
Abstract
The placenta, a pivotal organ in mammalian reproduction, allows nutrient exchange and hormonal signaling between the mother and the developing fetus. Understanding its molecular intricacies is essential for deciphering normal embryonic development and pathological conditions such as tumorigenesis. Here, we explore the multifaceted role of the tumor suppressor BRCA1-associated protein 1 (BAP1) in cancer and placentation. Initially recognized for its tumor-suppressive properties, BAP1 has emerged as a key regulator at the intersection of tumorigenesis and placental development. BAP1 influences crucial cellular processes such as cell death, proliferation, metabolism, and response to hypoxic conditions. By integrating insights from tumor and developmental biology, we illuminate the complex molecular pathways orchestrated by BAP1. This perspective highlights BAP1's significant impact on both cancer and placental development, and suggests novel therapeutic strategies that could improve outcomes for pregnancy disorders and cancer.
Similar content being viewed by others
Introduction
The precise genetic regulation governing cell proliferation and commitment is pivotal for initiating embryo formation and ensuring its proper development. The transition from pluripotency to commitment involves intricate signaling pathways and transcriptional factors, ultimately modulating gene expression and chromatin remodeling1. Embryogenesis, traditionally viewed as a progressive loss of developmental plasticity, sets the stage for the differentiation of distinct cell lineages crucial for embryonic and extraembryonic structures2.
However, deviations from normal differentiation pathways can lead to malignant transformation, often propelled by genome instability induced by various stressors affecting DNA replication3. This instability fosters intra-tumor heterogeneity, posing significant challenges in cancer comprehension and treatment3,4. Remarkably, cancer cells exhibit behaviors reminiscent of embryogenesis, including mechanisms to evade immune surveillance, akin to placental trophoblasts in mammalian reproduction5.
The intriguing parallels between cancer and placental trophoblast cell programs have sparked speculation about the involvement of dysregulated placental molecular circuits in tumorigenesis6,7,8. Notably, the tumor suppressor gene BAP1 has emerged as a crucial regulator in both placental development and embryogenesis9,10,11. BAP1, a deubiquitylase with widespread expression, regulates key cellular pathways involved in cell cycle control, cellular differentiation, and the DNA damage response12,13.
Genetic mutations or deletions in BAP1 are frequently observed in various cancers, including uveal melanoma, mesothelioma, and renal cell carcinoma, underscoring its significance in tumorigenesis14,15. In this review, we revisit the hypothesis that trophoblast and cancer cells exploit analogous molecular mechanisms for their proliferative, immunosuppressive, and invasive behaviors, with a particular focus on the role of BAP1 in cancer-related studies. We explore several molecular scenarios regulated by BAP1, aiming to elucidate the parallels between cancer and placental development, offering novel insights into pregnancy disorders and embryonic development.
Brief overview of placentation
The placenta stands as a vital intermediary organ facilitating the exchange of essential nutrients and oxygen between the mother and the developing embryo throughout gestation. The proper establishment and function of the placenta are fundamental for embryonic well-being, and any aberrations in its development can precipitate a spectrum of pregnancy complications, including preeclampsia, fetal growth restriction (FGR), preterm birth, and, in severe instances, miscarriage16.
In the murine model, placental development commences with the formation of the blastocyst at embryonic day (E) 3.5, comprising the outer trophectoderm (TE) which will give rise to all trophoblast cell types of the future placenta, and the inner cell mass (ICM), which later undergo differentiation into the epiblast (EPI) and the primitive endoderm or hypoblast16,17. Among the TE cells, those in direct contact with the ICM, termed polar TE, give rise to the extraembryonic ectoderm (ExE) and the ectoplacental cone (EPC)17,18. Conversely, mural TE cells, not in direct contact with the ICM, differentiate into primary trophoblast giant cells (TGCs), essential for successful embryo implantation17,18. During gastrulation (E6.5), the chorion, which is made up of trophoblast cells and an extra-embryonic mesodermal cell layer, as well as the amnion and the allantois, are formed. The allantois extends outward and connects with the chorion (chorio-allantoic fusion) around E8.5, a crucial step for development. By E9.5, blood vessels in the allantois begin to grow into the chorionic ectoderm, starting the development of the placental labyrinth, a complex network serving as the primary exchange interface between the embryo and the maternal circulation19.
The ExE, positioned closer to the EPI, contributes to the formation of the syncytiotrophoblast (SynT) layer, which elongates and undergoes fusion to generate the SynT-I and SynT-II layers, thereby establishing the main exchange surface of the placental labyrinth for nutrient transport16,17,19,20. Cells at the proximal tip of the ExE undergo further differentiation into the EPC, forming a cap-like structure atop the ExE19. Peripheral cells of the EPC differentiate into secondary TGCs, acquiring invasive characteristics necessary for penetrating the endometrial stroma and establishing contact with maternal arteries, while central cells give rise to the spongiotrophoblast (SpT) and glycogen cells (GCs), crucial constituents of the junctional zone (JZ) of the placenta. The JZ serves as the primary endocrine section of the placenta. It generates significant quantities of hormones, growth factors, and cytokines essential for the normal progression of pregnancy, influencing both maternal and fetal physiology16,19.
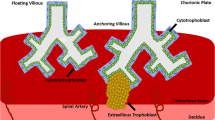
Despite notable morphological distinctions, comparative analysis with the human placenta underscores structural and functional resemblances (Fig. 1). The human placenta originates from the TE of the blastocyst, which in humans is formed around five days after fertilization (dpf). At this point, as in mice, the blastocyst is divided into two cell lineages: the ICM and the TE21.

The placenta serves as a crucial organ that facilitates the exchange of nutrients and oxygen between the mother and the developing embryo. Human and mouse hemochorial placentas exhibit functional similarities. In both species, a complex network (referred to as labyrinth in mice and chorionic villi in humans) acts as the primary interface for exchange between the embryo and the mother. Development of this structure involves similar invasive strategies into the maternal tissue; invasive mouse trophoblast giant cells and glycogen cells penetrate the endometrial stroma and establish contact with maternal arteries, whereas invasive human extravillous trophoblasts make contact with the uterine interstitium and remodel the maternal spiral arteries. A closer look shows that mouse labyrinth filled with maternal blood is constituted of sinusoidal giant cells surrounded by two layers of syncytiotrophoblast cells, SynT-I and SynT-II. These intricate extensions are neighboring fetal blood vessels, thus allowing the exchange of nutrients and oxygen. In turn, human chorionic villi, containing fetal capillaries, consist of a continuous layer of syncytiotrophoblasts cells enveloping a layer of villous cytotrophoblasts. CCT, cytotrophoblast column; vCTB, villous cytotrophoblasts; EVT, extravillous trophoblast; GlyT, glycogen trophoblast; SpT, spongiotrophoblast; STB and SynT, syncytiotrophoblast; TGC, trophoblast giant cell.
Embryo implantation occurs around 8-10 dpf, as the polar TE interfaces with the uterine luminal epithelium. After implantation, two trophoblast populations appear: early cytotrophoblasts (CTBs) and the primitive syncytium (PS), with the latter being the first invasive placental cell lineage to penetrate the maternal endometrium. Around 10 dpf, vacuoles originating from the PS merge to form the lacunar system, which serves as the precursor to the intervillous space. During this period, CTBs proliferate, invade the PS, and give rise to primary villi, which penetrate the maternal decidua and gradually disrupt uterine blood vessels and glands5,22. At this stage, the blastocyst is enveloped by three layers: the inner chorionic plate which borders the original cavity; the villi separated by the intervillous space; and the cytotrophoblast shell which contacts the decidua21.
As gestation progresses, extraembryonic mesenchymal cells infiltrate the villous core to form secondary villi. By day 18 dpf, fetal capillaries develop within the core, signaling the formation of tertiary villi. The villous tree grows swiftly by extending branches from the chorionic plate, forming a network of villous trees. At the junction between the maternal and fetal tissues, where the cytotrophoblast shell meets the decidua, individual cytotrophoblast cells separate from the shell and infiltrate the decidua as extravillous trophoblast (EVT), in a manner resembling epithelial-mesenchymal transition (EMT). This process sets up the fundamental structure of the placenta by the end of the first trimester. Once placental villi have fully developed, EVTs arise from the differentiation of CTBs located at the tips of anchoring villi22,23. In this villi, the proximal portion of the cell column trophoblast (CCT) acts as a reservoir for progenitor EVTs, which differentiate into invasive and migratory EVTs24. EVT subtypes include interstitial extravillous trophoblasts (iEVTs), which directly interact with decidual stromal cells, macrophages, and uterine natural killer (uNK) cells. The invasion of the uterine stroma by interstitial extravillous trophoblasts (iEVTs) plays a crucial role in establishing and maintaining maternal-fetal immune tolerance. These iEVTs extend into the inner third of the myometrium, where they are thought to fuse and form placental bed giant cells. They migrate toward the spiral arteries, surrounding them with a layer of cells and initiating arterial remodeling. The iEVTs infiltrate the spiral arteries and differentiate into endovascular extravillous trophoblasts (eEVTs), which travel along the artery lumen, inducing endothelial cell apoptosis and completely replacing the maternal endothelial cells within these vessels5. The conversion of these arteries into low-resistance, high-capacity vessels enhances placental perfusion by augmenting blood flow through the uterine arteries, an essential adaptation for normal pregnancy25. After the arteries undergo this transformation, the eEVTs create a plug that prevents blood from entering the intervillous space until the end of the first trimester, at which point the haemochorial circulation is established21,26.
