
Abstract
Despite decades of genetic studies on late-onset Alzheimer’s disease, the underlying molecular mechanisms remain unclear. To better comprehend its complex etiology, we use an integrative approach to build robust predictive (causal) network models using two large human multi-omics datasets. We delineate bulk-tissue gene expression into single cell-type gene expression and integrate clinical and pathologic traits, single nucleotide variation, and deconvoluted gene expression for the construction of cell type-specific predictive network models. Here, we focus on neuron-specific network models and prioritize 19 predicted key drivers modulating Alzheimer’s pathology, which we then validate by knockdown in human induced pluripotent stem cell-derived neurons. We find that neuronal knockdown of 10 of the 19 targets significantly modulates levels of amyloid-beta and/or phosphorylated tau peptides, most notably JMJD6. We also confirm our network structure by RNA sequencing in the neurons following knockdown of each of the 10 targets, which additionally predicts that they are upstream regulators of REST and VGF. Our work thus identifies robust neuronal key drivers of the Alzheimer’s-associated network state which may represent therapeutic targets with relevance to both amyloid and tau pathology in Alzheimer’s disease.
Similar content being viewed by others


Introduction
Late-onset Alzheimer’s disease (LOAD) is the leading cause of dementia, which is characterized by progressive impairments in memory, cognition, and executive functions, along with behavioral and psychiatric symptoms including agitation, aggression, mood disorders, and psychosis1. The hallmark features of Alzheimer’s disease (AD) include pathological aggregation of extracellular plaques, composed of amyloid-β (Aβ) peptides, and intracellular neurofibrillary tangles, composed of hyperphosphorylated tau (p-tau) protein2, which lead to neuron death. Genome-wide association studies have implicated over 30 loci associated with AD risk3,4,5,6,7,8,9,10,11,12,13,14,15,16. In previous studies, we and others have shown that LOAD is a complex pathological process involving an interactive network of pathways among multiple cell types in the brain (neurons, microglia, astrocytes, etc.) influenced by genetic variation, aging, and environmental factors17,18,19,20. Implicated pathways include those involved in mitochondrial metabolism, response to unfolded proteins, immune response, phagocytosis, and synaptic transmission21,22,23,24. The complexity of these multi-modal networks highlights the necessity to study networks of molecular interactions by cell type and to identify cell type-specific pathways and key drivers in AD. In this study, we developed a multi-step pipeline using advanced computational systems biology approaches to construct robust data-driven neuron-specific network models of gene regulatory programs in brain regions affected by LOAD. For these analyses, we utilized whole-genome gene expression and whole-genome genotyping data from two independent cohorts in the Accelerating Medicines Partnership—Alzheimer’s Disease (AMP-AD) consortium: the Mayo RNAseq Study (herein MAYO) and the Religious Orders Study and Memory and Aging Project (herein ROSMAP).
We applied a deconvolution method to deconvolve bulk-tissue RNA sequencing (RNAseq) data from post-mortem brain regions and then derive the neuron-specific gene expression signal. Although single-cell RNA sequencing (scRNAseq) studies, including those from the recent Human Cell Atlas endeavor, have greatly advanced our understanding of cellular heterogeneity and the discovery of novel cell populations25,26,27,28,29,30,31,32,33,34,35 as well as spurred developments of various computational analysis tools36, network inference performance using scRNAseq data is still very poor. Due to the high volume of missing gene expression measures and the immaturity of current network methods dealing with these missing data, inferred network models using scRNAseq data yield a large amount of uncertainty37,38, thus limiting the application of scRNAseq data in network inference. Alternatively, deconvolution of bulk-tissue RNAseq data has become increasingly popular in recent years as a complementary solution to the missing values in scRNAseq data39,40,41,42,43,44,45,46,47,48,49,50,51, based on the core assumption that gene expression in bulk-tissue data is equal to the averaged gene expression of each cell type weighted by its relative population in the tissue. Deconvolution methods decompose bulk-tissue RNAseq data into gene expression of individual cell types by using cell type-specific biomarker genes to implicitly estimate relative cell populations in the tissue. After deconvolution, the variances of the deconvoluted gene expression of each cell type become orthogonal to each other and can be analyzed independently52.
To derive neuron-specific gene expression signals from the bulk-tissue RNAseq data from the MAYO and ROSMAP cohorts, we employed the population-specific expression analysis (PSEA) method of deconvolution52. Whereas other popular deconvolution methods such as Cibersort43, dtangle40, DSA39, or NNLS53 can only estimate cell fraction in a bulk-tissue sample, the PSEA method directly estimates cell type-specific residuals from bulk-tissue RNAseq data. Here, we demonstrated the robustness of the PSEA deconvolution method using random selection of neuronal biomarkers derived from scRNAseq studies54,55,56,57,58.
After deconvolution, we applied a cutting-edge systems biology approach23,59,60 to build causal network models of the neuronal component of AD by integrating the deconvoluted neuron-specific RNAseq data with the whole-genome genotype data from the MAYO and ROSMAP datasets. Bayesian networks61 are a long-standing form of statistical network modeling used to reverse-engineer probabilistic causality among variables; with the development of high-throughput sequencing technology, Bayesian networks have been widely used to infer causal gene regulatory networks in different diseases62,63,64,65,66,67. Recent studies have applied Bayesian networks to infer molecular mechanisms and key drivers in Alzheimer’s disease24,68. However, Bayesian networks have substantial limitations with respect to inferring opposite causality given the symmetry of joint probability. Recent work has demonstrated that bottom-up causality inference can accurately distinguish true causality from opposite causality in equivalent classes69. Our group has developed a computational network model, called predictive network modeling, which integrates conventional (top-down) Bayesian networks with bottom-up causality inference in order to address the problem of opposite causality inference in Bayesian network modeling. In this study, we used our causal predictive network pipeline to incorporate multi-scale omics data, including genotypes and transcriptomic profiles, in the deconvoluted neuron-specific residuals of the MAYO and ROSMAP datasets in order to build causal predictive networks separately in both datasets.
We then agnostically identified neuron-specific gene regulatory network models and key genetic drivers predicted to modulate pathological Aβ and hyperphosphorylated tau accumulation in AD. To evaluate and ensure the robustness of our results, we performed the integrative analysis and key driver identification independently in the two cohorts and cross-validated the results at every step of the analysis. In total, we reconstructed 11 causal network models combined across the two separate analyses and predicted a total of 1563 potential key drivers modulating neuronal network states and AD pathology under LOAD.
To experimentally validate our network prediction, we then prioritized 19 targets which replicated across the two cohorts. We used shRNA-mediated knockdown in human induced pluripotent stem cell (iPSC)-derived neurons70,71,72 and measured levels of Aβ38, Aβ40, and Aβ42 as well as tau and p231-tau. Among the 19 targets, we identified 10 targets which affected Aβ (JMJD6, NSF, NUDT2, DCAF12, RBM4, YWHAZ, NDRG4, and STXBP1) and/or tau/p-tau levels (JMJD6, FIBP, and ATP1B1). Finally, to further validate our network models and to provide insights into network connectivity, we measured the whole-genome RNA expression of the iPSC-derived neurons after knocking down each of the 19 targets and compared the differential expression (DE) signature of each target to its downstream structure in the networks. We investigated pathways enriched by the gene knockdown DE signatures to shed light on the molecular mechanisms associated with LOAD, identifying the 10 validated targets as upstream regulators of master regulatory proteins REST and VGF.
Results
An integrative systems biology approach for constructing single cell-type regulatory networks of AD
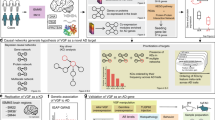
We developed an integrative network analysis pipeline to construct data-driven neuron-specific predictive networks of AD (Fig. 1). The overall strategy for elucidating the single cell-type gene network model depicted in Fig. 1 centers on the objective, data-driven construction of causal network models, which can be directly queried to identify the network components causally associated with AD as well as the master regulators (key drivers) of these AD-associated components. This model also predicts the impact of the key drivers on the biological processes and pathology involved in AD, moving us towards precision molecular models of disease. We previously developed this network reconstruction algorithm, i.e., predictive network, which statistically infers causal relationships between DNA variation, gene expression, protein expression, and clinical features measured in hundreds of individuals23,59,60.

a Discovery datasets include whole-genome genotype and RNAseq data of temporal cortex from the MAYO cohort and dorsolateral prefrontal cortex from the ROSMAP cohort in the AMP-AD consortium. Numbers in circles indicate the total number of subjects in each dataset with quality-controlled matched genotype and RNAseq data used in this study, whereas numbers in the table indicate the number of individuals of each phenotype used in the subsequent DE, co-expression module, and predictive network analyses. b Computational deconvolution of bulk-tissue RNAseq data into 5 single cell-type RNAseq sets per cohort dataset, followed by DE analysis and weighted gene co-expression network analysis in each cohort’s neuron-specific gene expression dataset. c mRNA expression and quantitative trait loci association analysis in each dataset provides a source of systematic perturbation for network reconstructions. d Construction of neuron-specific predictive network models and identification of key drivers (master regulators) from each dataset. e Prioritization of key driver targets from both datasets and experimental validation by shRNA-mediated gene knockdown in human iPSC-derived neurons. Venn diagrams on panels b–e indicate cross-validation at each step of the bioinformatics analyses performed independently in parallel for the MAYO and ROSMAP datasets, resulting in a single set of key driver targets. Statistical tests for each comparison are described in the text where relevant. Parts of this figure utilize graphics from Servier Medical Art (smart.servier.com) provided by Servier, licensed under a Creative Commons Attribution 3.0 Unported License. TCX temporal cortex, DLPFC dorsolateral prefrontal cortex, WGCNA weighted gene co-expression network analysis, eQTL expression quantitative trait locus. See also Supplementary Fig. 1.
The inputs required for our network analysis are the molecular and clinical data generated in the MAYO and ROSMAP populations, as well as first order relationships between these data such as quantitative trait loci (QTLs) associated with the molecular traits. These relationships are input as structure priors to the network construction algorithm as a source of perturbation, boosting the power to infer causal relationships at the network level, as we and others have previously shown21,23,24,60,73,74,75,76,77,78,79,80. To focus on the component of AD that is intrinsically encoded in neurons, we identified the neuron-specific expression component in each cohort by applying the PSEA deconvolution algorithm52 to the MAYO and ROSMAP transcriptomic datasets independently (Supplementary Fig. 1, Step 1). We further focused on the molecular traits associated with AD by identifying DE gene signatures—comprised of several thousands of gene expression traits—between AD and cognitively normal samples for each dataset (Supplementary Fig. 1, Step 2). To identify correlated gene expression traits associated with AD, we constructed gene co-expression networks for each dataset, and from these networks we identified highly interconnected sets of co-regulated genes (modules) that were significantly enriched for AD gene signatures (the significant DE genes) as well as for pathways previously implicated in AD (Supplementary Fig. 1, Step 3). To obtain a final set of genes for input into the causal network construction process for each dataset, we combined genes in the co-expression network modules enriched for AD signatures (Supplementary Fig. 1, Step 5—Module selection) and performed the pathFinder algorithm60 to enrich the seeding gene set by including genes upstream and downstream of this set from a compiled pathway database (Supplementary Fig. 1, Step 5—Seeding expansion). We note that during the expansion of seeding genes, we only include additional genes from the compiled pathway database as nodes in the network, and we discard the disease non-specific edges so as not to bias the process of data-driven network structure learning. The edges of the final extended networks are solely inferred from AD-specific data in each cohort.
