
Abstract
Background
Microcephaly with or without chorioretinopathy, lymphedema, or mental retardation syndrome (MCLMR) is a rare autosomal dominant disorder with variable expressivity. It is characterized by mild-to-severe microcephaly, often associated with intellectual disability, ocular defects and lymphedema. It can be sporadic or inherited. Eighty-seven patients have been described to carry a mutation in KIF11, which encodes a homotetrameric motor kinesin, EG5.
Methods
We tested 23 unreported MCLMR index patients for KIF11. We also reviewed the clinical phenotypes of all our patients as well as of those described in previously published studies.
Results
We identified 14 mutations, 12 of which are novel. We detected mutations in 12 affected individuals, from 6 out of 6 familial cases, and in 8 out of 17 sporadic patients. Phenotypic evaluation of patients (our 26 + 61 earlier published = 87) revealed microcephaly in 91%, eye anomalies in 72%, intellectual disability in 67% and lymphedema in 47% of the patients. Unaffected carriers were rare (4 out of 87: 5%). Family history is not a requisite for diagnosis; 31% (16 out of 52) were de novo cases.
Conclusions
All inherited cases, and 50% of sporadic cases of MCLMR are due to germline KIF11 mutations. It is possible that mosaic KIF11 mutations cause the remainder of sporadic cases, which the methods employed here were not designed to detect. On the other hand, some of them might have another mimicking disorder and genetic defect, as microcephaly is highly heterogeneous. In aggregate, KIF11 mutations likely cause the majority, if not all, of MCLMR.
Similar content being viewed by others


Background
Microcephaly with or without chorioretinopathy, lymphedema, or mental retardation syndrome (MCLMR, OMIM 152950, ORPHA2526) can occur as a sporadic or inherited (autosomal dominant) disease [1]. MCLMR is characterized by a broad nose, upslanting palpebral fissures, a rounded nasal tip, a long philtrum, a pointed chin, a thin upper lip, prominent ears, atrial septal defect and lymphedema of the dorsum of the feet (pedal œdema) (Figure 1) [2,3]. Ocular anomalies, the most common of which is chorioretinal dysplasia, require long-term ophthalmic follow-up due to slow development. Cerebral magnetic resonance imaging may reveal a smaller, but usually structurally normal brain [4,5].

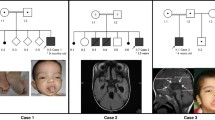
Clinical characteristics of patients. Clinical features of MCMLR (patients I-10 (A), IX-10 (B), X-10 (C) and XIV-10 (D). Note, broad nose (A, B), long philtrum (A, B), thin upper lip (A, D), prominent ears (C, D) and bilateral pedal lymphedema (A, B D).
Historically, MCLMR syndrome was divided into two distinct entities: microcephaly, lymphedema and chorioretinal dysplasia syndrome (MLCRD), and chorioretinal dysplasia, microcephaly and mental retardation syndrome (CDMMR). The discovery of KIF11 mutations in patients with MLCRD or CDMMR demonstrated that the phenotypes are part of a single entity with variable penetrance and expressivity [6]. KIF11 mutations were also discovered in 5 families with familial exudative vitreoretinopathy syndrome (FEVR), which thus belongs to the phenotypic spectrum caused by KIF11 mutations [7]. Thirty-one different KIF11 mutations have been described [6-10]. They include missense, nonsense, frameshift and splice-site mutations distributed across the gene, and are thought to cause loss-of-function. No genotype-phenotype correlation has been described. All of the mutations are autosomal-dominant or de novo; no autosomal recessive case with a KIF11 mutation has been described.
KIF11 is located on 10q23.33 (http://www.ensembl.org) and encodes the kinesin family member 11 (also known as EG5, KNSL1 or TRIP5). EG5 is a 1056-amino-acid protein, which forms homotetrameric kinesin motor complexes. The N-terminal part composes the motor domain (Gly16 – Phe363), containing a microtubule-binding domain that allows sliding along microtubules, an ATP-binding site with ATPase activity (between residues Gly105 and Thr112), and three internal domains that form coiled-coil structures responsible for multimerization into homotetramers (Figure 2). The C-terminal BimC domain also plays a role in multimerization [11].

