortium (http://exac.broadinstitute.org/gene/ENSG00000138160) or benign variants predicted by both PolyPhen-2 and SIFT were excluded. The remaining variants were considered as pathogenic candidates and were further searched in the HGMD database to determine whether they had been reported previously as pathogenic.
PCR and Sanger sequencing validation
Primer3 was used to design all of the PCR primers for the Sanger sequencing that was conducted to validate the potential pathogenic variants. The average amplicon size was 400 bp. The DNA was sequenced on the ABI 3130XL platform and subsequently analyzed using Mutation Surveyor.
Additional Information
How to cite this article: Li, J.-K. et al. Identification of novel KIF11 mutations in patients with familial exudative vitreoretinopathy and a phenotypic analysis. Sci. Rep. 6, 26564; doi: 10.1038/srep26564 (2016).
References
El-Nassan, H. B. Advances in the discovery of kinesin spindle protein (Eg5) inhibitors as antitumor agents. Eur. J. Med. Chem. 62, 614–631 (2013).
Pérez-Melero, C. KSP inhibitors as antimitotic agents. Curr. Top. Med. Chem. 14, 2286–2311 (2014).
Jarmas, A. L., Weaver, D. D., Ellis, F. D. & Davis, A. Microcephaly, microphthalmia, falciform retinal folds and blindness. A new syndrome. Am. J. Dis. Child. 1960 135, 930–933 (1981).
Crowe, C. A. & Dickerman, L. H. A genetic association between microcephaly and lymphedema. Am. J. Med. Genet. 24, 131–135 (1986).
Ostergaard, P. et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am. J. Hum. Genet. 90, 356–362 (2012).
Hazan, F. et al. A novel KIF11 mutation in a Turkish patient with microcephaly, lymphedema and chorioretinal dysplasia from a consanguineous family. Am. J. Med. Genet. A. 158A, 1686–1689 (2012).
Jones, G. E. et al. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur. J. Hum. Genet. 22, 881–887 (2014).
Mirzaa, G. M. et al. Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: five novel mutations and review of the literature. Am. J. Med. Genet. A. 164A, 2879–2886 (2014).
Mears, K., Bakall, B., Harney, L. A., Penticoff, J. A. & Stone, E. M. Autosomal dominant microcephaly associated with congenital lymphedema and chorioretinopathy due to a novel mutation in KIF11. JAMA Ophthalmol. 133, 720–721 (2015).
Schlögel, M. J. et al. No evidence of locus heterogeneity in familial microcephaly with or without chorioretinopathy, lymphedema, or mental retardation syndrome. Orphanet J. Rare Dis. 10, 25, 10.1186/s13023-015-0271-4 (2015).
Shuler, M. F., Sullivan, J. M., Hurley, B. R. & McNamara, J. A. In Pediatric Retina (eds Reynolds, J. & Olitsky, S. ) 315–344 (Springer, Berlin Heidelberg, 2011).
Criswick, V. G. & Schepens, C. L. Familial exudative vitreoretinopathy. Am. J. Ophthalmol. 68, 578–594 (1969).
Gilmour, D. F. Familial exudative vitreoretinopathy and related retinopathies. Eye Lond. Engl. 29, 1–14 (2015).
Salvo, J. et al. Next-generation sequencing and novel variant determination in a cohort of 92 familial exudative vitreoretinopathy patients. Invest. Ophthalmol. Vis. Sci. 56, 1937–1946 (2015).
Seo, S. H. et al. Molecular characterization of FZD4, LRP5 and TSPAN12 in familial exudative vitreoretinopathy. Invest. Ophthalmol. Vis. Sci. 56, 5143–5151 (2015).
Ohlmann, A. & Tamm, E. R. Norrin: molecular and functional properties of an angiogenic and neuroprotective growth factor. Prog. Retin. Eye Res. 31, 243–257 (2012).
Collin, R. W. J. et al. ZNF408 is mutated in familial exudative vitreoretinopathy and is crucial for the development of zebrafish retinal vasculature. Proc. Natl. Acad. Sci. USA 110, 9856–9861 (2013).
Robitaille, J. M. et al. Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol. 132, 1393–1399 (2014).
Hu, H. et al. KIF11 mutations are a common cause of autosomal dominant familial exudative vitreoretinopathy. Br. J. Ophthalmol. 100, 278–283 (2016).
Ranchod, T. M., Ho, L. Y., Drenser, K. A., Capone, A. & Trese, M. T. Clinical presentation of familial exudative vitreoretinopathy. Ophthalmology 118, 2070–2075 (2011).
Exertier, P. et al. Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5. Oncotarget 4, 2302–2316 (2013).
Kashani, A. H. et al. Diversity of retinal vascular anomalies in patients with familial exudative vitreoretinopathy. Ophthalmology 121, 2220–2227 (2014).
Acknowledgements
We would like to thank Dr. Hui Wang for the critical discussion and review of the manuscript. The study was supported by grants from the National Natural Science Foundation of China (81371063 to J.L., 81271045 and 81470642 to P.Z.), the Science and Technology Commission of Shanghai Municipality, China (15XD1502800 to P.Z.) and the National Key Basic Research Program of China (2012CB966901 to J.L.).
Author information
Authors and Affiliations
Contributions
Study conception and design: P.Z. Diagnosis of FEVR: P.Z. Data acquisition: J.-K.L., P.F., Q.-J.H., Y.-A.L., Q.Z., X.Z. and Y.-Q.R. Data analysis and interpretation: J.-K.L. and J.L. Manuscript preparation: J.L. and J.-K.L.
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Li, JK., Fei, P., Li, Y. et al. Identification of novel KIF11 mutations in patients with familial exudative vitreoretinopathy and a phenotypic analysis. Sci Rep 6, 26564 (2016). https://doi.org/10.1038/srep26564
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/srep26564
- Springer Nature Limited
This article is cited by
-
Kinesin-5 Eg5 is essential for spindle assembly, chromosome stability and organogenesis in development
Cell Death Discovery (2022)
-
An interphase pool of KIF11 localizes at the basal bodies of primary cilia and a reduction in KIF11 expression alters cilia dynamics
Scientific Reports (2020)
-
Integrin-linked kinase controls retinal angiogenesis and is linked to Wnt signaling and exudative vitreoretinopathy
Nature Communications (2019)
-
Rab18 Collaborates with Rab7 to Modulate Lysosomal and Autophagy Activities in the Nervous System: an Overlapping Mechanism for Warburg Micro Syndrome and Charcot-Marie-Tooth Neuropathy Type 2B
Molecular Neurobiology (2019)
-
Detection and quantification of a KIF11 mosaicism in a subject presenting familial exudative vitreoretinopathy with microcephaly
European Journal of Human Genetics (2018)