With our input set of neuron-centered genes for the AD network constructions defined, we mapped expression-QTLs (eQTLs) for neuron-specific gene expression traits in each dataset to incorporate the eQTLs as structure priors in the network reconstructions, given that they provide a systematic perturbation source that can boost the power to infer causal relationships (Supplementary Fig. 1, Step 4)24,73,74,76,77,78,79,80,81. The input gene set and eQTL data were then processed by an ensemble of causal network inference methods, including Bayesian networks and our recently developed top-down and bottom-up predictive networks23,59,60,69, in order to construct probabilistic causal network models of AD independently in the MAYO and ROSMAP cohorts (Supplementary Fig. 1, Step 6). We next applied a statistical algorithm to detect key driver genes in each given network structure82 and to identify and prioritize master regulators in the AD networks (Supplementary Fig. 1, Step 7). These key drivers derived from the individual networks across datasets were then pooled and prioritized based on ranking scores of impact and robustness (see ‘Methods’), resulting in a final group of 19 top-prioritized key drivers for which we performed functional validation in a human iPSC-derived neuron system. The entire analysis workflow for the independent datasets, resulting in this final group of replicated targets, is illustrated in Fig. 1.
MAYO and ROSMAP study populations and data processing
Our causal network pipeline starts by integrating whole-genome genotyping and RNAseq data generated from patients spanning the complete spectrum of clinical and neuropathological traits in AD. We used patient data from two separate cohorts within the AMP-AD consortium: temporal cortex data from 266 subjects in MAYO83,84,85 and dorsolateral prefrontal cortex data from 612 subjects in ROSMAP22,86,87,88 (Fig. 1a). We processed matched genotype and RNAseq data separately in each dataset (Fig. 1, Supplementary Fig. 1; see ‘Methods’).
Central nervous system (CNS) tissue consists of various cell types, including neurons, glia, and endothelial cells. To discover key network drivers specific to a single cell type in the CNS and study their contribution to AD in that specific cell type, we utilized verified single-cell marker genes to directly deconvolve bulk-tissue gene expression data into cell type-specific gene expression for the five major cell types in the CNS: neurons, microglia, astrocytes, endothelial cells, and oligodendrocytes (see ‘Methods’). In this study, we focused on investigating the role of neuronal cells in AD, as they are the primary cell type affected by AD pathogenesis89,90,91,92,93,94. After normalizing the bulk-tissue RNAseq data, we performed variance partition analysis (VPA)95 to evaluate the contributions of cell type-specific markers as well as demographic, clinical, and technical covariates (such as batch effects) to the gene expression variance before performing any covariate adjustment (Supplementary Fig. 2a, b). The cell type-specific marker genes used for neurons, microglia, astrocytes, endothelial cells, and oligodendrocytes were ENO296, CD6897, GFAP98, CD3499, and OLIG2100,101,102, respectively, as previously published having been obtained directly under the AD condition at the protein level. The VPA results reflect the prominent effect of CNS cell types on the variance of the brain RNAseq data. In the MAYO dataset, the additional covariates used in the VPA included exonic mapping rate, RNA integrity number, sequencing batch, diagnosis, age at death, tissue source, APOE genotype, and sex. In the ROSMAP dataset, we were able to include the same covariates with the exception of tissue source and the addition of age at first AD diagnosis, post-mortem interval, education, and study (ROS or MAP).
We then performed covariate adjustment and deconvolution using the PSEA method52 in each dataset, calculating gene expression residuals using a linear regression model to adjust the normalized bulk-tissue expression data with demographical and technical covariates as well as the cell type-specific markers. Cell type-specific gene expression, including the neuron-specific component, was directly derived by adding the estimated variance of each cell type to the residual (see ‘Methods’), avoiding the need to first estimate the cell population from bulk tissue data, which could induce approximation errors. We then repeated VPA in the neuron-specific residuals of each dataset to demonstrate that our deconvolution and covariate adjustment methods properly capture the neuronal component while removing potential confounds such as batch effect, age, and sex (Supplementary Fig. 2c, d). Finally, to justify the use of single cell type-specific markers for deconvolution by the PSEA method, we performed a set of analyses comparing multiple cell type-specific biomarker lists (derived from existing scRNAseq studies) to each other (Supplementary Fig. 3a), to our AD residuals (Supplementary Fig. 3b, c), and to the AMP-AD Agora list of potential therapeutic targets in AD (Supplementary Fig. 3j, k), as well as a robustness analysis demonstrating that our neuron-specific residual derived from ENO2 expression represents a robust neuronal component in the bulk-tissue RNAseq data when compared to random selections of multi-gene neuronal biomarkers derived from these scRNAseq datasets in AD (Supplementary Fig. 3b–i; see ‘Methods’).
Identifying AD-associated gene signatures in neurons and mapping their eQTLs
To identify an AD-centered set of neuronal gene expression traits, we performed DE analysis using the deconvoluted neuron-specific expression residuals in the MAYO and ROSMAP cohorts (see ‘Methods’). In comparing expression data between AD and cognitively normal controls (MAYO: 79 AD, 76 control; ROSMAP: 212 AD, 194 control), there were 3674 significant DE neuron-specific genes in the MAYO dataset (hereby MAYO-neuron) and 6626 neuron-specific DE genes in the ROSMAP dataset (hereby ROSMAP-neuron) (Fig. 2a, b; Supplementary Fig. 4; Supplementary Data 1). There were 2097 significant DE genes overlapping between the two datasets (Fisher’s exact test, odds ratio = 3.9784, p-value = 4.66E-242), thus cross-validating the neuron-specific DE signatures independently derived from the two cohorts.

DE analysis of deconvoluted neuron-specific residuals identifies a robust DE signature associated with the difference between AD patients and cognitively normal controls. a, b All significantly up- and down-regulated genes for MAYO-neuron (a) and ROSMAP-neuron (b). Significance was assessed by t-test with FDR < 0.05; thresholds of logFC shown in each volcano plot are for visualization only. MAYO (n = 79 and 76 for AD and cognitively normal) and ROSMAP (n = 212 and 194 for AD and cognitively normal). Gene symbols are highlighted in red text for the 19 key drivers later prioritized for experimental validation in vitro. See also Supplementary Fig. 4. c Pathway enrichment analysis on the neuron-specific DE expression signatures using human ConsensusPathDB reveals dysregulated biological processes associated with AD. Significance was assessed by Fisher’s exact test with p-value < 0.05. Source data for this panel is provided in Supplementary Data 13. Detailed statistical results of DE genes and enriched pathways are summarized in Supplementary Data 1 and 2, respectively, and overlapped pathways are summarized in Supplementary Data 13.
To examine the biological processes that are dysregulated in AD cases versus controls as reflected in the DE signatures, we performed pathway enrichment analysis on the MAYO-neuron and ROSMAP-neuron gene sets using Human ConsensusPathDB103,104,105,106,107. We identified 75 and 73 enriched pathways in each dataset, respectively, with 7 pathways that were significantly dysregulated in both datasets (Fig. 2c; Supplementary Data 2, 13). These signatures were enriched for a number of cellular/molecular pathways, including those involving CDC42108,109,110, IRAK/IKK111,112,113, EGFR/PLCG114, GAD115, Hippo116, and clock genes117,118, some of which have been implicated and/or interrogated in AD previously. Additional pathways of note implicated by a single cohort dataset with known relevance to amyloid and/or tau pathology include those related to NF-κB activation119,120 and N-cadherin signaling121,122.
We further validated our neuron-specific DE signatures in AD, which were derived from deconvoluted bulk-tissue RNAseq data, by comparing our MAYO-neuron and ROSMAP-neuron DE genes with the excitatory and inhibitory neuronal signatures identified by a separate study that generated scRNAseq data from the same ROSMAP cohort123. We employed the sampling-based method described in ref. 123 and first compared the pair-wise enrichment among the scRNAseq-derived DE gene signatures in the ROSMAP dataset123 for excitatory neurons, inhibitory neurons, astrocytes, oligodendrocytes, oligodendrocyte progenitor cells, and microglial cells (Supplementary Data 3). Briefly, we found that the excitatory neuron signature significantly overlaps with inhibitory neurons, astrocytes, oligodendrocytes, and oligodendrocyte progenitor cells (FDR = 1.24E-25, 2.26E-03, 2.69E-04, and 1.36E-02, respectively) but is not enriched for microglia (FDR = 0.58). We also found that the inhibitory neuron signature is significantly enriched for astrocytes (FDR = 3.83E-02) and oligodendrocytes (FDR = 9.30E-07), but not oligodendrocyte progenitor cells (FDR = 1) or microglia (FDR = 1). The significant overlap among scRNAseq-derived DE signatures of different cell types highlights the intrinsic biological interactions among different cell types in the AD brain. Next, we found a similar pattern of enrichment between our MAYO-neuron and ROSMAP-neuron DE signatures and scRNAseq-derived DE gene signatures from ROSMAP123 (Supplementary Data 4), i.e. their excitatory neuron signature (FDR = 3.41E-10 and 3.56E-17, respectively) as well as their inhibitory neuron signature (FDR = 0.0285 and 0.00429, respectively), demonstrating significant correlation between our deconvoluted neuron-specific DE signatures and scRNAseq-derived neuronal signatures in AD. The greater excitatory-neuronal enrichment among our deconvoluted neuron-specific DE signatures is consistent with ref. 123 and similarly suggests that our deconvoluted RNAseq datasets capture the aberrant increases in neuronal excitotoxicity associated with AD in humans124. Thus, overall, despite the inherent multi-cell type interactions revealed by these analyses, we argue that our deconvoluted neuron-specific AD signatures are robust and provide a complementary solution to single cell-type transcriptomics analysis.