Schematic representation of EG5 (1056 amino acids) with position of mutations causing MCLMR syndrome. Functional domains (colored), in vitro mutagenized residues (signs) and post-translationally modified amino acids (signs). Mutations found in this study, top; those published earlier, below (Ostergaard et al. 2012, Hazan et al. 2012, Jones et al. 2013, Mirzaa et al. 2014 and Robitaille et al. 2014). Splice-site alterations (¶: family II and family V) shown to result in r.211_308del; p.Thr71Argfs*8 and r.789_790insAG; p.Val264Argfs*26, respectively (Figure 4). N.B. positions based on amino acids in Mirzaa et al.
The aim of this study was to screen KIF11 in a large cohort of patients with signs of MCLMR in order to identify novel mutations, assess for locus heterogeneity and genotype-phenotype correlations, and better define the clinical phenotype associated with KIF11 mutations. Both sporadic and familial index patients were included, as well as additional family members when available.
Methods
Patient recruitment
DNA extracted from blood-samples of patients was collected from different countries (Argentina, Australia, Belgium, Israel, United-Kingdom, Spain and Sweden). Informed consent was obtained for all participants, as approved by the ethical committee of the Medical Faculty at the Université catholique de Louvain, Brussels, Belgium and the respective local committees. DNA was extracted using Wizard genomic DNA purification kit (Promega). Patients V-100, XI-100 and XIII-10 have previously been described in case reports [12-14]. Data on certain phenotypic characteristics, including level of intellectual disability, were not standardized; evaluation by the referring clinician was used. The average age of our patients at the time of conducting the study was 21 years.
Sanger sequencing of KIF11
To screen for KIF11 mutations, the 22 exons and their corresponding splice-sites were amplified, as described [6]. Purity and size of amplicons were evaluated by agarose gel electrophoresis using GeneRuler™ 100 bp (Thermo Scientific) DNA size ladder. Direct sequencing was performed on a 3130xl Genetic Analyzer (Life Technologies). All sequences were analyzed with CLC Main Workbench 6© (CLC Bio), using reference sequence NG_032580.
Mutation analysis
Mutations were named according to the guidelines of the Human Genome Variation Society (http://www.hgvs.org/mutnomen/), verified using Mutalyzer (https://mutalyzer.nl/) [15]. The mutations have been submitted to the Vascular Anomaly and Lymphedema Mutation Database (www.icp.ucl.ac.be/vikkula/VAdb). The dbSNP137 (http://www.ncbi.nlm.nih.gov/projects/SNP/) [16], 1000Genomes (http://browser.1000genomes.org), ESP6500 (http://evs.gs.washington.edu/EVS/), and GoNL databases (http://www.nlgenome.nl) of genomic variants were searched for each mutation identified, in order to eliminate any polymorphisms. The effects of amino acid changes on protein function were predicted using PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/) [17] and SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html) [18]. MutationTaster (http://www.mutationtaster.org/) [19] was used to detect potential splice-site changes. When DNA from additional family members was available, co-segregation of mutation with disease was assessed. If the mutation was not identified in either parent, the Powerplex HS16 genotyping kit (Promega) on a 3130xl Genetic Analyzer (Life Technologies) was used to confirm paternity and maternity.
Evaluation of splicing abnormalities associated with intronic mutations
RNA was extracted from EBV-transformed lymphoblasts or lymphocytes with TriPure (Roche) (patients II-10 and V-12 and 2 controls) and retro-transcribed using RevertAid H Minus First Strand cDNA Synthesis Kit (Fermentas) with random hexamers. For patient II-10 and a control, PCR-amplification was performed using primers 5’-GAGGATTGGCTGACAAGAGC and 5’-CGTGGAATTATACCAGCCAAG. After agarose gel electrophoresis, bands were extracted using QIAquick® Gel Extraction Kit (QIAGEN) and purified amplicons were sequenced and compared to the reference sequence NM_004523.3. For patient V-12 and controls, amplicons were generated with 5’-GGTTTAGAAGAAATTACAGTACAC and 5’-TGACCCTTCCCAAAGTCAAC. They were cloned into the pCR-II TOPO vector (Invitrogen) according to the manufacturer’s protocol and the plasmids were transfected into JM109 competent bacteria (Promega, Madison, WI, USA) by heat shock. For each cDNA (patient and control), 16 clones were picked and inserts were PCR-amplified and sequenced using the same primers (above).