The thin line between tumorigenesis and embryogenesis
The delicate equilibrium between cell proliferation and programmed cell death (PCD) stands as a cornerstone in both embryogenesis and tumorigenesis. In vertebrate development, non-apoptotic cell death serves as a normal process of eliminating damaged cells, a phenomenon observed in cells from the ICM before mouse embryo implantation27,28. This balance is crucial for the development and homeostasis of the placenta, which undergoes continuous tissue remodeling through trophoblast apoptosis29. Notably, the trophectoderm epithelium is responsible for phagocytosing apoptotic embryo cells30.
Throughout development, PCD also contributes to tissue maintenance, such as in the post-mitotic spinal cord of the mouse nervous system27. However, disruptions in the intrinsic apoptosis pathway are frequently encountered in cancer cells, compromising normal cellular regulatory mechanisms and fostering tumor progression, metastasis, and resistance to therapeutic interventions31,32.
Metabolism plays a pivotal role in embryonic development, providing the requisite energy for the developing embryo33. A significant increase in glucose consumption, indicative of highly active metabolism, is essential during initial development to sustain embryo viability 34. Perturbations in energy production during the preimplantation phase are associated with embryonic developmental impairments and reduced fetal viability35. Analogously, tumor cells undergo metabolic reprogramming to meet the heightened demands of ATP, NADPH, and NADH36,37,38. Similarly, the process of placentation demands a substantial energy supply to support its growth through exceptionally active metabolism, undergoing metabolic reprogramming to accommodate developmental changes and ensure proper growth and functionality39,40.
Uncontrolled cell proliferation and altered metabolism often lead to diminished oxygen supply, resulting in tumor hypoxia and exacerbating the risk of metastasis41. Paradoxically, the mammalian embryo develops in a low-oxygen environment, activating hypoxia-inducible factor (HIF) to facilitate anaerobic metabolism and induce modifications in surrounding vasculature42. Similarly, a low-oxygen environment is crucial for placental proliferation, invasion, and normal progression of placental angiogenesis during the early weeks after fertilization43.
Epithelial to mesenchymal transition (EMT), a cellular process integral to proper embryonic development, notably contributes to the establishment of germ layers during gastrulation44. Trophoblast cells share molecular invasion mechanisms with cancer cells45, exhibiting evidence of EMT during differentiation, granting them migratory and invasive capabilities46,47. However, unlike cancer cells, placental cells do not undergo tumorigenesis or metastasize to maternal organs during normal pregnancy45.
Nevertheless, trophoblast cells can occasionally evade regulatory mechanisms, leading to abnormal growth and the development of rare tumors known as Gestational Trophoblastic Disease (GTD)48,49. Beyond malignant transformation, excessive invasion can result in placenta accreta or inadequate deep placentation, both of which are linked to significant pregnancy disorders50,51,52. The remodeling of spiral arteries within the endometrium by EVTs plays a crucial role in pregnancy, with its failure implicated in conditions such as preeclampsia53.
Given the shared mechanisms between tumorigenesis and embryogenesis, studying tumor emergence and metastasis could offer valuable insights into placental development and malfunction (Fig. 2). This review explores the intricate interplay between tumorigenesis and embryogenesis, particularly focusing on parallels observed in placental development, to elucidate common regulatory pathways and potential therapeutic targets.

Tumorigenesis and human placentation share proliferative, metabolic, and invasive mechanisms. While cell death is finely regulated in the placenta but disrupted in tumors, both cancer and trophoblast cells display high rates of cell proliferation, which can be attributed to their unique metabolic strategies. Regardless of oxygen availability, cancer cells predominantly produce energy through aerobic glycolysis, a metabolic phenomenon known as the Warburg effect. Similarly, trophoblasts can modulate their metabolic activity in response to environmental changes. The increase in oxygen and glucose levels resulting from maternal blood circulation into the low-oxygen placental environment triggers metabolic adaptations crucial for trophoblast differentiation. In terms of invasion, both tumor and trophoblast cells possess the capacity to undergo epithelial-to-mesenchymal transition and invade into healthy tissues. This invasive behavior is critical for tumor progression and the establishment of the placenta during pregnancy. Additionally, both cell types modulate the host immune response to support their growth and development. Despite all similarities, trophoblast cells follow a well-controlled program of differentiation, avoiding uncontrolled growth or metastasis. CTB, cytotrophoblast; EMT, epithelial-to-mesenchymal transition; eEVTs, endovascular extravillous trophoblasts; iEVTs, interstitial extravillous trophoblasts; NK, natural-killer; STB, syncytiotrophoblast; TCA, tricarboxylic acid.
Molecular features of the tumor suppressor BAP1
The BRCA-1 associated protein 1 (BAP1) plays a pivotal role in various cellular processes, including cell growth, division, programmed cell death, DNA damage repair, and regulation of gene activity. Mutations in the BAP1 gene heighten the susceptibility to developing certain types of tumors, notably skin, eye, and kidney tumors54.
BAP1 functions as a deubiquitinase (DUB) within the ubiquitin C-terminal hydrolase (UCH) domain-containing protein family55. Serving as the catalytic subunit of the Polycomb Repressive-DUB (PR-DUB) complex, BAP1 interacts with ASXL1, ASXL2, or ASXL3 to ensure the complex’s functionality. The PR-DUB complex, which includes additional protein members such as HCF1, FOXK1/2, LSD2, OGT, and MBD5/6, induces conformational changes in BAP1, enhancing its affinity for ubiquitin56,57. This complex is responsible for deubiquitinating and maintaining the spatial distribution of Histone H2A at lysine 119 (H2AK119)56. BAP1’s deubiquitinating activity counteracts the function of the Polycomb repressive complex 1 (PRC1), which monoubiquitinates H2AK119, essential for repressing PRC1 target genes and recruiting the PRC2 complex in embryonic stem cells58,59. However, contradictory perspectives emerge regarding the accumulation of H2AK119ub resulting from BAP1 loss, suggesting a potential role in activating PRC genes60. The functional interplay between PR-DUB, PRC1, and PRC2 complexes orchestrates chromatin architecture, influencing crucial cellular functions such as stemness and differentiation60. In addition to the epigenetic changes induced by BAP1 activity, BAP1-targeted genes often exhibit histone marks associated with transcriptional activation, including H3K27ac, H3K4me1, and H3K4me3, suggesting its involvement in gene activation and broader regulation of genome transcriptional potential61.
The identification of inactivating mutations of BAP1 in various human cancers underscores its role as a critical tumor suppressor13. It has been reported a direct link between BAP1 germline mutations and increased susceptibility to multiple cancers, including mesothelioma, uveal melanoma (UM), clear cell renal cell carcinoma (ccRCC), cutaneous melanoma, and basal cell carcinoma62. In metastatic uveal melanoma, the loss of BAP1 function is strongly associated with increased metastatic potential and poor prognosis. Several mechanisms have been proposed to explain this such as induction of stem-like features. BAP1 loss induces stem-like features and loss of differentiation in UM cells, promoting a more aggressive phenotype. This dedifferentiation may enhance the cells’ ability to invade and metastasize63. BAP1 deficiency also leads to DNA methylation repatterning and transcriptional dysregulation, contributing to UM progression and metastasis. These epigenetic changes can alter the expression of genes involved in cell adhesion, migration, and invasion64. BAP1 mutations occur early in UM tumorigenesis, potentially enabling the seeding of micrometastases at an early stage. This early genetic alteration can set the stage for further genetic and epigenetic changes that drive metastasis65.
In mesothelioma, which is also strongly linked to BAP1 mutations, the loss of BAP1 function has been associated with increased cell proliferation, survival, and motility. BAP1 loss results in the dysregulation of gene expression programs involved in cell cycle control and apoptosis. This dysregulation can lead to unchecked cell proliferation and resistance to cell death66. The loss of BAP1 also impairs the DNA damage response and increases genomic instability. Cells with compromised genomic integrity are more likely to accumulate mutations that drive cancer progression67. BAP1 loss can lead to alterations in cellular metabolism and the management of metabolic stress. These metabolic changes can provide a survival advantage under the stressful conditions of the tumor microenvironment68. The concept of BAP1 cancer syndrome characterizes scenarios where individuals with hereditary BAP1-inactivating mutations manifest one or multiple cancers over their lifetime13.
It is important to note that BAP1, traditionally recognized as a tumor suppressor, demonstrates varied functions influenced by cellular contexts and specific molecular alterations in diverse cancer types. Recent studies have suggested potential oncogenic roles of BAP1 in settings such as breast cancer, small-cell lung carcinoma, melanoma, colon cancer, and myeloid neoplasms69,70,71,72,73,74. However, these findings primarily derive from preclinical research, highlighting the intricate and context-dependent nature of BAP1’s biological activities and underscoring the necessity for further clinical investigation into its physiological and pathological implications.