Another critical input for the construction of Bayesian network and causal predictive network models are the eQTLs, leveraged as a systematic source of perturbation to enhance causal inference among molecular traits. This is an approach we and others have demonstrated across a broad range of diseases and data types24,69,73,74,75,76,77,79,80,81,125,126,127,128,129,130,131,132,133,134,135,136,137,138. We mapped cis-eQTLs by examining the association of neuron-specific expression traits with genome-wide genotypes18,139,140,141 assayed in the MAYO and ROSMAP cohorts (see ‘Methods’). In the MAYO- and ROSMAP-neuron sets, 3331 (16.8%) and 5059 (25.0%), respectively, of the residual genes were significantly correlated with allele dosage (FDR < 0.01) (Supplementary Data 5). Of the cis-eQTLs detected in each cohort, 1569 genes were overlapping between the two sets (47% of MAYO cis-eQTLs and 31% of ROSMAP cis-eQTLs, Fisher’s exact test, p-value = 1.09E-209), providing further validation of the two independent cohorts.
Neuronal co-expression networks associated with LOAD
While DE analysis can reveal patterns of neuron-specific expression associated with AD, the power of such analysis to detect a small-to-moderate expression difference is low. To complement the DE analyses in identifying the input gene set for the causal network, we clustered the neuronal gene expression traits into data-driven, coherent biological pathways by constructing co-expression networks, which have enhanced power to identify co-regulated sets of genes (modules) that are likely to be involved in common biological processes under LOAD. We constructed co-expression networks on the AD patients within each dataset after filtering out lowly expressed genes (see ‘Methods’), resulting in the MAYO-neuron co-expression network consisting of 20 modules ranging in size from 30 to 6929 gene members and the ROSMAP-neuron co-expression network consisting of 14 modules ranging from 34 to 6604 gene members (Fig. 3a).

a Neuron-specific co-expression network analyses in the MAYO and ROSMAP cohorts identify gene modules associated with AD in each dataset. Module functions for each dataset were characterized by significantly enriched biological processes, with bold text indicating neuron-specific modules selected for further analysis. Each module was evaluated based on enrichment for neuron-specific DE genes, for scRNAseq derived neuron-specific biomarker genes, and for categories of available AD traits (MAYO and ROSMAP diagnosis by ANOVA; BRAAK, CERAD, and MMSE by linear regression). We also evaluated enrichment for scRNAseq-derived biomarker genes for microglia, astrocytes, endothelial cells, and oligodendrocytes to cross-validate that modules enriched for neuron-DE genes were not enriched for other cell types. Significance was assessed by Fisher’s exact test with FDR < 0.05 (n = 30 to 6929 for MAYO and 34 to 6604 for ROSMAP). b Among the neuron-specific modules in the two datasets, pathway enrichment analysis identifies robust pathways associated with AD which replicate in both cohorts. Source data is provided in Supplementary Data 2. c Cross-validation of neuron-specific co-expression modules between MAYO and ROSMAP identifies pairs of modules with significantly overlapping gene members. Significance was assessed by Fisher’s exact test with FDR < 0.05. See Supplementary Data 6 for further information on the biological processes significantly enriched in each module.
To evaluate the functional relevance of each cohort’s neuron-specific modules to AD pathology, we performed enrichment analysis of each module for its AD-associated neuronal DE signatures, known single-cell marker genes for the 5 major cell types in the CNS57, and categories of AD traits available from its respective cohort (Fig. 3a). From these enrichment results, we identified neuron-specific modules associated with AD DE genes: M1, M2, M10, M11, M15, and M16 from MAYO and M1, M5, and M10 from ROSMAP. It is worth noting that similar to the DE signatures (Supplementary Data 3), a small number of the MAYO- and ROSMAP-neuron co-expression modules are significantly enriched for astrocyte and oligodendrocyte biomarkers, highlighting the intrinsic cellular interactions between these cell types in the AD brain.
To further characterize the biological processes involved in the co-expression modules from each dataset, we performed pathway enrichment analysis to identify overrepresented biological processes within and across the modules (Fig. 3b; Supplementary Data 6). Out of the selected AD-associated modules from the MAYO- and ROSMAP-neuron co-expression networks, respectively, we found 36 and 16 significantly enriched pathways based on the Human ConsensusPathDB database, with 11 enriched pathways overlapping between the two datasets (Fisher’s exact test, odds ratio = 383.87, p-value = 5.41E-21; Supplementary Data 14). In comparing all pairs of modules between the datasets, we identified 17 module pairs with significant overlap of gene members (Fig. 3c), demonstrating the robustness of the two independent co-expression networks.
Ensemble of neuron-specific, causal gene regulatory networks identifies pathological pathways and key drivers for neuronal function in AD
The ultimate goal of this study was to identify upstream master regulators (key drivers) of neuronal pathways that contribute to AD. Following our DE, eQTL, and co-expression network analyses, we built an ensemble of causal network models—including standard Bayesian networks22,24 and state-of-the-art predictive network models21,23,60—by integrating the eQTLs and deconvoluted neuron-specific RNAseq residuals.
We first pooled all genes from the selected AD-associated modules per dataset (six MAYO-neuron modules and three ROSMAP-neuron modules, indicated in Fig. 3a) to create a seeding set of genes for each cohort for input into the network models. This resulted in 9361 seeding genes from the MAYO-neuron co-expression network and 7530 seeding genes from the ROSMAP-neuron co-expression network. We note an overlap of 4506 genes between the two seeding gene sets (48.1% of MAYO and 59.8% of ROSMAP, Fisher’s exact test, odds ratio = 2.875, p-value = 2.51E-157), indicating the reproducibility of these analyses across the two independent datasets. To further improve the robustness of our network models, we also expanded each set of seeding genes by including their known upstream and downstream genes in each cohort’s co-expression network, extracted from signaling pathway databases using the pathFinder algorithm60 (see ‘Methods’; note that we did not include the gene-gene interactions as prior edge information for network construction). Co-expression network modules are only sensitive to linear relationships between pairs of genes, whereas non-linear gene regulations will not be captured by co-expression analysis. This expansion step thus includes genes in the same pathways as the seeding genes which otherwise failed to be included in the same module derived from the co-expression networks, resulting in 14,683 expanded genes from MAYO-neuron, 13,681 expanded genes from ROSMAP-neuron, and an overlap of 11,952 genes between the two expanded gene sets (Fisher’s exact test, p-value = 0). The use of both the seeding gene set and the expanded gene set for analysis of the MAYO and ROSMAP datasets therefore increases the power to build robust networks and to discover high-confidence neuronal key drivers associated with AD pathology.
We also incorporated cis-eQTL genes into each network as structural priors. As cis-eQTLs causally affect the expression levels of neighboring genes, they can serve as a source of systematic perturbation to infer causal relationships among genes23,59,60,81. Of the 3331 and 5059 unique cis-eQTL genes identified in the MAYO- and ROSMAP-neuron datasets, respectively, 687 and 1978 overlapped with the seeding gene set and 2,162 and 2,998 overlapped with the expanded gene set. We finally proceeded to build Bayesian networks and predictive networks using the two sets of genes per dataset—i.e., 9361 seeding and 14,683 expanded genes for the MAYO dataset and 7530 seeding and 13,681 expanded genes for the ROSMAP dataset—and incorporating each dataset’s cis-eQTL genes as structural priors.
Since structure learning is a heuristic and stochastic process, we applied a wide range of cut-offs on the posterior probability of edges to derive sets of robust Bayesian and predictive network structures for each dataset. For the MAYO-neuron seeding gene set, we built Bayesian networks and applied two posterior probability cut-offs (0.4 and 0.5, see ‘Methods’) to get two MAYO-neuron Bayesian networks (MAYO-Neuron-BayesNet-Seed-1 and -2) which were comprised of 9111 and 9044 genes, respectively. In addition, we built predictive networks with the same two posterior probability cut-offs (0.4 and 0.5) to derive two MAYO-neuron predictive networks (MAYO-Neuron-PredNet-Seed-1 and -2), which also included 9111 and 9044 genes, respectively. For the MAYO-neuron expanded gene set, we built predictive networks and chose three posterior probability cut-offs (0.5, 0.6, 0.7) to get three MAYO-neuron predictive network models (MAYO-Neuron-PredNet-Expanded-1, -2, and -3), which were comprised of 14,238, 13,926, and 13,365 genes, respectively. For the ROSMAP-neuron seeding gene set, we built Bayesian networks and applied two cut-offs (0.3 and 0.4) to derive two Bayesian networks (ROSMAP-Neuron-BayesNet-Seed-1 and -2) which consisted of 6786 and 6756 genes, respectively. For the ROSMAP-neuron expanded gene set, we built two predictive networks and chose two cut-offs (0.3 and 0.4) to build two predictive networks (ROSMAP-Neuron-PredNet-Expanded-1 and -2) consisting of 12,147 and 12,074 genes, respectively. Thus, in total from the MAYO and ROSMAP datasets, we derived 11 networks for the inference of a robust set of key drivers, using several different network reconstruction methods, network gene sets, and posterior cut-offs. We demonstrate 2 of the final 11 causal network models in Fig. 4a, b (MAYO-Neuron-PredNet-Expanded-1 and ROSMAP-Neuron-PredNet-Expanded-1), and the remaining 9 causal networks are shown in Supplementary Fig. 5.

a, b Two predictive networks out of the final 11 neuron-specific causal Bayesian and predictive network models derived from the MAYO (a) and ROSMAP (b) seeding and expanded gene sets. The MAYO and ROSMAP networks shown here were built from their respective expanded gene sets with posterior probability cut-offs of 0.5 and 0.3, respectively. Close-up views show the 10 key driver targets which were validated in vitro (red) along with their neighboring downstream subnetworks. See also Supplementary Fig. 5. c, d The top 50 out of 1563 total key driver targets ranked individually according to impact (c) and robustness (d) scores across the 11 independent MAYO-neuron and ROSMAP-neuron Bayesian and predictive networks. Red text indicates prioritized key drivers; yellow highlights those which were validated in vitro. Source data is provided in Supplementary Data 15.
Identification and prioritization of neuronal key drivers regulating AD pathology
Having generated the causal predictive networks from the MAYO-neuron and ROSMAP-neuron datasets, we applied key driver analysis82 to derive a list of key driver genes from each network. Key driver analysis seeks to identify genes in a causal network which modulate network states; in the present analysis, we applied this analysis to identify genes causally modulating the network states of our neuron-specific Bayesian and predictive network models. In total, we identified 1563 key driver genes across the 11 independent networks.
To prioritize key drivers for further investigation, we first ranked the 1563 initial key driver targets according to two separate measures: an impact score and a robustness score (see ‘Methods’). Briefly, the impact score is a predicted value quantifying the regulatory impact of a given key driver on its downstream effector genes associated with AD pathology. Intuitively, the shorter a path from a key driver to its downstream effectors in a network—with less other parental co-regulators along the same path—the greater the impact of this target on its effectors in that network. The robustness score is reflective of the number of datasets (MAYO and/or ROSMAP), gene sets (seeding and/or expanded), and network models (Bayesian and/or predictive) by which a key driver is replicated. After ranking the total 1563 neuron key drivers according to each score, we focused on the top 50 key drivers in each ranked list (Fig. 4c, d; Supplementary Data 15).