Results
Sequencing of the 22 coding exons of KIF11 and their flanking splice-sites identified fourteen heterozygous mutations in the 23 index patients (Table 1 and Figure 2). All patients with family history (n = 6) had a mutation. These germline variants included two nucleotide substitutions and one single nucleotide deletion; all result in premature stop codons. Three splice-site alterations were also identified. The changes co-segregated with phenotype in the five families for which we had additional samples (n = 20) (Table 1 and Figure 3).

Pedigrees and phenotypes of screened patients. Upper panel, KIF11 mutation-positive; lower panel, KIF11 mutation-negative. Individuals with a bar, clinically examined; those with a number, sequenced. Patients with a star have a mutation.
Among the 17 sporadic patients, we found eight with a mutation in KIF11: one nonsense, four frameshifts and two canonical consensus splice-site changes (Table 1) that we consider pathogenic; and one missense alteration that we regarded as probably pathogenic, but requiring further functional validation (Table 1). DNA from parents was available for 7 of the 8 sporadic patients with a KIF11 mutation. All appeared to be de novo changes (100%) (Figure 3) absent in both parents, representing 41% (n = 7/17) of all our sporadic cases. In total, 30% of all the index cases studied (7/23) had de novo KIF11 mutations.
The mutations detected most likely result in loss of function of KIF11 due to nonsense-mediated mRNA decay or premature truncation of the protein (n = 13). Lymphoblast RNA from a patient with a splice-site alteration (Patient II-10: c.308+1G>T) showed skipping of exon 3 (r.211_308del) (Figure 4A), leading to a change in reading frame (p.Thr71Argfs*8) (Figure 4B). The mutant transcript was expressed at a lower level than the wild-type allele, suggesting mRNA loss by nonsense-mediated mRNA decay. In patient V-12, the splice-variant (c.790-3A>G) generates a new AG-acceptor site-like sequence. In lymphocytes, this led to the insertion of 2 nucleotides from the intronic splice site (r.789_790insAG) into the mRNA (Figure 4C and D), causing a reading frame shift and appearance of a premature stop codon 26 amino acids after (p.Val264Argfs*26). We identified only 1 mutation leading to a heterozygous amino acid substitution: Tyr82Phe (Patient I-10). The substitution occurs at a highly conserved amino acid position in the kinesin motor domain from fish to human being. It is predicted to alter protein function by PolyPhen-2, SIFT and Mutation Taster 2.0. The change is absent in the 1000Genomes, ESP6500, and GoNL databases.

Effect of splice site mutations. (A) RT-PCR using primers in exon 2 and 5 on cDNA from patient II-10 (c.308+1G>T) and two controls (c1 and c2). Wild-type amplicon, 250 bp; mutant, 152 bp. (B) (Sequencing of lower band unraveled skipping of exon 3 resulting in a premature stop-codon (r.211_308del: p.Thr71Argfs*8). (C) Sequencing of cDNA of patient V-12 (c.790-3A>G) for an amplicon covering exon 7/8 splice site revealed double-sequence. (D) Sequencing of cloned amplicon fragments revealed insertion of 2 nucleotides (AG) from the intronic acceptor site (r.790_791insAG) in mutant clones. This leads to a premature stop-codon (p.Val264Argfs*26).
Taking into account all the patients found to carry a mutation in this study (n = 26), as well as the 61 already reported [6-10], microcephaly was present in 79/87 patients (91%), eye anomalies in 63/87 patients (72%), intellectual disability in 58/87 (67%) and lymphedema in 41/87 patients (47%). Average SD for microcephaly was −5,1 (ranging from −1,1 to −9,5), on the basis of 64 patients with published data. None of the clinical signs was fully penetrant. Atypical features including cerebral and cardiac anomalies were present in 48 patients (55%). Taking all index patients with mutations into account (n = 52), sporadic cases represent 42% (22/52). It has been proven in 73% (16/22) of them that the mutation appeared de novo.
The nine KIF11 mutation-negative sporadic patients were included in KIF11 mutation screening due to the presence of signs and symptoms corresponding to MCLMR. However, certain patients showed additional features, not described for MCLMR: hydrops fetalis and lymphangiectasia (1 patient: XX-10), motor delay (2 patients: XVIII-10, XXIII-10), and Madelung deformity (1 patient: XIX-10). These patients probably reflect the clinical and genetic variability of microcephaly syndromes.