The role of BAP1 in regulating embryonic development
While the involvement of BAP1 in cancer remains a focal point of research, a comprehensive exploration of its functions in embryo development and placentation could offer a novel perspective. BAP1 plays pivotal roles in stem cell pluripotency and embryo development across various species, including Drosophila, Xenopus laevis, Artemia, and mice. Previous studies have examined the role of BAP1 in developmental lineages, providing insights into how BAP1 loss promotes cancer progression9. This avenue now presents an opportunity to enhance our understanding of BAP1-deficient tumors by elucidating its normal functions in development.
In Drosophila, the BAP1 orthologue Calypso is crucial for embryonic development and interacts with the Asx complex to protect transcriptionally active developmental genes against silencing by PRC175. Mutations affecting Calypso’s H2A deubiquitinase activity interfere with blastopore closure, leading to a delay or arrest in gastrulation76.
Moreover, BAP1 is involved in the formation and maintenance of diapause embryos in Artemia77. Knockdown of ArBAP1 by RNA interference results in the formation of embryos with split shells and abortive nauplii, indicating its essential role in embryo development77.
Transitioning to Xenopus laevis, targeted depletion of BAP1 protein causes a striking developmental phenotype affecting various lineages9. BAP1-deficient Xenopus embryos fail to express lineage-specific factors and show impaired differentiation, suggesting a possible role for BAP1 in mammalian embryogenesis.
Indeed, Bap1 knockout (KO) in mice leads to embryonic lethality due to developmental retardation between E8.5 and E9.5, highlighting its critical involvement in mammalian development11,78. BAP1 also plays a crucial role in the differentiation of mammalian progenitor cells, with its inactivation impairing normal differentiation and increasing progenitor proliferation78.
The multifaceted roles of BAP1 extend beyond its well-established anti-tumor functions, encompassing crucial contributions to embryonic development, stem cell pluripotency, and embryonic diapause. Insights from various species such as Drosophila, Artemia, Xenopus laevis, and mice underscore the significance of BAP1 in diverse biological processes, shedding light on its intricate involvement in transcriptional regulation, gastrulation, and lineage-specific differentiation.
The Role of BAP1 in regulating placentation
As part of the Deciphering the Mechanisms of Developmental Disorders (DMDD) program79, Perez-Garcia et al. identified a group of genes associated with placental defects, including Bap1, within novel molecular networks governing early placentation11. Deletion of Bap1 resulted in severe placental defects, leading to mid-gestational demise (E9.5), emphasizing its crucial role in placental development. During differentiation, Bap1-deficient mouse trophoblast stem cells (mTSCs) failed to upregulate markers associated with trophoblast lineages essential for placental function, such as SynT, glycogen cells, and sTGCs11.
Further investigation revealed that BAP1 downregulation at the protein level is essential for initiating epithelial-mesenchymal transition (EMT) during trophoblast differentiation and invasion. This process depends on its interaction with ASXL1/2 proteins to form the PR-DUB complex. The expression levels of the BAP1 protein decrease in human trophoblast stem cells (hTSCs) as they transition into invasive extravillous trophoblasts (EVTs), suggesting conservation of BAP1 PR-DUB complex modulation between mice and humans10. Recent insights from Mei et al. confirmed that BAP1 silencing in trophoblast cells promotes cell proliferation, invasion, and migration, accompanied by molecular changes indicative of EMT80.
Overall, the conservation of BAP1’s role in regulating trophoblast epithelial features and EMT during differentiation across species underscores its importance in placentation. Understanding BAP1’s role in placentation may offer insights into cancer research, as the downregulation of BAP1 levels in trophoblasts mirrors BAP1-deficient tumor behavior, potentially informing therapeutic interventions targeting common regulatory pathways in both fields.
Lessons from BAP1’s roles in tumorigenesis
The intricate processes of placentation, embryo development, and tumorigenesis share fundamental cellular mechanisms involving cell proliferation, apoptosis, metabolism, hypoxia response, and invasion, with BAP1 emerging as a central player (Fig. 2). While BAP1 deficiency in tumors leads to dysregulated cell behavior, its downregulation is crucial for placental formation9,10,58. Exploring the effects of BAP1 deficiency in cancer may illuminate early placentation processes and offer insights into managing pregnancy complications (Fig. 3).

The described effects of BAP1 deficiency in tumors could shed light into the mechanisms underlying placentation development and pregnancy complications. (1) Regulation of cell differentiation through BAP1 interaction with HDACs is disrupted in BAP1-deficient tumors, leading to upregulation of HDAC4 levels. Conversely, upregulation of BAP1 levels could result in the opposite effect; downregulation of HDAC4 in preeclamptic tissues suggests the potential implication of BAP1 in trophoblast invasion. (2) Similarly, the interaction between BAP1 and EZH2, which encompasses cell differentiation, is dysregulated in BAP1-deficient tumors, resulting in aberrant H3K27me3 levels. Contrasting EZH2 and H3K27me3 levels in pregnancy complications could result from upregulated placental BAP1 levels. (3) The potential involvement of BAP1-mediated cell death regulation (ER stress, Ca2+ apoptosis, and ferroptosis) and cell survival (suppressing anti-apoptotic genes) has been described through BAP1-deficient tumor examination. Pregnancy complications display similar dysregulation of these cell-death mechanisms, suggesting the potential implication of BAP1 in trophoblast cell death regulation. (4) BAP1’s activity also regulates cell cycle machinery through interactions with HCF-1, YY1, and KLF5 transcription factors, which have been associated with trophoblast invasion. BAP1-deficient tumors show altered expression levels of these cell-death regulators, offering a novel avenue for exploring BAP1 interaction with cell-cycle regulators in pregnancy disorders. Elevated levels of PTEN, which inhibits PI3K-AKT pathway, have been associated with BAP1-deficient tumors, whereas pregnancy complications show downregulated levels. In this sense, BAP1 could play a crucial role in modulating PI3K-AKT pathway during placental development. (5) BAP1 interaction with OGT/HCF-1 complex and PGC-1α is involved in regulation of cell metabolism. Considering that disruption of placental PGC-1α and OGT levels have been observed in pregnancy complications, investigating the impact of BAP1 in trophoblast cells may provide insights into the metabolism of pathogenic placenta. Additionally, BAP1 has been implicated in the regulation of LKB1 and ASS1, regulators of lipid metabolism and nitric oxide production, respectively. Dysregulation of LKB1 and ASS1 levels in pregnancy outcomes raises the question of the possible BAP1 involvement in trophoblast metabolism. (6) Mutations in BAP1 disrupt HIF-1α stabilisation through deubiquitination, thus decreasing its levels. Given the elevated levels of HIF-1α observed in placental dysfunction, BAP1-mediated regulation of HIF-1α may play a critical role in modulating placental development under low oxygen levels. GDM, gestational diabetes mellitus; PE, preeclampsia; PRC2, polycomb repressive complex 2.
BAP1 expression mediates epigenetic regulation
One significant aspect of BAP1’s influence lies in its modulation of histone deacetylases (HDACs) balance. HDACs, pivotal in embryonic development and cell differentiation, regulate chromatin structure by removing acetyl groups from histones, thus repressing transcription81,82. In cancer, HDACs govern critical cellular processes, making them promising targets for therapeutic intervention83. Remarkably, decreased HDAC4 expression in preeclamptic tissues triggers autophagy and apoptosis in trophoblast cells, suggesting its role in pregnancy complications84. Similarly, aberrant HDAC expression and autophagy levels are associated with recurrent spontaneous abortion, emphasizing the importance of HDAC regulation in maintaining placental health85.
BAP1 deficiency renders tumors vulnerable to HDAC depletion, as observed in various cancer types86,87. The intricate interplay between BAP1 and HDACs influences gene-environment interactions implicated in carcinogenesis, particularly in asbestos-induced cancers88. BAP1 combines into a trimer alongside high-mobility group box 1 protein (HMGB1) and histone deacetylase 1 (HDAC1), influencing gene-environment interactions in the presence of asbestos. Decreased levels of BAP1 result in heightened ubiquitylation and degradation of HDAC1. This process enhances the acetylation of HMGB1, prompting its active secretion and consequently facilitating the malignant transformation of mesothelial cells88. Moreover, in mesothelioma and uveal melanoma, BAP1 loss results in dysregulated HDACs expression, rendering cells sensitive to HDAC inhibitors and highlighting the therapeutic potential of targeting this interaction9,87.
Interestingly, while BAP1-deficient tumors exhibit upregulated HDAC4 levels9, preeclamptic tissues show a contrasting downregulation, potentially affecting trophoblast invasion and leading to pregnancy complications84. Given the parallels between tumor and trophoblast invasion, further investigation into the BAP1-HDAC4 interaction in placental development and pregnancy pathologies is warranted.