We then performed a series of steps to prioritize a final group of key driver targets for in vitro experimentation out of the ensemble of the top 50 ranked candidates for each score. We first calculated the replication frequency across the two ranked lists and identified 11 replicated targets, indicating robustness across these two independent ranking scores, and 39 unique targets in each ranked list (78 total). For the 11 replicated targets, we removed any which ranked lower than 15 in both scores, resulting in 7 top-ranked targets (ICA1, NSF, FSCN3, HP1BP3, DCAF12, JMJD6, and SLC25A45) which were replicated in both lists and ranked within the top 15 in one or both scores. Next, for the remaining 78 unique targets, we first selected the top 3 unique targets from each ranked list (CIRBP, NUDT2, and FIBP for impact score; YWHAZ, NDRG4, and RHBDD2 for robustness score). To further select targets from the remaining 36 neuron-specific targets in each ranked list (72 total), we identified 4 targets (GABARAPL1, ATP1B1, ATP6V1A, and RAB3A) which were previously nominated to the AMP-AD Agora list based on separate data-driven network analysis using the bulk-tissue RNAseq data in the MAYO and ROSMAP datasets with the same approach as this study142. Finally, to balance our selection strategy, we selected an additional 4 targets (RBM4, RAB9A, FMNL2, and STXBP1) out of the lower-to-middle ranked top 50 unique targets based on the availability of proper constructs.
In summary, we prioritized a group of 19 targets for experimental validation in vitro (Fig. 4c, d, highlighted in red) by selecting the top-ranked replicated targets across the two scores (we note that SLC25A45 and FSCN3 were excluded at this stage due to lack of proper constructs), 6 top-ranked unique targets (top 3 from each score), 4 targets overlapping with prior data-driven nominations to the AMP-AD Agora list, and 4 lower-to-middle ranked targets.
Validation of AD-associated function of neuronal key drivers by knockdown in human neurons
We next aimed to test the functional consequences of perturbation of the top candidate driver genes in human neurons. Healthy control human iPSCs were differentiated to a neuronal fate using a modified version of the well-established NGN2 differentiation protocol71, which rapidly generates induced neurons (iNs) which are most similar to layer 2/3 glutamatergic neurons of the cerebral cortext71,72,143. By 2 weeks in culture, iNs are post-mitotic, electrically active, and express a full array of synaptic markers71,143. In order to perturb the expression of the top 19 candidate key driver genes, we obtained sets of validated short hairpin RNA (shRNA) constructs packaged in lentivirus, with each set containing three constructs against each selected gene. At day 17 of differentiation, iNs were transduced with lentivirus encoding a single shRNA, alongside control cells which either received empty virus or were not transduced. Media were exchanged on all cells 18 h later. Five days following transduction (day 22 of differentiation), conditioned media were collected, and cells were lysed either to collect RNA for RNAseq or to harvest protein for analyses of Aβ and p-tau/tau, similar to our previous study of LOAD genome-wide association study hits144. All Aβ and tau data were normalized to total protein in the cell lysate per well, and all data for each shRNA knockdown were additionally normalized to the average of control conditions (empty vector and no transduction) (Fig. 5a–g; Supplementary Data 16).

a–c Secretion of Aβ42 (a), Aβ40 (b), and Aβ38 (c) was measured in conditioned media by ELISA, normalized to the average of controls (no transduction and empty vector) as well as to total protein in the neuronal cell lysate. The ratio of Aβ40:42 was also calculated (d). e, f p231-tau (e) and total tau (f) were measured in cell lysates by ELISA, normalized to the average of controls (no transduction and empty vector) as well as to total protein in the cell lysate. The ratio of p231-tau to total tau was also calculated (g). For all panels, a black dashed line indicates the median for control conditions for that measurement. For each gene knockdown condition, 3 shRNA constructs were used, each shRNA construct was used in duplicate wells, and each dot represents data from one well. For each boxplot, the box contains the middle quartiles of the data and black bar denotes the median value, and the upper and lower quartiles contain the maximum and minimum values and the remaining 50% of the data. Source data is provided in Supplementary Data 16. For each measured parameter, we first performed a Welch’s ANOVA with unequal variance to detect significant differences across conditions, with an additional non-parametric Kruskal-Wallis ANOVA to confirm the significance; p-values are indicated on each panel. We then used a Dunnett’s T3 multiple comparisons test to compare each target shRNA to the control condition for each parameter; *adj-p < 0.05, **0.001 < adj-p < 0.05, ***0.0001 < adj-p < 0.001, ****adj-p < 0.0001. (n = 5–29). h Circus plot summarizing the effects of the 10 key driver targets found to modulate levels of Aβ42, 40, 38, Aβ42:40, tau, p231-tau and/or p231-tau:tau. Significance was assessed by -log10(Dunnett’s T3 adjusted p-value). Red indicates that knockdown of the target significantly increased the given measurement value, whereas blue indicates that knockdown significantly decreased the value. Data frequency distributions and detailed statistical results for this figure are provided in Supplementary Fig. 6 and Supplementary Data 7, respectively.
Aβ38, 40, and 42 levels were measured in conditioned media from the transduced and control iNs using the Meso Scale Discovery Triplex ELISA platform. Of the 19 genes tested, knockdown of 11 genes had no significant effect on the levels of any Aβ peptides measured nor the ratio of Aβ42 to Aβ40 (Fig. 5a–d). However, targeted knockdown of YWHAZ significantly raised Aβ42 peptide levels, knockdown of DCAF12 and YWHAZ increased Aβ38 levels, and knockdown of NSF and NUDT2 significantly increased levels of all three Aβ peptides measured (Aβ38, 40, and 42) (Fig. 5a–c). On the other hand, knockdown of RBM4 significantly reduced levels of both Aβ42 and Aβ40 (Fig. 5a, b). Lastly, knockdown of NDRG4, STXBP1, YWHAZ, and JMJD6 resulted in a significant elevation of the putatively neurotoxic Aβ42 to 40 ratio145,146(Fig. 5d).
We also examined levels of tau species in the transduced and control iN lysates using a Meso Scale Discovery ELISA measuring both total tau and phospho-tau (Thr231). Knockdown of 16 of the 19 candidate genes tested had no significant effect on the levels of tau, p231-tau, or the neurotoxic ratio of p231-tau to tau (Fig. 5e–g). However, targeted knockdown of JMJD6 significantly decreased the levels of both p231-tau and tau (Fig. 5e, f). We also note that knockdown of NSF approached significance of increased levels of p231-tau (Fig. 5e; Dunnett’s T3 adjusted p-value = 0.075). Finally, knockdown of FIBP and JMJD6 resulted in significant elevation of the p231-tau to tau ratio, while knockdown of ATP1B1 significantly lowered this ratio (Fig. 5g).
Thus, we confirm modulation of AD endophenotypes in human iNs following independent reduction of the expression of 10 different genes out of the top 19 predicted key driver targets (Fig. 5h). Data frequency distributions and detailed statistical results of Aβ and tau measurements are included in Supplementary Fig. 6 and Supplementary Data 7. We additionally analyzed the overlap of these 10 targets with our DE and cis-eQTL analyses in MAYO and ROSMAP (Supplementary Data 8 and 9, respectively). As not all of the targets are significant DE or cis-eQTL genes, we conclude that our network analysis adds a critical value to the identification and prioritization of targets which cannot be achieved by DE and eQTL analyses alone.
Validation of AD-associated networks and pathways by RNAseq of human neurons following targeted gene knockdown
To validate the network structure, we repeated shRNA-mediated knockdown of each of the 19 target key drivers in another set of cultured control iNs and subsequently measured gene expression by RNAseq. For each of the 10 AD endophenotype-modulating targets, we derived a DE signature from the RNAseq data (Fig. 6a–j, Supplementary Data 10). Next, we extracted the downstream (sub)network of each of those 10 targets from the MAYO- and ROSMAP-neuron networks and evaluated the enrichment of the knockdown DE signature by the downstream subnetworks for each target. We found that 8 out of the 10 DE signatures were enriched by the downstream subnetworks of their corresponding target (Fig. 6k), validating that our network models capture a large portion of molecular processes and pathways at the neuron level.

a–j RNAseq analysis showing significantly up- and down-regulated DE genes after shRNA-mediated knockdown of each of the 10 validated targets. Significance was assessed using the two-stage linear step-up procedure of Benjamini, Krieger, and Yekutieli with q-value < 0.05, indicated by the black dashed line. Red gene symbols indicate any of the prioritized 19 target genes that were significantly affected. k Network validation by enrichment analysis of significant DE genes following shRNA knockdown of the 10 validated targets in the 11 subnetwork networks. We compared the DE genes after knockdown of each target to the individual downstream subnetwork of that target extracted from the 11 reconstructed networks. Significance was assessed by Fisher’s exact test with p < 0.05. l Significant overlap in DE genes resulting from knockdown of each of the 10 validated targets in human iPSC-derived neurons. Significance was assessed by Kruskal-Wallis ANOVA with Dunnett’s T3 multiple comparisons test with FDR < 0.05. Detailed results are summarized in Supplementary Data 10.
We then further examined the gene expression changes resulting from knockdown of the 10 validated targets. Following JMJD6 knockdown, which significantly altered ratios of both Aβ and tau in iNs, 656 genes were significantly upregulated and 419 genes significantly downregulated (Fig. 6a). Interestingly, among those significantly upregulated genes were 3 of our other 19 key driver candidates (NDRG4, ATP6V1A, and NSF), indicating that their expression is affected by the reduction of JMJD6 in neurons. Volcano plots in Fig. 6b–j highlight additional key driver candidates whose expression was affected by knockdown of each of the 9 other validated targets. Moreover, we found certain common genes affected by the perturbation of multiple validated targets: 6 genes (FGF11, GIT2, KLHL28, PLCB3, SEPSECS, and SLC48A1) were affected by knockdown of NDRG4, STXBP1, YWHAZ, and JMJD6, and 9 genes (SEPTIN3, ABR, AOC2, CTFIP2, ZGTF2H1, MRPL17, NIIPSNAP1, RIMS4, and TMEM246) were affected by perturbation of DCAF12, NSF, and NUDT2. This observation indicates that there may be unique and common molecular pathways among these validated AD endophenotype-modulating targets; we illustrate the significant overlap of DE genes after each target knockdown in Fig. 6l.
To investigate possible mechanisms underlying these observations, we extracted regulatory pathways among the 10 validated targets in each of the 11 MAYO- and ROSMAP-neuron networks. We found that these 10 targets tightly regulate each other, and, interestingly, are all upstream regulators of the prominent proteins REST and VGF (Fig. 7a, b). REST (restrictive element 1-silencing transcription factor) is a known master regulator of neurogenesis via epigenetic mechanisms, apoptosis, and oxidative stress;147,148 VGF (nerve growth factor inducible) is a recently identified AD target whose overexpression in a mouse model reversed AD phenotypes68. In particular, our networks identified FIBP as a direct upstream regulator of VGF. Our findings thus indicate that these 10 targets may modulate AD-related pathology partially through REST and VGF pathways.