Discussion
We discovered 14 KIF11 mutations, 12 of which are novel, in 26 MCLMR patients (Figure 2). A mutation was detected in all patients with a family history of MCLMR, similar to previously reported series [6-10]. We also discovered a heterozygous germline de novo mutation in 7 sporadic patients and in 1 patient without family history of MCLMR, but for whom no parental DNA was available for testing. Thus, 87,5% of our sporadic patients with a KIF11 mutation had a proven de novo mutation. This represents 41% (7/17) of sporadic index patients in this study. Taking into account all index patients with a KIF11 mutation, sixteen sporadic patients have been reported to have a de novo mutation (7 in the current study, and 9 in previously published works), indicating that KIF11 has a high de novo mutation rate: 16 de novo/52 mutation-positive index patients; i.e. 31% across studies. Several de novo mutations have also been identified in other genes mutated in microcephaly (i.e. CTNNB1 [20] and TUBA1A [21]) and in primary lymphedema, such as SOX18, FOXC2 and VEGFR3 [22-24].
No KIF11 mutation-negative familial cases were identified; however, about half of the sporadic cases were KIF11 mutation-negative. These may be explained by several hypotheses: (1) Deletions or deep-intronic mutations in KIF11, which were not assessed in the study; (2) Somatic/mosaic KIF11 mutations that are not detectable in the blood by Sanger sequencing: mosaicism as a cause of MCLMR can be investigated by sequencing tissue and blood-DNA using targeted deep sequencing; (3) Existence of a mimicking disorder: many different genes can cause microcephaly, including those implicated in autosomal recessive primary microcephaly and syndromic microcephaly [25], or primary lymphedema [26]. The presence of motor delay in patients XVIII-10 and XIII-10 may suggest Cohen syndrome (OMIM 216550) [27]. The Madelung deformity and hydrops fetalis observed in two other patients may also be indicative of a distinct syndrome, as they have never been described in MCLMR. Eight out of nine of our negative cases had microcephaly and lymphedema; (4) Novel genetic cause for MCLMR in mutation-negative patients. Overall, our results suggest that KIF11 is the major causative gene for MCLMR, with no evidence of locus heterogeneity in familial cases.
The most frequent clinical signs and symptoms in patients with a KIF11 mutation are microcephaly (91%), eye anomalies (72%) and intellectual disability (67%) (Figure 3 and Table 1). MCLMR patients may have a structurally abnormal brain i.e. pachygyria (Table 1: Patients VII-10 & XI-100) [9]. Lymphedema is present in 47% of mutation-positive patients (Table 1).
Although none of the individual symptoms is fully penetrant, the overall penetrance of MCLMR is high (83/87; 95%). Only 4 asymptomatic mutation carriers have been identified amongst 87 individuals tested. Patients in the same family (Family II, IV, V, VI, and XI), with the same mutation may however have different phenotypes, demonstrating variable expressivity.
The 44 mutations identified so far in KIF11 (nine missense, twenty-five premature stop codons and ten splice-site changes) are scattered across the length of gene (Figure 2). There are no clear phenotypic differences between missense mutation carriers and those with premature stop codon mutations. As the most 5’ premature truncation codon occurs at the very 5’ end (amino acid 47), and lymphoblasts had a significantly lower expression of the mutant allele of patient II-10 (c.308+1G>T), all mutations most likely cause loss of EG5 function.
KIF11 mutations have pleiotropic effects with variable penetrance. One possible explanation could be the occurrence of somatic second-hit mutation(s), as we have proven for glomuvenous malformation [28,29] and mucocutaneous venous malformation [30]. Random somatic changes inevitably occur in the cells of all individuals, and result in a double-hit situation in some cells in patients with a germline mutation. The resulting phenotypes would depend on the types and developmental stages of cells affected.