The regulatory dynamics of BAP1 and EZH2 (Enhancer of Zeste Homolog 2), the catalytic subunit of the PCR2, underscore their pivotal roles in governing cell differentiation processes. EZH2, a key player in epigenetic regulation, orchestrates gene repression via H3K27 trimethylation (H3K27me3), crucial for maintaining cellular differentiation and proliferation89. Dysregulation of EZH2 is implicated in both embryonic lethality and cancer progression, making it a significant therapeutic target90,91.
In tumor development, EZH2 overexpression promotes tumor progression and metastasis, emphasizing its oncogenic potential91. Similarly, EZH2’s involvement in placentation, where it regulates trophoblast differentiation and invasiveness, highlights its multifaceted roles beyond tumorigenesis92. Moreover, dysregulation of EZH2 expression is associated with pregnancy complications such as recurrent miscarriage and preeclampsia, suggesting its critical role in placental development and function92,93.
Emerging evidence suggests a reciprocal relationship between BAP1 and EZH2 levels in cell differentiation processes across various cancer types. BAP1 loss leads to upregulated EZH2 expression, contributing to aberrant H3K27me3 levels and disrupted differentiation pathways, as observed in hematopoietic stem cells (HSC) and mesothelioma78,94,95. Notably, inhibition of EZH2 rescues normal H3K27me3 levels and mitigates tumor growth in BAP1-deficient contexts, suggesting a potential therapeutic strategy78,94,95.
The possible common convergence of BAP1 loss and EZH2 levels between cancer and placentation presents an intriguing nexus of epigenetic regulation. Considering the association between EZH2 upregulation and invasiveness in BAP1-deficient mesothelioma, a similar interaction may occur in the placenta, where EZH2 upregulation and BAP1 downregulation are two conditions needed for trophoblast differentiation and invasiveness10,92. Accordingly, reduced expression of EZH292 and potential increased expression of BAP1 in preeclamptic tissues support the possible tight regulation and interaction of both proteins in placental development.
Cell death-related BAP1 target genes in tumors and placenta
Cell death plays a pivotal role in ensuring tissue homeostasis and proper development during embryogenesis and placental remodeling. The intricate mechanisms governing cell death pathways have garnered significant attention, particularly in understanding pregnancy-related disorders such as preeclampsia. Recent insights into the multifaceted roles of BAP1 in tumor cell survival and cell death regulation offer valuable perspectives on trophoblast biology and pregnancy complications12.
ATF3 and CHOP transcription factors are activated by glucose deprivation and regulate the unfolded protein response (UPR), which causes ATP depletion and ROS production96. Moreover, ATF3 has been described to inhibit tumor proliferation and migration in different types of cancers97,98. Intriguingly, ATF3 has been identified as a promoter of proliferation and migration in trophoblast cells, an opposed role to that observed in tumors99. Accordingly, reduction in ATF3 levels is apparent in preeclamptic placentas, highlighting the possibility of a dual role of this gene in promoting or inhibiting cell invasion100. Considering that BAP1 inhibits transcription of ATF3 and CHOP genes by binding to their corresponding promoters96, BAP1 downregulation in normal trophoblast differentiation10 may release ATF3 promotors from inhibition, thus contributing to trophoblast invasion. In the same manner, the observed reduction of ATF3 in preeclamptic placentas could result from the abnormal expression of BAP1, which seems to be higher in comparison to controls101.
CHOP serves as a placental ER stress marker, with its expression rising in placental explants from non-PE pregnant women treated with serum samples from women with PE102. Similar detection of placental ER stress via elevated CHOP levels has been documented in placental injuries and in placentae from pregnant women of advanced age103,104. Contrary to the notion that BAP1 expression in PE tissues may lead to the downregulation of CHOP, the current findings indicate an opposing scenario. This could imply that CHOP regulation involves complex signaling pathways and is influenced by other inducers beyond BAP1, warranting further investigation.
Type 3 inositol-1,4,5-triphosphate receptor (IP3R3) activation facilitates Ca+2 release from the ER, a phenomenon that, in turn, triggers mitochondria to absorb the excess of Ca+2, subsequently instigating the release of cytochrome C and culminating in cell death105,106. IP3R3 integral role in vasculature development at the mouse embryonic-maternal interface has garnered attention to its involvement in placentation107. In view of that, placentas from women diagnosed with severe intrahepatic cholestasis of pregnancy (ICP), which cause adverse fetal outcomes and even fatal death, exhibited diminished IP3R3 levels in the mitochondria-associated membranes108. The dysregulated IP3R3 levels were associated to elevated p-AKT levels, triggering a disrupted flow of Ca+2 from the ER to the mitochondria108.
Suggesting a mechanism in which BAP1-deficient tumors escape from cell death, BAP1 has been observed to deubiquitinate IP3R3, thus regulating apoptosis106. A noteworthy implication of BAP1 inhibition is its dampening effect on cellular sensitivity to Ca+2-dependent apoptosis, offering potential respite for stressed cells seeking survival106. Given BAP1’s pivotal role in early placentation10, the interaction with IP3R3 might also be replicated in trophoblast cells. The possible stabilisation of IP3R3 by BAP1 deubiquitination could imply a novel mechanism of cell death regulation in placentation that may be disrupted in pregnancy complications.
Ferroptosis, a stress-induced form of cell death, offers another intriguing avenue linking BAP1 to pregnancy complications. SLC7A11 is a transmembrane protein that participates in the cysteine/glutamate antiporter system, initiating a metabolic pathway that results in ferroptosis inhibition109. The repression of SLC7A11 by BAP1 unveils a potential ferroptosis axis implicated in placental physiology and pathology, shedding light on novel mechanisms underlying pregnancy disorders110,111. Additionally, the transcriptional regulation of anti-apoptotic genes Bcl2 and Mcl1 by BAP1 underscores its broader role in modulating cell death pathways from embryogenesis to placentation80,112,113,114. The intersection between BAP1-mediated cell death regulation and pregnancy complications provides a fertile ground for further exploration, holding promise for improving our understanding and management of pregnancy-related disorders.
Cell proliferation is regulated by BAP1 activity
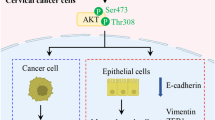
Emerging evidence suggests that BAP1, plays a pivotal role in modulating cell proliferation through its interaction with key regulatory proteins115,116. BAP1’s activity is intertwined with the cell cycle machinery, particularly through its interactions with HCF-1 and YY1, which promote cell cycle progression by facilitating the transcription of E2F-target genes117,118. This intricate partnership suggests that BAP1 may exert its proliferative effects by promoting the expression of genes involved in cell cycle progression. YY1, a transcription factor implicated in trophoblast invasion and adhesion, emerges as a central player in early placentation119,120. Dysregulation of YY1 has been associated with pregnancy complications such as preeclampsia121. Given BAP1’s crucial role in early placentation10, it is plausible that BAP1 may interact with YY1 to regulate trophoblast proliferation and invasion, offering a novel avenue for exploring the mechanisms underlying pregnancy disorders.
Furthermore, BAP1’s involvement in modulating the ERK1/2 signaling pathway adds another layer of complexity to its role in cell proliferation regulation122. Dysregulation of the ERK1/2 pathway has been implicated in both trophoblast biology and pregnancy complications such as preeclampsia123,124. Understanding how BAP1 influences trophoblast differentiation through modulation of the ERK1/2 pathway may provide valuable insights into the pathogenesis of pregnancy-related disorders.
Moreover, BAP1 has been implicated in the regulation of PTEN, a tumor suppressor that controls the PI3K-AKT pathway and plays a crucial role in trophoblast cell proliferation and invasion125,126. Dysregulation of PTEN has been associated with pregnancy complications such as spontaneous abortion127. Finally, the interaction between BAP1 and KLF5, a transcription factor implicated in various biological processes including cell proliferation and differentiation, further underscores BAP1’s role in regulating cellular functions69,128. Understanding how BAP1 influences KLF5 expression in trophoblasts may provide novel insights into the mechanisms governing placental development and function.
In conclusion, unraveling the intricate network of interactions involving BAP1 offers valuable insights into the regulation of cell proliferation during embryonic development and placental formation. Further investigation into the molecular mechanisms underlying BAP1’s role in trophoblast biology may lead to the development of novel therapeutic strategies for managing pregnancy-related disorders.
BAP1 expression mediates cell metabolism
Cellular metabolism plays a fundamental role in supporting the high-energy demands of developing embryos, the placenta, and cancer cells39,129,130. Dysregulation of metabolic pathways can have profound consequences, influencing both embryonic development and pregnancy outcomes. Understanding the intricate connections between metabolism and cellular processes holds promise for elucidating the pathogenesis of pregnancy complications such as preeclampsia and may offer novel therapeutic avenues.
The OGT/HCF-1 complex is known to regulate gluconeogenesis by modulating the stability of PGC-1α, a key regulator of mitochondrial biogenesis and energy metabolism131. Disruption of placental OGT has been associated with impaired mitochondrial function and reduced offspring body weights, highlighting the importance of OGT in regulating metabolic processes during development132. Interestingly, dysregulation of PGC-1α has been implicated in abnormal glucose metabolism and impaired placental angiogenesis in conditions such as gestational diabetes mellitus (GDM) and obesity133,134,135. BAP1 interaction with the OGT/HCF-1 complex and PGC-1α, suggests a potential role for BAP1 in regulating cellular metabolism9,115,131,136. Given the importance of metabolic regulation in placental development, it is plausible that BAP1 may influence trophoblast metabolism through its interactions with these proteins. Investigating the impact of BAP1 on metabolic pathways in trophoblast cells may provide insights into the pathogenesis of pregnancy complications.