a The downstream members of each validated target were extracted from every network model, and edges from each downstream sub-network were pooled together into a consensus subnetwork of the 10 validated targets (yellow nodes). The 10 targets tightly regulate each other and are upstream regulators of both REST and VGF (green nodes). Blue, red, and purple edges, respectively, indicate the shortest paths from any of the 10 validated targets to REST, VGF, or both. Edge thickness indicates the frequency of corresponding edges appearing across all the networks. b The shortest paths from each of the 10 targets to REST and VGF were extracted from each network and pooled together into a hierarchical structure. The coloring of each target node annotates its representative ConsensusPathDB pathways enriched by significant DE genes in the shRNA knockdown experiments. c The overall ConsensusPathDB pathways significantly enriched by each of the 10 target genes (assessed by Fisher’s exact test with p-value < 0.05) were pooled and ranked in descending order by the frequency of enrichment by any of the targets. Detailed statistical results and descriptions of all pathways affected by these targets are provided in Supplementary Data 11.
Finally, we performed pathway enrichment analysis (see ‘Methods’) on the DE signatures derived from the RNAseq data in order to identify the unique and shared pathways affected by the knockdown of the 10 AD endophenotype-modulating targets (JMJD6, NSF, NUDT2, DCAF12, RBM4, YWHAZ, NDRG4, STXBP1, FIBP and ATP1B1). We compared significant pathways enriched by the DE signature of each of the targets and found that 1 pathway is shared by 9/10 targets, 4 pathways are shared by 8/10 targets, 2 pathways are shared by 7/10 targets, 18 pathways are shared by 6/10 targets, and 40 pathways are shared by 5/10 targets (Fig. 7c). Comprehensive descriptions of all pathways affected by these targets are included in Supplementary Data 11. Moreover, we found an interesting association between JMJD6 (as well as NUDT2 and NDRG4 among the 10 validated targets) and allele dosage. These 3 key driver genes are significantly associated with SNPs in their promoter regions (cis-eQTLs) in the MAYO and ROSMAP cohorts, further indicating that these genes may be actionable targets for AD therapeutic development.
Discussion
AD is the most common neurodegenerative disease in the world, affecting millions of people worldwide. In the United States alone, an estimated 5.8 million Americans are currently living with AD dementia and this number is anticipated to reach 13.8 million by 2025149. Previous studies of LOAD pathogenesis using multi-omic data have identified numerous targets21,22,23,24,68. However, although neurons are the principal cell type affected by AD etiology, the molecular mechanisms and therapeutic targets for AD revealed by these studies are not specific to neurons due to a lack of large-scale scRNAseq data on neurons in AD. Thus, a comprehensive characterization of neuron-specific gene regulatory networks with association to AD is crucial to provide insight into the underlying causes of this disorder.
Here, we employed a self-developed computational systems biology approach to model AD neuronal gene regulatory networks, with which we identified upstream regulators (key drivers) in neurons that contribute to AD pathology. In our pipeline, we employed PSEA to deconvolute RNAseq data from brain region-specific tissue in the MAYO and ROSMAP cohorts into the five major cell types in the CNS including neurons, microglia, astrocytes, endothelial cells, and oligodendrocytes. In this study, we focused on the neuron-specific gene expression data and performed basic bioinformatics analyses including DE analysis, eQTL identification, co-expression module networks, and pathway enrichment analysis, followed by construction of causal network models and key driver gene identification.
From the network models, we identified a total of 1563 neuronal key drivers which may represent potential therapeutic targets. We used an unbiased ranking approach to prioritize 19 predicted key drivers for in vitro experimentation and tested the effects of their knockdown on the central components of the pathological hallmarks of AD, Aβ peptides (Aβ38, Aβ40, Aβ42) and phosphorylated tau protein, in a human iN system. We validated 10 targets which affected Aβ (JMJD6, NSF, NUDT2, DCAF12, RBM4, YWHAZ, NDRG4, and STXBP1) and/or tau/p-tau levels (JMJD6, FIBP, and ATP1B1). Only YWHAZ has been previously linked to AD through expression and mechanistic studies150,151,152,153,154, while others have not yet been studied. Our findings of alterations to the neurotoxic ratios of both Aβ42 to Aβ40 and p231-tau to tau suggest therapeutic potential to both early and later stages of disease considering known patterns of pathology development in AD155.
Most interestingly, we identified that knockdown of JMJD6 (Jumonji Domain Containing 6, Arginine Demethylase and Lysine Hydroxylase) significantly increased both Aβ42 to 40 and p231-tau to tau ratios, suggesting therapeutic relevance to multiple stages of AD pathology. JMJD6 belongs to the JmjC domain-containing family, catalyzes protein hydroxylation and histone demethylation, and appears to interact with distinct molecular pathways through epigenetic modifications of the genome156,157. JMJD6 is expressed in many tissues throughout the body, including the brain according to the Human Protein Atlas158, but very little is known about its role in the brain or in neurodegenerative disease. However, based on its known role in epigenetic regulation, it is expected that reduction of JMJD6 expression may result in widespread changes in gene expression. Indeed, consistent with this prediction, we observed expression changes in a large number of genes following neuronal knockdown of JMJD6, including alteration of the expression of 3 other key driver targets of interest highlighted in this study (NDRG4, ATP6V1A, and NSF).
We recognize that one caveat of our experimental system is that neurons in a dish are not the same as neurons present in the aged AD brain; however, neurons in vitro do represent a powerful system for interrogating molecular connections between gene expression and proteins relevant to AD (namely, Aβ and tau). We recently showed that neurons derived from >50 different individuals show concordance between their levels of specific Aβ peptides and p-tau species and levels of these same proteins expressed in the brains of the same individuals72. Further, we showed concordance between protein and RNA module expression between the iPSC-derived neurons and the brain tissue of the same people. Taken together, these results suggest that in spite of the reductionist nature of the system and the lack of aging, molecular networks are captured within the cells in vitro that are reflected in changes in Aβ and tau. Here, we employ this same experimental system to show that targeted reduction of JMJD6 levels in human neurons induces effects on Aβ ratios and tau levels and phosphorylation.
Through our network models, we also discovered REST and VGF as two shared downstream effectors of the 10 validated targets, which may potentially explain the observed modulation of AD pathology. REST is a known master regulator of neurogenesis via epigenetic mechanisms, apoptosis, and oxidative stress147,148 whose loss has been causally linked to Alzheimer’s disease159,160. Additionally, recent studies have identified an association between changes in the epigenome, such as DNA methylation and histone modification, with changes in cognitive functions such as learning and memory161,162,163,164,165,166,167,168,169,170. Thus, dysregulation of epigenetic mechanisms through modulation of the targets may play a role in the pathogenesis of AD162,171. VGF is also a target of interest which was recently found to partially rescue memory impairment and neuropathology in 5xFAD mice68. Overexpression of VGF increased activated BDNF receptor levels as well as adult hippocampal neurogenesis, which in turn regulated postsynaptic protein PSD-95 and improved cognition in the 5xFAD mice68. Our pathway enrichment analysis confirmed that all 10 key drivers and their downstream genes in the network models were also significantly enriched for a variety of convergent and unique downstream cellular processes and functions which may explain additional molecular mechanisms at play, including vesicle-mediated membrane trafficking (common downstream of 8 targets); axon guidance, intra-Golgi trafficking, and retrograde Golgi-to-ER trafficking (common of 7 targets); and signaling pathways for sphingolipids, prolactin, BDNF/NTRKs, EGF-EGFR, TNFα, RHO GTPases, TP53, receptor tyrosine kinases (RTKs), and ER-to-Golgi transport (common of 6 targets).
In summary, our innovative computational systems biology approach using predictive network modeling has identified 10 targets which significantly modulate AD pathology via regulation of a variety of downstream pathways. These processes involve a wide spectrum of cellular pathways and possible mechanisms, and our results offer insights into potential therapeutic targets for drug discovery in AD.
Methods
Obtaining RNAseq and genome-wide genotype datasets
MAYO temporal cortex RNAseq data (id: syn3163039) and genome-wide genotype data (id: syn8650953) were downloaded from the AMP-AD knowledge portal hosted on Synapse.org (doi:10.7303/syn2580853). The ROSMAP dorsolateral prefrontal cortex RNAseq data (id: syn4164376), genotypes (id: syn3157325), and clinical covariates (id: syn3191087) were downloaded from Synapse.org using the synapseClient R library172.
Study participants and ethical statements
The MAYO dataset includes 278 subjects: 84 with AD, 84 with progressive supranuclear palsy (PSP), 80 cognitively normal controls, and 30 with pathologic aging83,84,85,173 (see Supplementary Note 1 for more information on diagnostic criteria relevant to this study). All AD and PSP subjects along with 65 control subjects were from the Mayo Clinic Brain Bank; all pathologic aging and remaining control subjects were from the Banner Sun Health Institute. The Mayo RNAseq Study was approved by the Mayo Clinic Institutional Review Board. All human subjects or their next of kin provided informed consent. All subjects were North American Caucasians. In this study, we analyzed a total of 266 MAYO subjects with matched RNAseq and genome-wide genotype data, including 79 AD subjects and 76 cognitively normal subjects. All disease subjects had ages at death ≥60 years; a more relaxed age cutoff of ≥50 years was applied for control subjects to achieve a sample size similar to that of the AD subjects, but we note there were only two additional control subjects with age at death below 60. We performed rigorous statistical testing to demonstrate that these samples are well balanced with respect to age at death (p-value = 0.57) as well as sex (p-value = 0.24) (Supplementary Fig. 7a, c).
The ROSMAP dataset includes the Religious Orders Study (ROS) and the Memory and Aging Project (MAP)174, which are both longitudinal clinical-pathologic cohort studies of aging and dementia run by the Rush Alzheimer’s Disease Center in Chicago, IL. All participants enroll without known dementia and agree with informed consent to annual clinical evaluation and brain donation. Each sample is associated with a cognitive diagnosis of: not cognitively impaired, mild cognitive impairment, or AD (see Supplementary Note 2 for more information on diagnostic criteria). The ROS and MAP studies were each approved by an Institutional Review Board of Rush University Medical Center. In this study, we analyzed a total of 612 ROSMAP subjects with matched RNAseq and genome-wide genotype data, including 212 AD subjects and 194 cognitively normal subjects.