There are nine amino acid substitutions identified in MCLMR patients. One of them affects the initiating methionine at position 1 (p.Met1Thr) most likely causing non-translation of KIF11. Seven of the remaining eight amino acid substitutions occur in the kinesin motor domain (p.Tyr82Phe, p.Phe144Leu, p.Arg221Gly, p.Arg234Cys, p.Ser235Cys, p.His244Tyr, and p.Glu344Lys) and the last one in the BimC Box (p.Arg944Cys), implicated in multimerization (Figure 2). Both domains have been shown to be important for KIF11 function during mitosis [11]. The kinesin motor domain contains amino acids important for tertiary structure. Arg234 makes a salt bridge with Glu270 to close the active site essential for ATP hydrolysis [31]. Substitution of Arg234 for a cysteine likely perturbs closure of the pocket and ATP hydrolysis. The impact of Ser235Cys is likely similar due to its vicinity to Arg234. In addition, the serine-to-cysteine change would affect polarity and thus modify the stabilization of a molecule of water in the nucleotide-binding pocket. Moreover, in the 3D structure (PDB ID: 3hqd), Arg221 is located in front of Ser235. Substitution for glycine could influence the structure of the nucleotide-binding pocket. Tyr82, Phe144 and Glu344 are located in an alpha-helix and His244 in a central beta-sheet in the kinesin domain. It is unclear whether these mutations, and the BimC Box mutation, affect stability, structure and/or function.
In vitro mutagenesis (Pro131Ala, Thr926Ala and Ser1033Ala) showed that the N-tail and the BimC domain are important for intra-cellular localization of the protein [32]. Thr926 of the BimC domain is phosphorylated by CDK1 (Cyclin-dependent kinase 1), whereas Ser1033 is partially phosphorylated by NEK6 (Never in mitosis gene a -related kinase 6). Phosphorylation of these sites is important for the mitotic function of EG5 [33,34]. Depletion of KIF11 causes mitotic arrest. This can be rescued by re-expression of the protein, but not by a Thr926Ala mutant, which lacks the ability to interact with microtubules [33]. The Arg944Cys mutation likely causes dysfunction of this same domain.
EG5 is a plus end-directed microtubule motor. It contributes to the establishment and maintenance of bipolar spindles during mitosis and meiosis that push the poles apart [35]. This is required for the separation of duplicated centrosomes. EG5 is considered as a slow kinesin, which slows down the spindle separation during mitosis. It is dispersed in the cytoplasm during the interphase, regroups at the spindle poles during prophase and stays alongside the spindle in metaphase. In mice, Eg5 is expressed in proliferative tissues. Disruption by homozygous deletion of Kif11 in mice resulted in embryonic lethality due to defective implantation in the uterine wall [36]. Microcephaly, ocular anomalies and œdema were not reported in heterozygous embryos. Morpholino-based knock-down of Kif11 also led to embryonic lethality, or severe œdema and circulation defects [37]. Inhibition of EG5 by dimethylenastron (DMN) or ispinesib diminished endothelial cell proliferation (HUVEC, hCMEC/D3 and LEC) and embryonic angiogenesis [37].
EG5 disruption is implicated in neuronal development and the phenotype observed in MCLMR adds KIF11 to the long list of genes implicated in intellectual disability. Inhibition of EG5 in cultures of synaptic neurons led to faster growth of axons, with an abnormal maturation [38]. Intellectual disability due to EG5 dysfunction could thus be explained by neuronal inability to grow and/or to connect correctly. The etiopathogenesis of the eye anomalies could be due to defective neurogenesis or angiogenesis.
EG5 could cause lymphedema by a microtubule-dependent function in trafficking the major lymphangiogenic tyrosine kinase receptor, VEGFR3, in endocytotic vesicles [39]. On the other hand, it could be that the endothelial cell function of EG5 is independent of the kinesin-motor domain, as is the case for KIF26A (kinesin family member 26A), KIF4 (kinesin family member 4A/B) and MAP2 (microtubule-associated protein 2) [40-42]. In fact, EG5 plays also a role in protein translation [43]. The expression of KIF11 in LECs is regulated by FOXC2, a major lymphangiogenic transcription factor [44]. It is however not known if MCLMR patients have lymphatic hyperplasia as observed in FOXC2-mutated patients (lymph and venous reflux and failure or absence of lymphatic and venous valves) or hypoplasia as seen in VEGFR3-mutation caused Nonne-Milroy lymphedema [45,46]. Lymphoscintigraph of one MCLMR patient showed no significant main-tract filling suggesting a peripheral lymphatic-vessel dysfunction [6].