Furthermore, BAP1 has been implicated in the regulation of LKB1, a key regulator of glucose and lipid metabolism137. Loss of LKB1 results in defective placental development and midgestation lethality in mice, highlighting its essential role in trophoblast function138. BAP1-mediated stabilization of LKB1 suggests a potential link between BAP1 expression and trophoblast metabolism, warranting further investigation into the molecular mechanisms underlying this interaction.
Additionally, the expression of argininosuccinate synthase (ASS1), a key enzyme involved in nitric oxide production, has been implicated in placental development and pregnancy outcomes139. ASS1 appears to localise in CTBs, whereas EVTs display a lack of ASS1 during the early stages of placental development140. These findings suggest that ASS1 expression within CTBs might be linked to proliferation maintenance, whereas the absence of ASS1 could potentially contribute to the differentiation of CTB into EVTs during the first trimester of pregnancy140. Recently, it has been observed that BAP1 loss leads to ASS1 overexpression in epithelioid mesothelioma, raising questions about the potential interplay between BAP1 and ASS1 in trophoblast cells141. Exploring the relationship between BAP1 expression and ASS1 levels in the placenta may provide valuable insights into the metabolic regulation of placental development.
In summary, unraveling the connections between BAP1 expression and cellular metabolism offers a promising avenue for understanding the molecular mechanisms underlying embryonic development and pregnancy complications. Investigating the role of BAP1 in regulating metabolic pathways in trophoblast cells may reveal novel therapeutic targets for managing pregnancy-related disorders and improving maternal and fetal health.
BAP1 regulation of cellular adaptations to hypoxia
Hypoxia-inducible factor 1-alpha (HIF-1α) and Hypoxia-inducible factor 2-alpha (HIF-2α) serve as pivotal transcriptional regulators in response to oxygen fluctuations, orchestrating cellular and developmental adaptations to hypoxic conditions142,143. In the context of tumorigenesis, HIF-1α activation promotes cancer cell survival and metastasis by enabling adaptation to low-oxygen environments144. Similarly, HIF-2α promotes tumor development and growth in response to hypoxic conditions143. During early pregnancy, oxygen levels play a crucial role in modulating trophoblast differentiation, with HIF-dependent pathways governing cell adhesion and morphology-related processes145.
Notably, aberrant expression of HIF-1α and HIF-2α in trophoblasts has been implicated in placental dysfunction and the development of pregnancy complications such as preeclampsia146,147. Prolonged elevation of HIF-1α levels in trophoblasts has been associated with impaired fetal growth and alterations in placental differentiation, mirroring key features of preeclampsia in animal models146. Consistent with these findings, elevated placental HIF-1α levels have been identified as potential biomarkers for predicting preeclampsia in clinical settings148,149,150. Analogously, HIF-2α protein expression is selectively increased in preeclamptic placentas compared to normal term placentas, primarily located in the nuclei of syncytiotrophoblasts and the fetoplacental vascular endothelium147.
Recent advances have shed light on the role of BAP1 in stabilizing HIF-1α through deubiquitination under hypoxic conditions151. Mutations in BAP1 that disrupt its function result in decreased levels of HIF-1α, implicating BAP1 as a key regulator of HIF-1α stability in response to low oxygen levels151. Given the dynamic nature of oxygen levels during pregnancy, particularly during early placentation, BAP1-mediated regulation of HIF-1α may play a critical role in modulating trophoblast function and placental development.
The interplay between BAP1 and HIF-1α in response to oxygen fluctuations highlights a novel mechanism underlying placental development and suggests potential implications for understanding and managing pregnancy complications.
Insights from clear cell renal cell carcinoma (ccRCC) studies suggest a role for BAP1 and HIF-2α in activating interferon-stimulated gene factor 3 (ISGF3), a tumor suppressor in ccRCC152. Due to the similar HIF-1α and HIF-2α aberrant levels in preeclamptic placentas, it is tempting to speculate that BAP1 may also regulate HIF-2α during low oxygen conditions in early placentation.
Further investigation into the regulatory pathways involving BAP1, HIF-1α and HIF-2α may uncover novel therapeutic strategies for mitigating pregnancy complications such as preeclampsia and improving maternal and fetal health.
Concluding remarks
In conclusion, the multifaceted roles of the tumor suppressor BAP1 transcend tumorigenesis, extending into vital functions in embryo development and placentation. While much research has centered on BAP1’s implications in cancer progression, recent insights have illuminated its significance in early placentation, revealing shared mechanisms between cancer and embryo development. This convergence underscores the potential for harnessing cancer research to deepen our understanding of placental biology and pregnancy complications.
One notable area of overlap lies in the epithelial-to-mesenchymal transition (EMT), a process pivotal for cancer metastasis and trophoblast invasion during placental development. Understanding how BAP1 influences EMT in both contexts may unveil novel insights into trophoblast invasion and its dysregulation in pregnancy complications. Moreover, BAP1’s modulation of cell differentiation through HDACs and EZH2 expression levels holds promise for understanding and potentially addressing pregnancy complications characterized by aberrant epigenetic modifications. The intricate interplay between BAP1 and cell death components, as well as regulators of cell proliferation and metabolism, further underscores the potential for leveraging insights from cancer research to elucidate mechanisms underlying placental development and pregnancy complications. Additionally, BAP1’s role in stabilizing HIF-1α under hypoxic conditions underscores its importance in orchestrating cellular responses to oxygen fluctuations, with implications for both tumorigenesis and placental development. Further exploration of the relationship between BAP1 and the HIF proteins may offer new avenues for understanding and managing pregnancy complications associated with hypoxia.
In summary, the lessons gleaned from studying BAP1 in the context of tumorigenesis provide valuable insights into placental biology and pregnancy complications. By embracing interdisciplinary approaches and fostering collaborative efforts, future research endeavors hold the promise of unraveling the complexities of BAP1’s roles in embryogenesis, placentation and tumorigenesis, thereby paving the way for innovative therapeutic strategies and advancements in maternal and fetal health.
References
Hough, S. R., Laslett, A. L., Grimmond, S. B., Kolle, G. & Pera, M. F. A continuum of cell states spans pluripotency and lineage commitment in human embryonic stem cells. PLoS One 4, e7708 (2009).
Nichols, J. & Smith, A. Pluripotency in the embryo and in culture. Cold Spring Harb. Perspect. Biol. 4, a008128–a008128 (2012).
Negrini, S., Gorgoulis, V. G. & Halazonetis, T. D. Genomic instability — an evolving hallmark of cancer. Nat. Rev. Mol. Cell Biol. 11, 220–228 (2010).
Marusyk, A., Almendro, V. & Polyak, K. Intra-tumour heterogeneity: A looking glass for cancer? Nat. Rev. Cancer 12, 323–334 (2012).
Knöfler, M. et al. Human placenta and trophoblast development: Key molecular mechanisms and model systems. Cell. Mol. Life Sci. 76, 3479–3496 (2019).
Ferretti, C., Bruni, L., Dangles-Marie, V., Pecking, A. P. & Bellet, D. Molecular circuits shared by placental and cancer cells, and their implications in the proliferative, invasive and migratory capacities of trophoblasts. Hum. Reprod. Update 13, 121–141 (2007).
Costanzo, V., Bardelli, A., Siena, S. & Abrignani, S. Exploring the links between cancer and placenta development. Open Biol. 8, 180081 (2018).
Pang, H. et al. Three categories of similarities between the placenta and cancer that can aid cancer treatment: Cells, the microenvironment, and metabolites. Front. Oncol. 12, 977618 (2022).
Kuznetsov, J. N. et al. BAP1 regulates epigenetic switch from pluripotency to differentiation in developmental lineages giving rise to BAP1-mutant cancers. Sci. Adv. 5, 1–11 (2019).
Perez-Garcia, V. et al. BAP1/ASXL complex modulation regulates epithelial-mesenchymal transition during trophoblast differentiation and invasion. Elife 10, e63254 (2021).
Perez-Garcia, V. et al. Placentation defects are highly prevalent in embryonic lethal mouse mutants. Nature 555, 463–468 (2018).
Masclef, L. et al. Roles and mechanisms of BAP1 deubiquitinase in tumor suppression. Cell Death Differ 28, 606–625 (2021).
Kwon, J., Lee, D. & Lee, S. A. BAP1 as a guardian of genome stability: implications in human cancer. Exp. Mol. Med. 55, 745–754 (2023).
Masoomian, B., Shields, J. A. & Shields, C. L. Overview of BAP1 cancer predisposition syndrome and the relationship to uveal melanoma. J. Curr. Ophthalmol. 30, 102–109 (2018).