MAYO RNAseq, data processing, and quality control
RNA extraction, library preparation, and sequencing of the temporal cortex samples were conducted at the Mayo Clinic Medical Genome Facility Genome Analysis Core, as previously described175 (see also Supplementary Note 3 for more information). Only samples with an RNA integrity number ≥5.0 were included in this study. Briefly, all samples underwent 101 base-pair, paired-end sequencing on Illumina HiSeq2000 instruments. Base-calling was performed using Illumina’s Real-Time Analysis 1.17.21.3. FASTQ sequence reads were aligned to the human reference genome using TopHat 2.0.12176 and Bowtie 1.1.0177, and Subread 1.4.4 was used for gene counting178. FastQC179 was used for quality control (QC) of raw sequence reads, and RSeQC180 was used for QC of mapped reads.
All MAYO RNAseq samples had percentage of mapped reads ≥85%. Raw read counts were transformed to counts per million (CPM), log2 normalized, and normalized using Conditional Quantile Normalization (CQN) via the Bioconductor package181, accounting for sequencing depth (calculated as the sum of reads mapped to genes), gene length, and GC content (calculated via Repitools182 in the Bioconductor package). Genes with non-zero counts across all samples were retained and principal component analysis was performed using the prcomp function in R. Principal components 1 and 2 were plotted and no outliers (>6 SD from mean) were identified.
ROSMAP RNAseq, data processing, and quality control
BAM files174 were sorted using samtools183 and converted to FASTQ files using the SamToFastq function184. RAPiD185 was used to generate a count matrix for the gene expression data and a vcf file for each sample aligned to hg19 from the FASTQ files. Read count expression data was normalized using log2 counts per million (CPM) and the TMM method186 was implemented in edgeR187. Genes with over 1 CPM in at least 30% of the experiments were retained. We then used precision weights as implemented in the voom function from the limma188 R package to further normalize the gene counts.
Regarding the ROSMAP cohort, it has been noted that the range of age at death is broad but restricted to the older segment of the age distribution of the North American population and that age and sex are important confounders when performing any analyses of ROS and MAP data88. We observed this variance in the age at death (p-value < 0.05) but found no significant difference in sex among the ROSMAP subjects used in our analysis (p-value = 0.072) (Supplementary Fig. 7b, d). To address the imbalanced age distribution, we later performed covariate adjustment for age (together with other covariates, see section ‘Deconvolution of RNAseq data into neuron-specific expression residuals’ below), and we confirmed removal of the effects of age and other confounding variables by variance partition analysis before and after covariate adjustment (Supplementary Fig. 2b, d).
Genome-wide genotype data and quality control
Whole-genome genotyping of MAYO subjects was performed at the Mayo Clinic Medical Genome Facility Genome Analysis Core using the Illumina Infinium HumanOmni2.5-8 Kit (see also Supplementary Note 4 for more information). Whole genome genotype calls were made using the auto-calling algorithm in Illumina’s BeadStudio 2.0 software, after which they were converted into PLINK formats for analysis189. Samples were removed if they had discordant sex, heterozygosity rates >3 SD from the mean, or apparent relation. The dataset was filtered to include only autosomal SNPs and to remove complex genomic regions regions (chr8:1-12,700,000; chr2:129,900,001-136,800,000; chr17:40,900,001-44,900,000; and chr6:32,100,001-33,500,000). Linkage disequilibrium was pruned using the SNPRelate (v1.4.2) package in R190, implementing a linkage disequilibrium threshold of 0.15 and a sliding window of 1E-07 base pairs. Remaining SNPs and subjects were analyzed using EIGENSOFT191 for population outliers. See Supplementary Note 5 for more details regarding MAYO sample exclusion.
ROSMAP subject genotype data was processed using PLINK 2.0192. Positions were converted from hg18 to hg19 (http://genome.ucsc.edu/cgi-bin/hgLiftOver) and resulting genotype files were sorted using Picard184. Samples were removed using PLINK 2.0 if they had variants with >2% missing values, minor allele frequency <1%, Hardy-Weinberg equilibrium <10E-6, or inbreeding coefficient >0.15.
Genotype data imputation
1000 Genomes Project193 data and IMPUTEv2194 were used to impute untyped variants. Imputed variants were removed if they failed any of the previously listed quality control criteria or had information scores <0.6. After imputation, we had 7,132,687 variants in MAYO and 9,333,139 variants in ROSMAP.
Deconvolution of RNAseq data into neuron-specific expression residuals
Residuals were obtained for each RNAseq dataset by adjusting for covariates using the limma R package188. For MAYO, expression residuals were obtained by correcting for the effects of technical confounding factors (i.e., sequencing batch), sample-specific variables (RNA integrity number, exonic mapping rate, source of tissue), and patient-specific covariates (sex, age at death, APOE genotype). For ROSMAP, we adjusted for a slightly different set of covariates due to a greater number of recorded measurements available: study (ROS or MAP), sequencing batch, post-mortem interval, RNA integrity number, exonic mapping rate, sex, educational attainment, APOE genotype, and age at death. For both MAYO and ROSMAP data, we computed the exonic mapping rate using RNAseQC195.
We additionally adjusted for previously published single-gene biomarkers derived at the protein level under AD for the five major cell types in the CNS: neurons, ENO296; microglia, CD6897; endothelial cells, CD3499; astrocytes, GFAP98; and oligodendrocytes, OLIG2100,101,102. To obtain expression residuals that mimic expression patterns seen in neurons, for every gene, we added the ENO2 effects estimated by the linear regression models back to the expression residuals. Comparing the variance of normalized gene expression before and after covariate adjustment, we confirmed removal of the effects from confounding variables (Supplementary Fig. 2), allowing us to conclude that the residual results are unbiased and robust against these adjusted covariates.
The final neuron-specific expression residual data available for further analysis included 19,885 genes from 155 individuals in MAYO (79 AD, 76 cognitively normal) and 20,276 genes from 406 individuals in ROSMAP (212 AD, 194 cognitively normal), with 18,408 genes common to both datasets (Fig. 1b, Fisher’s exact test, p-value = 0); this is comparable to processed residuals of the same cohorts on the AMP-AD knowledge portal (https://adknowledgeportal.synapse.org).
Rationalization and validation of single-gene biomarkers for bulk-tissue RNAseq deconvolution
Our rationale for using single-gene biomarkers over multi-gene biomarkers derived from scRNAseq data was manifold. First, multi-gene biomarkers derived from various scRNAseq studies in control human brains54,55,56,57,58 (Supplementary Data 12) show no significant overlap among themselves, indicating a lack of robustness and consensus in these biomarkers derived from scRNAseq studies (Supplementary Fig. 3a). Second, PCA analysis shows a prominent overlap of scRNAseq biomarker expression across different cell types in MAYO and ROSMAP AD data, indicating that the majority of scRNAseq-derived biomarker gene expression is convoluted and reflecting potential interactions between different cell types under the AD condition (Supplementary Fig. 3b, c; Supplementary Data 12). Furthermore, there is significant overlap between scRNAseq-derived biomarkers and AD therapeutic targets in the AMP-AD Agora list142 (Supplementary Fig. 3j, k). This overlap is more significant than randomly selected genes from the background overlapping with the Agora list, indicating that scRNAseq-derived biomarkers may play a role in AD pathology. For these reasons, all or a random subset of scRNAseq-derived cell type biomarkers are not ideal for adjusting the bulk-tissue gene expression variance by PSEA.
By contrast, our single-gene biomarkers are derived directly at the protein level under AD conditions and have been validated by other groups83,96,97,98,99,100,101,102. They show no overlap with AD therapeutic targets in the Agora list, thus making them good candidates for PSEA. Furthermore, our neuron-specific residual derived from the single-gene biomarker ENO2 is significantly correlated with pseudo neuron-specific residuals derived from a randomly selected subset of scRNAseq biomarkers (Supplementary Fig. 3h, i; Supplementary Note 6), indicating that our neuron-specific residual represents a robust neuronal component in the bulk-tissue RNAseq data for neuron-specific therapeutic target discovery in LOAD.
Computational analyses of neuron-specific gene expression data
eQTL analysis was performed using the R package MatrixEQTL v2.1.1196 using quality-controlled genotypes and normalized and covariate-adjusted cell type-specific expression residuals. cis-eQTL analysis considered markers within 1 Mb of the transcription start site of each gene. False discovery rates (FDR) were computed using the Benjamini–Hochberg procedure197.
DE analysis to interrogate the cell type-specific residual expression data for genes differentially expressed between AD cases and healthy controls was performed using linear models implemented in the limma R package188. Significance was assessed using FDR < 0.05. We note that the log-fold change thresholds in Fig. 2a, b are for visualization only and were not used in the analysis in any way.
For pathway enrichment analyses, we downloaded pathways from ConsensusPathDataBase105. For each given set of genes, we performed enrichment analysis of each pathway over the set by Fisher’s exact test with p < 0.05.
Co-expression networks were constructed using the coexpp R package198. A soft thresholding parameter value of 6.5 was used to power the expression correlations. Seeding gene lists for the predictive networks were obtained by selecting genes in co-expression modules that were statistically enriched (FDR < 0.05) for DE genes or neuronal cell markers57.
To perform key driver analysis, we used the KDA R package82 (version 0.1, available at http://research.mssm.edu/multiscalenetwork/Resources.html). The package first defines a background sub-network by looking for a neighborhood k-step away from each node in the target gene list in the network. Then, stemming from each node in this sub-network, it assesses the enrichment in its k-step (k varies from 1 to K) downstream neighborhood for the target gene list. In this analysis, we used K = 6. Prioritization of key drivers for subsequent assessment was determined by ranking their impact score and robustness score (described in Supplementary Notes 7 and 8, respectively).
Predictive network modeling was performed according to detailed methods described in our recent publications23,59,60,199,200 as well as methodology patent US11068799B2.