Conclusions
KIF11 mutations are identified in all familial cases of MCLMR. De novo mutation rate is high for KIF11: 16 out of 22 (73%) sporadic cases. Mutations most likely cause loss of EG5 function. There is no genotype-phenotype correlation. The phenotype of the KIF11 mutation-negative sporadic patients may be due to non-detected germline or mosaic KIF11 mutations and/or defects in (a) gene(s) causing (a) mimicking disorder(s), as microcephaly and primary lymphedema are genetically highly heterogeneous. A better elucidation of the pathophysiological mechanisms is needed to enable development of targeted therapies for MCLMR.
Availability of supporting data
The mutations were submitted to the Vascular Anomaly and Lymphedema Mutation Database (www.icp.ucl.ac.be/vikkula/VAdb).
Abbreviations
- MCLMR:
-
Microcephaly with or without chorioretinopathy, lymphedema, or mental retardation
- KIF11:
-
Kinesin family member 11
- CDMMR:
-
Chorioretinal dysplasia, microcephaly and mental retardation syndrome
- MLCRD:
-
Microcephaly, lymphedema and chorioretinal dysplasia syndrome
- FEVR:
-
Familial exudative vitreoretinopathy syndrome
- EBV:
-
Epstein-barr virus
- SOX18:
-
SRY (sex determining region Y)-box 18
- FOXC2:
-
Forkhead box C2 (MFH-1, mesenchyme forkhead 1)
- VEGFR3:
-
Fms-related tyrosine kinase 4
- CTNNB1:
-
Catenin beta 1
- TUBA1A:
-
Tubulin alpha 1a
References
McKusick VA, Stauffer M, Knox DL, Clark DB. Chorioretinopathy with hereditary microcephaly. Arch Ophthalmol. 1966;75:597–600.
Vasudevan PC, Garcia-Minaur S, Botella MP, Perez-Aytes A, Shannon NL, Quarrell OW. Microcephaly-lymphoedema-chorioretinal dysplasia: three cases to delineate the facial phenotype and review of the literature. Clin Dysmorphol. 2005;14:109–16.
Eventov-Friedman S, Singer A, Shinwell ES. Microcephaly, lymphedema, chorioretinopathy and atrial septal defect: a case report and review of the literature. Acta Paediatr. 2009;98:758–9.
Gupta A, Vasudevan P, Biswas S, Smith JC, Moore AT, Lloyd C, et al. Microcephaly with chorioretinal dysplasia: two case reports and a review of the literature. Ophthalmic Genet. 2009;30:157–60.
Lee BJ, Kim JH, Yu YS. Lissencephaly and mild cerebellar vermis hypoplasia in a case of microcephaly and chorioretinal dysplasia. Ophthalmic Genet. 2010;31:89–93.
Ostergaard P, Simpson MA, Mendola A, Vasudevan P, Connell FC, van Impel A, et al. Mutations in KIF11 cause autosomal-dominant microcephaly variably associated with congenital lymphedema and chorioretinopathy. Am J Hum Genet. 2012;90:356–62.
Robitaille JM, Gillett RM, LeBlanc MA, Gaston D, Nightingale M, Mackley MP, et al. Phenotypic overlap between familial exudative vitreoretinopathy and microcephaly, lymphedema, and chorioretinal dysplasia caused by KIF11 mutations. JAMA Ophthalmol. 2014;132:1393–9.
Hazan F, Ostergaard P, Ozturk T, Kantekin E, Atlihan F, Jeffery S, et al. A novel KIF11 mutation in a Turkish patient with microcephaly, lymphedema, and chorioretinal dysplasia from a consanguineous family. Am J Med Genet A. 2012;158A:1686–9.
Jones GE, Ostergaard P, Moore AT, Connell FC, Williams D, Quarrell O, et al. Microcephaly with or without chorioretinopathy, lymphoedema, or mental retardation (MCLMR): review of phenotype associated with KIF11 mutations. Eur J Hum Genet. 2013;22:881–7.
Mirzaa GM, Enyedi L, Parsons G, Collins S, Medne L, Adams C, et al. Congenital microcephaly and chorioretinopathy due to de novo heterozygous KIF11 mutations: Five novel mutations and review of the literature. Am J Med Genet A. 2014;164A:2879–86.
Le Guellec R, Paris J, Couturier A, Roghi C, Philippe M. Cloning by differential screening of a Xenopus cDNA that encodes a kinesin-related protein. Mol Cell Biol. 1991;11:3395–8.