Wang, A., Papneja, A., Hyrcza, M., Al-Habeeb, A. & Ghazarian, D. BAP1: Gene of the month. J. Clin. Pathol. 69, 750–753 (2016).
Hemberger, M., Hanna, C. W. & Dean, W. Mechanisms of early placental development in mouse and humans. Nat. Rev. Genet. 21, 27–43 (2020).
Maltepe, E. & Fisher, S. J. Placenta: The forgotten organ. Annu. Rev. Cell Dev. Biol. 31, 523–552 (2015).
Ozguldez, H. O. et al. Polarity inversion reorganizes the stem cell compartment of the trophoblast lineage. Cell Rep. 42, 112313 (2023).
Woods, L., Perez-Garcia, V. & Hemberger, M. Regulation of placental development and its impact on fetal growth—new insights from mouse models. Front. Endocrinol. (Lausanne). 9, 1–18 (2018).
Álvarez-Sánchez, A. et al. The GPI-anchor biosynthesis pathway is critical for syncytiotrophoblast differentiation and placental development. Cell. Mol. Life Sci. 81, 246 (2024).
Turco, M. Y. & Moffett, A. Development of the human placenta. Dev 146, 1–14 (2019).
Velicky, P., Knöfler, M. & Pollheimer, J. Function and control of human invasive trophoblast subtypes: Intrinsic vs. maternal control. Cell Adh. Migr. 10, 154–162 (2016).
Knöfler, M. & Pollheimer, J. Human placental trophoblast invasion and differentiation: A particular focus on Wnt signaling. Front. Genet. 4, 190 (2013).
Pollheimer, J., Vondra, S., Baltayeva, J., Beristain, A. G. & Knöfler, M. Regulation of Placental Extravillous Trophoblasts by the Maternal Uterine Environment. Front. Immunol. 9, 2597 (2018).
Albrecht, E. D. & Pepe, G. J. Regulation of uterine spiral artery remodeling: A review. Reprod. Sci. 27, 1932–1942 (2020).
Meakin, C., Barrett, E. S. & Aleksunes, L. M. Extravillous trophoblast migration and invasion: Impact of environmental chemicals and pharmaceuticals. Reprod. Toxicol. 107, 60–68 (2022).
Kutscher, L. M. & Shaham, S. Non-apoptotic cell death in animal development. Cell Death Differ 24, 1326–1336 (2017).
Ishizaki, Y., Cheng, L., Mudge, A. W. & Raff, M. C. Programmed cell death by default in embryonic cells, fibroblasts, and cancer cells. Mol. Biol. Cell 6, 1443–1458 (1995).
Gong, J.-S. & Kim, G. J. The role of autophagy in the placenta as a regulator of cell death. Clin. Exp. Reprod. Med. 41, 97 (2014).
Hoijman, E. et al. Cooperative epithelial phagocytosis enables error correction in the early embryo. Nature 590, 618–623 (2021).
Shortt, J. & Johnstone, R. W. Oncogenes in cell survival and cell death. Cold Spring Harb. Perspect. Biol. 4, a009829–a009829 (2012).
Wong, R. S. Y. Apoptosis in cancer: From pathogenesis to treatment. J. Exp. Clin. Cancer Res. 30, 87 (2011).
Folmes, C. D. L. & Terzic, A. Metabolic determinants of embryonic development and stem cell fate. Reprod. Fertil. Dev. 27, 82 (2015).
de Lima, C. B. et al. The dynamics between in vitro culture and metabolism: embryonic adaptation to environmental changes. Sci. Rep. 10, 15672 (2020).
Vergaro, P. & Sturmey, R. G. Embryo metabolism and what does the embryo need? In Manual of Embryo Culture in Human Assisted Reproduction 30–41 (Cambridge University Press, 2021).
Romero-Garcia, S., Lopez-Gonzalez, J. S., B´ez-Viveros, J. L., Aguilar-Cazares, D. & Prado-Garcia, H. Tumor cell metabolism. Cancer Biol. Ther. 12, 939–948 (2011).
Martínez-Reyes, I. & Chandel, N. S. Cancer metabolism: Looking forward. Nat. Rev. Cancer 21, 669–680 (2021).
Bergers, G. & Fendt, S.-M. The metabolism of cancer cells during metastasis. Nat. Rev. Cancer 21, 162–180 (2021).
Aye, I. L. M. H., Aiken, C. E., Charnock-Jones, D. S. & Smith, G. C. S. Placental energy metabolism in health and disease—significance of development and implications for preeclampsia. Am. J. Obstet. Gynecol. 226, S928–S944 (2022).
Díaz, P., Powell, T. L. & Jansson, T. The role of placental nutrient sensing in maternal-fetal resource allocation. Biol. Reprod. 91, 82 (2014).
Muz, B., de la Puente, P., Azab, F. & Azab, A. K. The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl., N. Z.) 3, 83–92 (2015).
Skuli, N. et al. Endothelial deletion of hypoxia-inducible factor–2α (HIF-2α) alters vascular function and tumor angiogenesis. Blood 114, 469–477 (2009).
Tuuli, M. G., Longtine, M. S. & Nelson, D. M. Review: Oxygen and trophoblast biology – A source of controversy. Placenta 32, S109–S118 (2011).
Kim, D. et al. Epithelial mesenchymal transition in embryonic development, tissue repair and cancer: A comprehensive overview. J. Clin. Med. 7, 1 (2017).
Lala, P. K., Nandi, P., Hadi, A. & Halari, C. A crossroad between placental and tumor biology: What have we learnt? Placenta 116, 12–30 (2021).
Davies, J. E. et al. Epithelial-mesenchymal transition during extravillous trophoblast differentiation. Cell Adh. Migr 10, 310–321 (2016).
Jena, S. K., Das, S., Chakraborty, S. & Ain, R. Molecular determinants of epithelial mesenchymal transition in mouse placenta and trophoblast stem cell. Sci. Rep. 13, 10978 (2023).
Kaur, B. Pathology of gestational trophoblastic disease (GTD). Best. Pract. Res. Clin. Obstet. Gynaecol. 74, 3–28 (2021).
Seckl, M. J., Sebire, N. J. & Berkowitz, R. S. Gestational trophoblastic disease. Lancet 376, 717–729 (2010).
Hecht, J. L. et al. Classification and reporting guidelines for the pathology diagnosis of placenta accreta spectrum (PAS) disorders: recommendations from an expert panel. Mod. Pathol. 33, 2382–2396 (2020).
Brosens, I., Pijnenborg, R., Vercruysse, L. & Romero, R. The “Great Obstetrical Syndromes” are associated with disorders of deep placentation. Am. J. Obstet. Gynecol. 204, 193–201 (2011).
Carter, A. M., Enders, A. C. & Pijnenborg, R. The role of invasive trophoblast in implantation and placentation of primates. Philos. Trans. R. Soc. B Biol. Sci. 370, 20140070 (2015).
Brosens, I., Puttemans, P. & Benagiano, G. Placental bed research: I. The placental bed: from spiral arteries remodeling to the great obstetrical syndromes. Am. J. Obstet. Gynecol. 221, 437–456 (2019).
Kobrinski, D. A., Yang, H. & Kittaneh, M. BAP1: Role in carcinogenesis and clinical implications. Transl. Lung Cancer Res 9, S60–S66 (2020).
Sahtoe, D. D., van Dijk, W. J., Ekkebus, R., Ovaa, H. & Sixma, T. K. BAP1/ASXL1 recruitment and activation for H2A deubiquitination. Nat. Commun. 7, 10292 (2016).
Hauri, S. et al. A high-density map for navigating the human polycomb complexome. Cell Rep. 17, 583–595 (2016).
Kolovos, P. et al. PR-DUB maintains the expression of critical genes through FOXK1/2- and ASXL1/2/3-dependent recruitment to chromatin and H2AK119ub1 deubiquitination. Genome Res 30, 1119–1130 (2020).
Szczepanski, A. P. & Wang, L. Emerging multifaceted roles of BAP1 complexes in biological processes. Cell Death Discov. 7, 20 (2021).
Tamburri, S. et al. Histone H2AK119 mono-ubiquitination is essential for polycomb-mediated transcriptional repression. Mol. Cell 77, 840–856.e5 (2020).
Conway, E. et al. BAP1 enhances Polycomb repression by counteracting widespread H2AK119ub1 deposition and chromatin condensation. Mol. Cell 81, 3526–3541.e8 (2021).
Parreno, V., Martinez, A.-M. & Cavalli, G. Mechanisms of Polycomb group protein function in cancer. Cell Res 32, 231–253 (2022).
Carbone, M. et al. Biological mechanisms and clinical signifi cance of BAP1 mutations in human cancer. Cancer Discov 10, 1103–1120 (2020).
Matatall, K. A. et al. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer 13, 371 (2013).
Caporali, S., Butera, A. & Amelio, I. BAP1 in cancer: Epigenetic stability and genome integrity. Discov. Oncol. 13, 117 (2022).
Uner, O. E., See, T. R. O., Szalai, E., Grossniklaus, H. E. & Stålhammar, G. Estimation of the timing of BAP1 mutation in uveal melanoma progression. Sci. Rep. 11, 8923 (2021).