For pathway analysis, we used the PathFinder method60 which is based on the classical Depth-First Search algorithm201. The goal of PathFinder is to expand the initial target gene set by including genes in the background network located in the paths connecting input genes. Since the background network could contain directed and undirected edges, we transformed the undirected edges into two edges with the same two end nodes but different directions. We did not allow these two edges to form a loop and simultaneously appear in one path. The Depth-First Search algorithm starts from one input gene and stops if the length of path it explores reaches K or if the path arrives at a node without a valid child node. Whenever any of the stop criteria above was satisfied, we checked whether the path contained at least two input genes. If not, the path was discarded. Otherwise, among all the input genes in the path, we determined the target gene with the maximum distance to the starting input gene, and all the nodes between this gene and the starting input gene were then included in the seeding gene list for the network. In practice, we ran Depth-First Search for each input gene and combined the results to obtain the final network seeding gene list.
iPSC maintenance and induced neuron differentiation
The human control iPSC line YZ1 was obtained from the University of Connecticut Stem Cell Core facility and was maintained in StemFlex Medium (Thermo Fisher Scientific, Waltham, MA). Induced neurons (iNs) were generated as described71,72,143, with minor modifications. Briefly, iPSCs were plated in mTeSR1 media (STEMCELL Technologies, Vancouver, Canada) at a density of 95 K cells/cm2 on Matrigel-coated plates (Corning Inc., Corning, NY) for viral transduction. Media was changed from StemFlex to mTeSR1 as we found better transduction viability with mTeSR1. Viral plasmids were obtained from Addgene (plasmids #19780, 52047, 30130; Watertown, MA). FUdeltaGW-rtTA was a gift from Konrad Hochedlinger (Addgene plasmid #19780), and Tet-O-FUW-EGFP (Addgene plasmid #30130) and pTet-O-Ngn2-puro (Addgene plasmid #52047) were gifts from Marius Wernig. Lentiviruses were obtained from ALSTEM (Richmond, CA) with ultra-high titers and used at the following concentrations: pTet-O-NGN2-puro: 0.1 μl/50 K cells; Tet-O-FUW-eGFP: 0.05 μl/50 K cells; Fudelta GW-rtTA: 0.11 μl/50 K cells. Transduced cells were dissociated with Accutase (Gibco, Thermo Fisher Scientific) and plated onto Matrigel-coated plates at 50 K cells/cm2 in mTeSR1 (day 0). On day 1, media was changed to KSR media with doxycycline (2 μg/ml, Sigma-Aldrich, St. Louis, MO). Doxycycline was maintained in the media for the remainder of the differentiation. On day 2, media was changed to 1:1 KSR:N2B media with puromycin (5 μg/ml, Gibco). On day 3, media was changed to N2B media with 1:100 B27 supplement and puromycin (10 μg/ml). Puromycin was maintained at this concentration in the media for the remainder of the differentiation. From day 4 onwards, cells were cultured in NBM media with 1:50 B27 and BDNF, GDNF, CNTF (10 ng/ml each, PeproTech, Rocky Hill, NJ), plus doxycycline and puromycin as described. iNs were not co-cultured with human or primary rodent astrocytes in this study. See Supplementary Note 9 for media formulations.
Lentiviral transduction of induced neurons
At day 17 of differentiation, neurons were transduced with lentiviruses encoding shRNA constructs against selected targets (Broad Institute, Cambridge, MA), as described in ref. 22. For each round of experiments, two controls were included: a lentivirus expressing the pLKO vector without an shRNA (empty) or else not transduced (fresh media only). iNs were transduced with a 1:1 ratio of media to lentivirus. Following ~18 h of incubation, media containing virus was removed and replaced with fresh media, and cells were incubated for an additional 96 h. On day 22 of differentiation, conditioned media was then collected and stored at −20 °C for Aβ analyses, and cells were lysed either for RNA purification or protein harvest. Gene knockdowns were confirmed by qPCR.
Aβ ELISA
Aβ present in the conditioned media was measured by the 6E10 Aβ Peptide Panel Multiplex ELISA (Meso Scale Discovery, Rockville, MD) following manufacturer instructions. Briefly, conditioned media from transduced cells were incubated in pre-blocked wells along with detection antibody solution. Plates were read using an MSD SECTOR Imager 2400 and resulting peptide concentrations were normalized to total protein in the cell lysate per well measured using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific). Data for each shRNA knockdown were additionally normalized to the average of control conditions for each parameter measured. To then compare each target shRNA to the control condition for each parameter, Dunnett’s T3 tests for multiple comparisons were performed in Prism 9.0.
Tau ELISA
Protein was extracted from iNs by lysis in NP-40 lysis buffer (1% NP40, 0.5 M EDTA, 5 M NaCl, 1 M Tris) containing cOmplete protease inhibitors and phosSTOP (Roche, Penzberg, Germany). Lysates were analyzed using the Multi-Spot Phospho (Thr 231)/Total Tau ELISA (Meso Scale Discovery) following manufacturer instructions. Briefly, lysates were incubated in pre-blocked wells for 1 hr prior to detection antibody application for 1 hr. Plates were read using an MSD SECTOR Imager 2400 and resulting concentrations were again normalized to total protein in the cell lysate per well (Pierce BCA Protein Assay Kit) and data for each shRNA knockdown were normalized to the average of control concentrations for each parameter. To then compare each target shRNA to the control condition for each parameter, Dunnett’s T3 tests for multiple comparisons were performed in Prism 9.0.
RNA sequencing of induced neurons
For iNs, at least 250 ng of total RNA input was oligo(dT) purified using the PureLink RNA Mini Kit (Invitrogen), then double-stranded cDNA was synthesized using SuperScript III Reverse Transcriptase (Invitrogen) with random hexamers. RNA integrity number >9 was confirmed using the Agilent 4200 TapeStation system (Agilent Technologies). RNAseq on the shRNA-treated iNs was performed by Functional Genomics Core at the University of Arizona at a depth of 30 million single-end reads (100 bp long). The RNAseq data (GSE228156) was QCed and processed with the same steps as outlined in the Methods section “ROSMAP RNAseq, data processing, and quality control”.
Statistics and reproducibility
All statistical analyses were performed in R Foundation for Statistical Computing, version 3.2.3, unless otherwise noted. Genotyping and RNAseq data was pre-processed and normalized (range of N = 266 for MAYO and N = 612 for ROSMAP). The normalized gene expression counts were then corrected for covariates as described in ‘Methods’. The residual values were then used to perform DE, co-expression module, and network analysis.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The datasets analyzed in this study are available on the AMP-AD knowledge portal hosted on Synapse.org (doi:10.7303/syn2580853) with the following accessions: MAYO temporal cortex RNAseq data (syn3163039), MAYO genome-wide genotype data (syn8650953), ROSMAP DLPFC RNAseq data (syn4164376), ROSMAP genotypes (syn3157325), and ROSMAP clinical covariates (syn3191087). Requests for ROSMAP data can also be made at www.radc.rush.edu/. The RNAseq data generated from target knockdown experiments can be downloaded from GEO (GSE228156). Source data underlying Figs. 2–5 are presented in Supplementary Data 13–16, respectively.
Code availability
The code for network construction can be obtained at labs.icahn.mssm.edu/zhulab/software/. The software Co-expp for co-expression module construction can be obtained from bitbucket at https://bitbucket.org/multiscale/coexpp/src/master/. The network software can be obtained from https://labs.icahn.mssm.edu/zhulab/?s=rimbanet.
References
Cerejeira, J., Lagarto, L. & Mukaetova-Ladinska, E. B. Behavioral and psychological symptoms of dementia. Front. Neurol. 3, 73 (2012).
Murphy, M. P. & LeVine, H. III Alzheimer’s disease and the amyloid-beta peptide. J. Alzheimers Dis. 19, 311–323 (2010).
Wang, R. Z. et al. Genome-wide association study of brain Alzheimer’s disease-related metabolic decline as measured by [18F] FDG-PET imaging. J. Alzheimers Dis. 77, 401–409 (2020).
Tosto, G. & Reitz, C. Genome-wide association studies in Alzheimer’s disease: a review. Curr. Neurol. Neurosci. Rep. 13, 381 (2013).
Sherva, R. et al. Genome-wide association study of the rate of cognitive decline in Alzheimer’s disease. Alzheimers Dement 10, 45–52 (2014).
Sherva, R. et al. Genome-wide association study of rate of cognitive decline in Alzheimer’s disease patients identifies novel genes and pathways. Alzheimers Dement 16, 1134–1145 (2020).
Shen, L. & Jia, J. An overview of genome-wide association studies in Alzheimer’s disease. Neurosci. Bull. 32, 183–190 (2016).
Shang, Z. et al. Genome-wide haplotype association study identify TNFRSF1A, CASP7, LRP1B, CDH1 and TG genes associated with Alzheimer’s disease in Caribbean Hispanic individuals. Oncotarget 6, 42504–42514 (2015).
Moreno-Grau, S. et al. Genome-wide association analysis of dementia and its clinical endophenotypes reveal novel loci associated with Alzheimer’s disease and three causality networks: The GR@ACE project. Alzheimers Dement 15, 1333–1347 (2019).
Liu, C. & Yu, J. Genome-wide association studies for cerebrospinal fluid soluble TREM2 in Alzheimer’s disease. Front. Aging Neurosci. 11, 297 (2019).
Lee, Y. H. & Song, G. G. Genome-wide pathway analysis of a genome-wide association study on Alzheimer’s disease. Neurol. Sci. 36, 53–59 (2015).
Elsheikh, S. S. M., Chimusa, E. R., Mulder, N. J. & Crimi, A. Genome-wide association study of brain connectivity changes for Alzheimer’s disease. Sci. Rep. 10, 1433 (2020).
Deters, K. D. et al. Genome-wide association study of language performance in Alzheimer’s disease. Brain Lang. 172, 22–29 (2017).
Deming, Y. et al. Genome-wide association study identifies four novel loci associated with Alzheimer’s endophenotypes and disease modifiers. Acta Neuropathol. 133, 839–856 (2017).
Chung, J. et al. Genome-wide association study of Alzheimer’s disease endophenotypes at prediagnosis stages. Alzheimers Dement 14, 623–633 (2018).
Beecham, G. W. et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet 10, e1004606 (2014).
Hamshere, M. L. et al. Genome-wide linkage analysis of 723 affected relative pairs with late-onset Alzheimer’s disease. Hum. Mol. Genet 16, 2703–2712 (2007).
Webster, J. A. et al. Genetic control of human brain transcript expression in Alzheimer disease. Am. J. Hum. Genet. 84, 445–458 (2009).
Wan, Y. W. et al. Meta-analysis of the Alzheimer’s disease human brain transcriptome and functional dissection in mouse models. Cell Rep. 32, 107908 (2020).
Milind, N. et al. Transcriptomic stratification of late-onset Alzheimer’s cases reveals novel genetic modifiers of disease pathology. PLoS Genet 16, e1008775 (2020).
Kruti Rajan Patel, K. Z. et al. Single cell-type integrative network modeling identified novel microglial-specific targets for the phagosome in Alzheimer’s disease. bioRxiv https://doi.org/10.1101/2020.06.09.143529 (2020).
Mostafavi, S. et al. A molecular network of the aging human brain provides insights into the pathology and cognitive decline of Alzheimer’s disease. Nat. Neurosci. 21, 811–819 (2018).
Petyuk, V. A. et al. The human brainome: network analysis identifies HSPA2 as a novel Alzheimer’s disease target. Brain 141, 2721–2739 (2018).
Zhang, B. et al. Integrated systems approach identifies genetic nodes and networks in late-onset Alzheimer’s disease. Cell 153, 707–720 (2013).
Panina, Y., Karagiannis, P., Kurtz, A., Stacey, G. N. & Fujibuchi, W. Human cell Atlas and cell-type authentication for regenerative medicine. Exp. Mol. Med. 52, 1443–1451 (2020).
Lindeboom, R. G. H., Regev, A. & Teichmann, S. A. Towards a human cell Atlas: taking notes from the past. Trends Genet 37, 625–630 (2021).