Casteels I, Devriendt K, Van Cleynenbreugel H, Demaerel P, De Tavernier F, Fryns JP. Autosomal dominant microcephaly–lymphoedema-chorioretinal dysplasia syndrome. Br J Ophthalmol. 2001;85:499–500.
Young ID, Fielder AR, Simpson K. Microcephaly, microphthalmos, and retinal folds: report of a family. J Med Genet. 1987;24:172–4.
Fryns JP, Smeets E, Van den Berghe H. On the nosology of the “primary true microcephaly, chorioretinal dysplasia, lymphoedema” association. Clin Genet. 1995;48:131–3.
Wildeman M, van Ophuizen E, den Dunnen JT, Taschner PE. Improving sequence variant descriptions in mutation databases and literature using the Mutalyzer sequence variation nomenclature checker. Hum Mutat. 2008;29:6–13.
Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM, et al. dbSNP: the NCBI database of genetic variation. Nucleic Acids Res. 2001;29:308–11.
Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, et al. A method and server for predicting damaging missense mutations. Nat Methods. 2010;7:248–9.
Ng PC, Henikoff S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003;31:3812–4.
Schwarz JM, Rodelsperger C, Schuelke M, Seelow D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat Methods. 2010;7:575–6.
Kuechler A, Willemsen MH, Albrecht B, Bacino CA, Bartholomew DW, van Bokhoven H, et al. De novo mutations in beta-catenin (CTNNB1) appear to be a frequent cause of intellectual disability: expanding the mutational and clinical spectrum. Hum Genet. 2015;134:97–109.
Sohal AP, Montgomery T, Mitra D, Ramesh V. TUBA1A mutation-associated lissencephaly: case report and review of the literature. Pediatr Neurol. 2012;46:127–31.
Brouillard P, Boon L, Vikkula M. Genetics of lymphatic anomalies. J Clin Invest. 2014;124:898–904.
Ghalamkarpour A, Debauche C, Haan E, Van Regemorter N, Sznajer Y, Thomas D, et al. Sporadic in utero generalized edema caused by mutations in the lymphangiogenic genes VEGFR3 and FOXC2. J Pediatr. 2009;155:90–3.
Irrthum A, Devriendt K, Chitayat D, Matthijs G, Glade C, Steijlen PM, et al. Mutations in the transcription factor gene SOX18 underlie recessive and dominant forms of hypotrichosis-lymphedema-telangiectasia. Am J Hum Genet. 2003;72:1470–8.
von der Hagen M, Pivarcsi M, Liebe J, von Bernuth H, Didonato N, Hennermann JB, et al. Diagnostic approach to microcephaly in childhood: a two-center study and review of the literature. Dev Med Child Neurol. 2014;56:732–41.
Schlögel MJ, Brouillard P, Boon LM, Vikkula M. Genetic Causes of Lymphedema. In: Arin K, editor. Lymphedema. Greene SAS, Håkan Brorson. Switzerland: Springer International Publishing; 2015. p. 19–31.
Douzgou S, Petersen MB. Clinical variability of genetic isolates of Cohen syndrome. Clin Genet. 2011;79:501–6.
Brouillard P, Boon LM, Mulliken JB, Enjolras O, Ghassibe M, Warman ML, et al. Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet. 2002;70:866–74.
Amyere M, Aerts V, Brouillard P, McIntyre BA, Duhoux FP, Wassef M, et al. Somatic uniparental isodisomy explains multifocality of glomuvenous malformations. Am J Hum Genet. 2013;92:188–96.
Limaye N, Wouters V, Uebelhoer M, Tuominen M, Wirkkala R, Mulliken JB, et al. Somatic mutations in angiopoietin receptor gene TEK cause solitary and multiple sporadic venous malformations. Nat Genet. 2009;41:118–24.
Parke CL, Wojcik EJ, Kim S, Worthylake DK. ATP hydrolysis in Eg5 kinesin involves a catalytic two-water mechanism. J Biol Chem. 2010;285:5859–67.
Sawin KE, Mitchison TJ. Mutations in the kinesin-like protein Eg5 disrupting localization to the mitotic spindle. Proc Natl Acad Sci U S A. 1995;92:4289–93.