Singh, A. et al. BAP1 loss induces mitotic defects in mesothelioma cells through BRCA1-dependent and independent mechanisms. Oncogene 42, 572–585 (2023).
Huang, R. & Zhou, P.-K. DNA damage repair: historical perspectives, mechanistic pathways and clinical translation for targeted cancer therapy. Signal Transduct. Target. Ther. 6, 254 (2021).
Han, A., Purwin, T. J. & Aplin, A. E. Roles of the BAP1 tumor suppressor in cell metabolism. Cancer Res 81, 2807–2814 (2021).
Qin, J. et al. BAP1 promotes breast cancer cell proliferation and metastasis by deubiquitinating KLF5. Nat. Commun. 6, 8471 (2015).
Luo, X. et al. BAP1 deletion abrogates growth and metastasis of murine cutaneous melanoma. Melanoma Res 31, 119–129 (2021).
Tsuboyama, N. et al. Therapeutic targeting of BAP1/ASXL3 sub-complex in ASCL1-dependent small cell lung cancer. Oncogene 41, 2152–2162 (2022).
Balasubramani, A. et al. Cancer-associated ASXL1 mutations may act as gain-of-function mutations of the ASXL1-BAP1 complex. Nat. Commun. 6, 1–15 (2015).
Kang, M. et al. Targeting BAP1 with small compound inhibitor for colon cancer treatment. Sci. Rep. 13, 2264 (2023).
Asada, S. et al. Mutant ASXL1 cooperates with BAP1 to promote myeloid leukaemogenesis. Nat. Commun. 9, 2733 (2018).
Campagne, A. et al. BAP1 complex promotes transcription by opposing PRC1-mediated H2A ubiquitylation. Nat. Commun. 10, 348 (2019).
Scheuermann, J. C. et al. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 465, 243–247 (2010).
Yang, F. et al. Deubiquitinating enzyme BAP1 is involved in the formation and maintenance of the diapause embryos of Artemia. Cell Stress Chaperones 17, 577–587 (2012).
Dey, A. et al. Loss of the tumor suppressor BAP1 causes myeloid transformation. Science 337, 1541–1546 (2016).
Mohun, T. et al. Deciphering the mechanisms of developmental disorders (DMDD): A new programme for phenotyping embryonic lethal mice. Dis. models mechanisms 6, 562–566 (2013).
Mei, X., Hu, Y., Pan, Q. & Li, H. BAP1 overexpression inhibited the PI3K-AKT-mTOR pathway via deubiquitinating PTEN and suppressing trophoblastic EMT. J. Hard Tissue Biol. 32, 21–28 (2023).
Zhou, J. J. et al. Histone deacetylase 1 maintains lineage integrity through histone acetylome refinement during early embryogenesis. Elife 12, e79380 (2023).
Milazzo, G. et al. Histone deacetylases (HDACs): Evolution, specificity, role in transcriptional complexes, and pharmacological actionability. Genes (Basel) 11, 556 (2020).
Ropero, S. & Esteller, M. The role of histone deacetylases (HDACs) in human cancer. Mol. Oncol. 1, 19–25 (2007).
Du, J., Ji, Q., Dong, L., Meng, Y. & Xin, G. HDAC4 knockdown induces preeclampsia cell autophagy and apoptosis by miR-29b. Reprod. Sci. 28, 334–342 (2021).
Wang, P. et al. Dysregulation of histone deacetylases inhibits trophoblast growth during early placental development partially through TFEB-dependent autophagy-lysosomal pathway. Int. J. Mol. Sci. 24, 11899 (2023).
Landreville, S. et al. Histone deacetylase inhibitors induce growth arrest and differentiation in uveal melanoma. Clin. Cancer Res. 18, 408–416 (2012).
Sacco, J. J. et al. Loss of the deubiquitylase BAP1 alters class I histone deacetylase expression and sensitivity of mesothelioma cells to HDAC inhibitors. Oncotarget 6, 13757–13771 (2015).
Novelli, F. et al. BAP1 forms a trimer with HMGB1 and HDAC1 that modulates gene × environment interaction with asbestos. Proc. Natl. Acad. Sci. USA. 118, e2111946118 (2021).
Mu, W., Starmer, J., Yee, D. & Magnuson, T. EZH2 variants differentially regulate polycomb repressive complex 2 in histone methylation and cell differentiation. Epigenetics Chromatin 11, 71 (2018).
Huang, X.-J. et al. EZH2 is essential for development of mouse preimplantation embryos. Reprod. Fertil. Dev. 26, 1166 (2014).
Duan, R., Du, W. & Guo, W. EZH2: a novel target for cancer treatment. J. Hematol. Oncol. 13, 104 (2020).
Qian, X. & Zhang, Y. EZH2 enhances proliferation and migration of trophoblast cell lines by blocking GADD45A-mediated p38/MAPK signaling pathway. Bioengineered 13, 12583–12597 (2022).
Lv, S. et al. The attenuation of trophoblast invasion caused by the downregulation of EZH2 is involved in the pathogenesis of human recurrent miscarriage. Mol. Ther. Nucleic Acids 14, 377–387 (2019).
LaFave, L. M. et al. BAP1 loss results in EZH2-dependent transformation in myelodysplastic syndromes. Blood 126, 713–713 (2015).
Sun, C. et al. EZH2 Expression is increased in BAP1-mutant renal clear cell carcinoma and is related to poor prognosis. J. Cancer 9, 3787–3796 (2018).
Dai, F. et al. BAP1 inhibits the ER stress gene regulatory network and modulates metabolic stress response. Proc. Natl. Acad. Sci. 114, 3192–3197 (2017).
Chen, C. et al. ATF3 inhibits the tumorigenesis and progression of hepatocellular carcinoma cells via upregulation of CYR61 expression. J. Exp. Clin. Cancer Res. 37, 263 (2018).
Hackl, C. et al. Activating transcription factor-3 (ATF3) functions as a tumor suppressor in colon cancer and is up-regulated upon heat-shock protein 90 (Hsp90) inhibition. BMC Cancer 10, 668 (2010).
Wang, M., Zhang, L., Huang, X. & Sun, Q. Ligustrazine promotes hypoxia/reoxygenation-treated trophoblast cell proliferation and migration by regulating the microRNA-27a-3p/ATF3 axis. Arch. Biochem. Biophys. 737, 109522 (2023).
Kaitu’u-Lino, T. J. et al. Activating transcription factor 3 is reduced in preeclamptic placentas and negatively regulates sFlt-1 (Soluble fms-Like Tyrosine Kinase 1), soluble endoglin, and proinflammatory cytokines in placenta. Hypertension 70, 1014–1024 (2017).
Herse, F. et al. Dysregulation of the circulating and tissue-based renin-angiotensin system in preeclampsia. Hypertension 49, 604–611 (2007).
Castro, K. R. et al. Serum from preeclamptic women triggers endoplasmic reticulum stress pathway and expression of angiogenic factors in trophoblast cells. Front. Physiol. 12, 799653 (2021).
Pasha, M. et al. The effect of tauroursodeoxycholic Acid (TUDCA) treatment on placental endoplasmic reticulum (ER) stress in a rat model of advanced maternal age. PLoS One 18, e0282442 (2023).
Zhao, Y. et al. Gestational exposure to BDE-209 induces placental injury via the endoplasmic reticulum stress-mediated PERK/ATF4/CHOP signaling pathway. Ecotoxicol. Environ. Saf. 233, 113307 (2022).
Bauer, T. M. & Murphy, E. Role of mitochondrial calcium and the permeability transition pore in regulating cell death. Circ. Res. 126, 280–293 (2020).
Bononi, A. et al. BAP1 regulates IP3R3-mediated Ca2+ flux to mitochondria suppressing cell transformation. Nature 546, 549–553 (2017).
Uchida, K., Nakazawa, M., Yamagishi, C., Mikoshiba, K. & Yamagishi, H. Type 1 and 3 inositol trisphosphate receptors are required for extra-embryonic vascular development. Dev. Biol. 418, 89–97 (2016).
Li, Y., Chen, D., Xu, J., Wang, X. & Zhou, F. The spatial expression of mTORC2-AKT-IP3R signal pathway in mitochondrial combination of endoplasmic reticulum of maternal fetal interface trophoblast in intrahepatic cholestasis of pregnancy. J. Perinat. Med. 51, 1032–1039 (2023).
Lei, P., Bai, T. & Sun, Y. Mechanisms of ferroptosis and relations with regulated cell death: A review. Front. Physiol. 10, 139 (2019).
Zhang, Y. et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat. Cell Biol. 20, 1181–1192 (2018).
El-Khalik, S. R. A., Ibrahim, R. R., Ghafar, M. T. A., Shatat, D. & El-Deeb, O. S. Novel insights into the SLC7A11-mediated ferroptosis signaling pathways in preeclampsia patients: identifying pannexin 1 and toll-like receptor 4 as innovative prospective diagnostic biomarkers. J. Assist. Reprod. Genet. 39, 1115–1124 (2022).
He, M. et al. Intrinsic apoptosis shapes the tumor spectrum linked to inactivation of the deubiquitinase BAP1. Science. 364, 283–285 (2019).
Soleymanlou, N. et al. Hypoxic switch in mitochondrial myeloid cell leukemia factor-1/Mtd apoptotic rheostat contributes to human trophoblast cell death in preeclampsia. Am. J. Pathol. 171, 496–506 (2007).
Sakuragi, N. et al. Bcl-2 expression and apoptosis in human trophoblast. Placenta 18, 63–74 (1997).
Machida, Y. J., Machida, Y., Vashisht, A. A., Wohlschlegel, J. A. & Dutta, A. The deubiquitinating enzyme BAP1 regulates cell growth via interaction with HCF-1. J. Biol. Chem. 284, 34179–34188 (2009).
Daou, S. et al. The BAP1/ASXL2 histone H2A deubiquitinase complex regulates cell proliferation and is disrupted in cancer. J. Biol. Chem. 290, 28643–28663 (2015).
Pan, H. et al. BAP1 regulates cell cycle progression through E2F1 target genes and mediates transcriptional silencing via H2A monoubiquitination in uveal melanoma cells. Int. J. Biochem. Cell Biol. 60, 176–184 (2015).
Yu, H. et al. The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol. Cell. Biol. 30, 5071–5085 (2010).
Wang, Y. et al. YY1/ITGA3 pathway may affect trophoblastic cells migration and invasion ability. J. Reprod. Immunol. 153, 103666 (2022).
Tian, F. et al. The YY1/MMP2 axis promotes trophoblast invasion at the maternal–fetal interface. J. Pathol. 239, 36–47 (2016).
Li, H., Yu, L., Ding, Y., Nie, Y. & Yang, M. Yin Yang 1 impacts upon preeclampsia by regulating Treg/TH17 cells and PI3K/AKT pathway. J. Immunotoxicol. 20, 2228420 (2023).
Chen, X.-X. et al. BAP1 acts as a tumor suppressor in intrahepatic cholangiocarcinoma by modulating the ERK1/2 and JNK/c-Jun pathways. Cell Death Dis. 9, 1036 (2018).
Fang, L. et al. TGF-β1 inhibits human trophoblast cell invasion by upregulating kisspeptin expression through ERK1/2 but not SMAD signaling pathway. Reprod. Biol. Endocrinol. 20, 22 (2022).
D’Oria, R. et al. PKB/Akt and MAPK/ERK phosphorylation is highly induced by inositols: Novel potential insights in endothelial dysfunction in preeclampsia. Pregnancy Hypertens. 10, 107–112 (2017).
Deng, R. et al. BAP1 suppresses prostate cancer progression by deubiquitinating and stabilizing PTEN. Mol. Oncol. 15, 279–298 (2021).
Xue, P. et al. Up-regulation of PTEN via LPS/AP-1/NF-κB pathway inhibits trophoblast invasion contributing to preeclampsia. Mol. Immunol. 118, 182–190 (2020).
Tian, S. et al. Overexpression of PTEN regulated by miR-19b and miR-494 in the villous of recurrent spontaneous abortion patients. J. Reprod. Immunol. 140, 103133 (2020).
Jia, X. et al. BAP1 antagonizes WWP1-mediated transcription factor KLF5 ubiquitination and inhibits autophagy to promote melanoma progression. Exp. Cell Res. 402, 112506 (2021).
Lee, S. H., Liu, X., Jimenez-Morales, D. & Rinaudo, P. F. Murine blastocysts generated by in vitro fertilization show increased Warburg metabolism and altered lactate production. Elife 11, e79153 (2022).
Dang, C. V. Links between metabolism and cancer. Genes Dev 26, 877–890 (2012).
Ruan, H.-B. et al. O-GlcNAc transferase/host cell factor C1 complex regulates gluconeogenesis by modulating PGC-1α stability. Cell Metab. 16, 226–237 (2012).
Moore, M., Avula, N., Jo, S., Beetch, M. & Alejandro, E. U. Disruption of O-linked N-acetylglucosamine signaling in placenta induces insulin sensitivity in female offspring. Int. J. Mol. Sci. 22, 6918 (2021).
Peng, H. et al. Maternal obesity inhibits placental angiogenesis by down-regulating the SIRT1/PGC-1α pathway. Ann. Transl. Med. 10, 446–446 (2022).
Keleher, M. R. et al. Placental insulin/IGF-1 Signaling, PGC-1α, and inflammatory pathways are associated with metabolic outcomes at 4–6 years of age: The ECHO Healthy start cohort. Diabetes 70, 745–751 (2021).
Wang, L. et al. Altered expression of PGC-1 α and PDX1 and their methylation status are associated with fetal glucose metabolism in gestational diabetes mellitus. Biochem. Biophys. Res. Commun. 501, 300–306 (2018).
Moon, S., Lee, Y.-K., Lee, S.-W. & Um, S.-J. Suppressive role of OGT-mediated O-GlcNAcylation of BAP1 in retinoic acid signaling. Biochem. Biophys. Res. Commun. 492, 89–95 (2017).
Yang, C. et al. BAP1 regulates AMPK-mTOR signalling pathway through deubiquitinating and stabilizing tumoursuppressor LKB1. Biochem. Biophys. Res. Commun. 529, 1025–1032 (2020).
Ylikorkala, A. et al. Vascular abnormalities and deregulation of VEGF in Lkb1 -deficient mice. Science. 293, 1323–1326 (2001).
Sladek, S. M., Magness, R. R. & Conrad, K. P. Nitric oxide and pregnancy. Am. J. Physiol. Integr. Comp. Physiol. 272, R441–R463 (1997).
Fantone, S. et al. Downregulation of argininosuccinate synthase 1 (ASS1) is associated with hypoxia in placental development. Hum. Cell 36, 1190–1198 (2023).
Barnett, S. E. et al. BAP1 loss is associated with higher ASS1 expression in epithelioid mesothelioma: Implications for therapeutic stratification. Mol. Cancer Res. 21, 411–427 (2023).
Lee, J.-W., Bae, S.-H., Jeong, J.-W., Kim, S.-H. & Kim, K.-W. Hypoxia-inducible factor (HIF-1)α: Its protein stability and biological functions. Exp. Mol. Med. 36, 1–12 (2004).
Gordan, J. D., Bertout, J. A., Hu, C.-J., Diehl, J. A. & Simon, M. C. HIF-2α promotes hypoxic cell proliferation by enhancing c-Myc transcriptional activity. Cancer Cell 11, 335–347 (2007).
Jun, J. C., Rathore, A., Younas, H., Gilkes, D. & Polotsky, V. Y. Hypoxia-inducible factors and cancer. Curr. Sleep. Med. Rep. 3, 1–10 (2017).
Wakeland, A. K. et al. Hypoxia directs human extravillous trophoblast differentiation in a hypoxia-inducible factordependent manner. Am. J. Pathol. 187, 767–780 (2017).
Albers, R. E. et al. Trophoblast-specific expression of Hif-1α results in preeclampsia-like symptoms and fetal growth restriction. Sci. Rep. 9, 2742 (2019).
Rajakumar, A. et al. Selective overexpression of the hypoxia-inducible transcription factor, HIF-2α, in placentas from women with preeclampsia1. Biol. Reprod. 64, 499–506 (2001).
Tianthong, W. & Phupong, V. Serum hypoxia-inducible factor-1α and uterine artery Doppler ultrasound during the first trimester for prediction of preeclampsia. Sci. Rep. 11, 6674 (2021).
Tal, R. et al. Effects of hypoxia-inducible factor-1α overexpression in pregnant mice. Am. J. Pathol. 177, 2950–2962 (2010).
Rath, G., Aggarwal, R., Jawanjal, P., Tripathi, R. & Batra, A. HIF-1 alpha and placental growth factor in pregnancies complicated with preeclampsia: A qualitative and quantitative analysis. J. Clin. Lab. Anal. 30, 75–83 (2016).
Bononi, A. et al. BAP1 is a novel regulator of HIF-1α. Proc. Natl. Acad. Sci. 120 https://doi.org/10.1073/pnas.2217840120 (2023).
Langbein, L. E. et al. BAP1 maintains HIF-dependent interferon beta induction to suppress tumor growth in clear cell renal cell carcinoma. Cancer Lett. 547, 215885 (2022).
Acknowledgements
This work was supported by the Fundación Ramón Areces and the grants RYC2019-026956-I and PID2020-114459RA-I00, funded by the MICIU/AEI/10.13039/501100011033, and by ESF investing in your future.
Author information
Authors and Affiliations
Contributions
P.D.-B., and V.P.-G., contributed to the conceptualization and writing of the manuscript. PD-B assembled the figures. All authors reviewed and/or edited the article before submission.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Xuan Shao and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Primary Handling Editors: Kaliya Georgieva. [A peer review file is available.]
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Doria-Borrell, P., Pérez-García, V. Understanding the intersection between placental development and cancer: Lessons from the tumor suppressor BAP1. Commun Biol 7, 1053 (2024). https://doi.org/10.1038/s42003-024-06689-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-024-06689-2
- Springer Nature Limited