Rapley J, Nicolas M, Groen A, Regue L, Bertran MT, Caelles C, et al. The NIMA-family kinase Nek6 phosphorylates the kinesin Eg5 at a novel site necessary for mitotic spindle formation. J Cell Sci. 2008;121:3912–21.
Bertran MT, Sdelci S, Regue L, Avruch J, Caelles C, Roig J. Nek9 is a Plk1-activated kinase that controls early centrosome separation through Nek6/7 and Eg5. Embo J. 2011;30:2634–47.
Ferenz NP, Gable A, Wadsworth P. Mitotic functions of kinesin-5. Semin Cell Dev Biol. 2010;21:255–9.
Castillo A, Justice MJ. The kinesin related motor protein, Eg5, is essential for maintenance of pre-implantation embryogenesis. Biochem Biophys Res Commun. 2007;357:694–9.
Exertier P, Javerzat S, Wang B, Franco M, Herbert J, Platonova N, et al. Impaired angiogenesis and tumor development by inhibition of the mitotic kinesin Eg5. Oncotarget. 2013;4:2302–16.
Haque SA, Hasaka TP, Brooks AD, Lobanov PV, Baas PW. Monastrol, a prototype anti-cancer drug that inhibits a mitotic kinesin, induces rapid bursts of axonal outgrowth from cultured postmitotic neurons. Cell Motil Cytoskeleton. 2004;58:10–6.
Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol. 2011;12:551–64.
Zhou R, Niwa S, Homma N, Takei Y, Hirokawa N. KIF26A is an unconventional kinesin and regulates GDNF-Ret signaling in enteric neuronal development. Cell. 2009;139:802–13.
Midorikawa R, Takei Y, Hirokawa N. KIF4 motor regulates activity-dependent neuronal survival by suppressing PARP-1 enzymatic activity. Cell. 2006;125:371–83.
Harada A, Teng J, Takei Y, Oguchi K, Hirokawa N. MAP2 is required for dendrite elongation, PKA anchoring in dendrites, and proper PKA signal transduction. J Cell Biol. 2002;158:541–9.
Bartoli KM, Jakovljevic J, Woolford Jr JL, Saunders WS. Kinesin molecular motor Eg5 functions during polypeptide synthesis. Mol Biol Cell. 2011;22:3420–30.
Ivanov KI, Agalarov Y, Valmu L, Samuilova O, Liebl J, Houhou N, et al. Phosphorylation regulates FOXC2-mediated transcription in lymphatic endothelial cells. Mol Cell Biol. 2013;33:3749–61.
Brice G, Mansour S, Bell R, Collin JR, Child AH, Brady AF, et al. Analysis of the phenotypic abnormalities in lymphoedema-distichiasis syndrome in 74 patients with FOXC2 mutations or linkage to 16q24. J Med Genet. 2002;39:478–83.
Brice G, Child AH, Evans A, Bell R, Mansour S, Burnand K, et al. Milroy disease and the VEGFR-3 mutation phenotype. J Med Genet. 2005;42:98–102.
Acknowledgements
We are grateful to all family members for their participation. This work was supported by Belgian Science Policy Office Interuniversity Attraction Poles (BELSPO-IAP) programme through the project IAP P7/43-BeMGI; and the F.R.S.-FNRS (Fonds de la Recherche Scientifique). We also acknowledge the Fédération de Wallonie-Bruxelles and the Lotterie nationale of Belgium for their support. MJS and EF are doctoral students supported by the Fonds pour la Formation à la Recherche dans l’Industrie et dans l’Agriculture (FRIA).
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
The authors declare that they have no competing interests.
Authors’ contributions
PB and MV designed the research. MJS, AM and EF carried out the molecular genetic studies. PV, KD, TJLR, HVE, IC, AC, FC, KF, KJ, ML, IB, AS, MS, MMV, NR and LMB collected medical information and blood samples. MJS, PB and MV wrote the manuscript. All authors read and approved the final manuscript.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

Cite this article
Schlögel, M.J., Mendola, A., Fastré, E. et al. No evidence of locus heterogeneity in familial microcephaly with or without chorioretinopathy, lymphedema, or mental retardation syndrome. Orphanet J Rare Dis 10, 52 (2015). https://doi.org/10.1186/s13023-015-0271-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13023-015-0271-4