Eraslan, G. et al. Single-nucleus cross-tissue molecular reference maps toward understanding disease gene function. Science 376, eabl4290 (2022).
Grubman, A. et al. A single-cell atlas of entorhinal cortex from individuals with Alzheimer’s disease reveals cell-type-specific gene expression regulation. Nat. Neurosci. 22, 2087–2097 (2019).
Yang, A. C. et al. A human brain vascular atlas reveals diverse mediators of Alzheimer’s risk. Nature 603, 885–892 (2022).
Jiang, J., Wang, C., Qi, R., Fu, H. & Ma, Q. scREAD: A single-cell RNA-seq database for Alzheimer’s disease. iScience 23, 101769 (2020).
Wagner, D. E. et al. Single-cell mapping of gene expression landscapes and lineage in the zebrafish embryo. Science 360, 981–987 (2018).
Plass, M. et al. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science https://doi.org/10.1126/science.aaq1723 (2018).
Briggs, J. A. et al. The dynamics of gene expression in vertebrate embryogenesis at single-cell resolution. Science https://doi.org/10.1126/science.aar5780 (2018).
Plasschaert, L. W. et al. A single-cell atlas of the airway epithelium reveals the CFTR-rich pulmonary ionocyte. Nature 560, 377–381 (2018).
Montoro, D. T. et al. A revised airway epithelial hierarchy includes CFTR-expressing ionocytes. Nature 560, 319–324 (2018).
Rostom, R., Svensson, V., Teichmann, S. A. & Kar, G. Computational approaches for interpreting scRNA-seq data. FEBS Lett. 591, 2213–2225 (2017).
Chen, S. & Mar, J. C. Evaluating methods of inferring gene regulatory networks highlights their lack of performance for single cell gene expression data. BMC Bioinforma. 19, 232 (2018).
Luecken, M. D. & Theis, F. J. Current best practices in single-cell RNA-seq analysis: a tutorial. Mol. Syst. Biol. 15, e8746 (2019).
Zhong, Y., Wan, Y. W., Pang, K., Chow, L. M. & Liu, Z. Digital sorting of complex tissues for cell type-specific gene expression profiles. BMC Bioinforma. 14, 89 (2013).
Hunt, G. J., Freytag, S., Bahlo, M. & Gagnon-Bartsch, J. A. dtangle: accurate and robust cell type deconvolution. Bioinformatics 35, 2093–2099 (2019).
Baron, M. et al. A single-cell transcriptomic map of the human and mouse pancreas reveals inter- and intra-cell population structure. Cell Syst. 3, 346–360.e344 (2016).
Rusk, N. Expanded CIBERSORTx. Nat. Methods 16, 577 (2019).
Newman, A. M. et al. Robust enumeration of cell subsets from tissue expression profiles. Nat. Methods 12, 453–457 (2015).
Newman, A. M. et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat. Biotechnol. 37, 773–782 (2019).
Chiu, Y. J., Hsieh, Y. H. & Huang, Y. H. Improved cell composition deconvolution method of bulk gene expression profiles to quantify subsets of immune cells. BMC Med Genom. 12, 169 (2019).
Menden, K. et al. Deep learning-based cell composition analysis from tissue expression profiles. Sci. Adv. 6, eaba2619 (2020).
Kang, K. et al. CDSeq: A novel complete deconvolution method for dissecting heterogeneous samples using gene expression data. PLoS Comput. Biol. 15, e1007510 (2019).
Diaz-Mejia, J. J. et al. Evaluation of methods to assign cell type labels to cell clusters from single-cell RNA-sequencing data. F1000 Res. https://doi.org/10.12688/f1000research.18490.3 (2019).
Dong, M. et al. SCDC: bulk gene expression deconvolution by multiple single-cell RNA sequencing references. Brief. Bioinform 22, 416–427 (2021).
Tsoucas, D. et al. Accurate estimation of cell-type composition from gene expression data. Nat. Commun. 10, 2975 (2019).
Wang, X., Park, J., Susztak, K., Zhang, N. R. & Li, M. Bulk tissue cell type deconvolution with multi-subject single-cell expression reference. Nat. Commun. 10, 380 (2019).
Kuhn, A., Thu, D., Waldvogel, H. J., Faull, R. L. & Luthi-Carter, R. Population-specific expression analysis (PSEA) reveals molecular changes in diseased brain. Nat. Methods 8, 945–947 (2011).
Abbas, A. R., Wolslegel, K., Seshasayee, D., Modrusan, Z. & Clark, H. F. Deconvolution of blood microarray data identifies cellular activation patterns in systemic lupus erythematosus. PLoS One 4, e6098 (2009).
Darmanis, S. et al. A survey of human brain transcriptome diversity at the single cell level. Proc. Natl Acad. Sci. USA 112, 7285–7290 (2015).
Zhang, Y. et al. Purification and characterization of progenitor and mature human astrocytes reveals transcriptional and functional differences with mouse. Neuron 89, 37–53 (2016).
Lake, B. B. et al. Neuronal subtypes and diversity revealed by single-nucleus RNA sequencing of the human brain. Science 352, 1586–1590 (2016).
Zeisel, A. et al. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science 347, 1138–1142 (2015).
Zhang, Y. et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 34, 11929–11947 (2014).
Carcamo-Orive, I. et al. Predictive network modeling in human induced pluripotent stem cells identifies key driver genes for insulin responsiveness. PLoS Comput Biol. 16, e1008491 (2020).
Carcamo-Orive, I. et al. Analysis of transcriptional variability in a large human iPSC library reveals genetic and non-genetic determinants of heterogeneity. Cell Stem Cell 20, 518–532 (2017).
Jensen, F. V. An Introduction to Bayesian Networks. Vol. 92 1215–1216 (Springer, 1996).
Sinha, S. Reproducibility of parameter learning with missing observations in naive Wnt Bayesian network trained on colorectal cancer samples and doxycycline-treated cell lines. Mol. Biosyst. 11, 1802–1819 (2015).
Oh, J. H. et al. A Bayesian network approach for modeling local failure in lung cancer. Phys. Med. Biol. 56, 1635–1651 (2011).
Myte, R. et al. Untangling the role of one-carbon metabolism in colorectal cancer risk: a comprehensive Bayesian network analysis. Sci. Rep. 7, 43434 (2017).
Jiang, X. H. et al. Optimal nutrition formulas for patients undergoing surgery for colorectal cancer: a Bayesian network analysis. Nutr. Cancer. 73, 1–10 (2020).
Feng, F. et al. Efficacy and safety of targeted therapy for metastatic HER2-positive breast cancer in the first-line treatment: a Bayesian network meta-analysis. Onco Targets Ther. 12, 959–974 (2019).
Liu, F., Zhang, S. W., Guo, W. F., Wei, Z. G. & Chen, L. Inference of gene regulatory network based on local Bayesian networks. PLoS Comput. Biol. 12, e1005024 (2016).
Beckmann, N. D. et al. Multiscale causal networks identify VGF as a key regulator of Alzheimer’s disease. Nat. Commun. 11, 3942 (2020).
Chang, R., Karr, J. R. & Schadt, E. E. Causal inference in biology networks with integrated belief propagation. Pac. Symp. Biocomput. 1, 359–370 (2015).
Muratore, C. R. et al. Cell-type dependent Alzheimer’s disease phenotypes: probing the biology of selective neuronal vulnerability. Stem Cell Rep. 9, 1868–1884 (2017).
Zhang, Y. et al. Rapid single-step induction of functional neurons from human pluripotent stem cells. Neuron 78, 785–798 (2013).
Lagomarsino, V. N. et al. Stem cell-derived neurons reflect features of protein networks, neuropathology, and cognitive outcome of their aged human donors. Neuron 109, 3402–3420.e3409 (2021).
Franzen, O. et al. Cardiometabolic risk loci share downstream cis- and trans-gene regulation across tissues and diseases. Science 353, 827–830 (2016).
Peters, L. A. et al. A functional genomics predictive network model identifies regulators of inflammatory bowel disease. Nat. Genet 49, 1437–1449 (2017).
Doss, S., Schadt, E. E., Drake, T. A. & Lusis, A. J. Cis-acting expression quantitative trait loci in mice. Genome Res. 15, 681–691 (2005).
Yang, X. et al. Validation of candidate causal genes for obesity that affect shared metabolic pathways and networks. Nat. Genet 41, 415–423 (2009).
Zhu, J. et al. An integrative genomics approach to the reconstruction of gene networks in segregating populations. Cytogenet Genome Res. 105, 363–374 (2004).
Zhu, J. et al. Stitching together multiple data dimensions reveals interacting metabolomic and transcriptomic networks that modulate cell regulation. PLoS Biol. 10, e1001301 (2012).
Zhu, J. et al. Increasing the power to detect causal associations by combining genotypic and expression data in segregating populations. PLoS Comput. Biol. 3, e69 (2007).
Zhu, J. et al. Integrating large-scale functional genomic data to dissect the complexity of yeast regulatory networks. Nat. Genet 40, 854–861 (2008).
Schadt, E. E. et al. An integrative genomics approach to infer causal associations between gene expression and disease. Nat. Genet 37, 710–717 (2005).
Zhang Bin, Z. J. Identification of key causal regulators in gene. Netw. Lect. Notes Eng. Comput. Sci. 2, 1309–1312 (2013).
Allen, M. et al. Conserved brain myelination networks are altered in Alzheimer’s and other neurodegenerative diseases. Alzheimers Dement 14, 352–366 (2018).
Allen, M. et al. Divergent brain gene expression patterns associate with distinct cell-specific tau neuropathology traits in progressive supranuclear palsy. Acta Neuropathol. 136, 709–727 (2018).
Allen, M. et al. Human whole genome genotype and transcriptome data for Alzheimer’s and other neurodegenerative diseases. Sci. Data 3, 160089 (2016).
Bennett, D. A. et al. Overview and findings from the rush memory and aging project. Curr. Alzheimer Res. 9, 646–663 (2012).
Bennett, D. A., Schneider, J. A., Arvanitakis, Z. & Wilson, R. S. Overview and findings from the religious orders study. Curr. Alzheimer Res. 9, 628–645 (2012).
De Jager, P. L. et al. A multi-omic atlas of the human frontal cortex for aging and Alzheimer’s disease research. Sci. Data 5, 180142 (2018).
Henstridge, C. M., Hyman, B. T. & Spires-Jones, T. L. Beyond the neuron-cellular interactions early in Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 20, 94–108 (2019).
Wightman, D. P. et al. A genome-wide association study with 1,126,563 individuals identifies new risk loci for Alzheimer’s disease. Nat. Genet. 53, 1276–1282 (2021).
Ghatak, S. et al. Mechanisms of hyperexcitability in Alzheimer’s disease hiPSC-derived neurons and cerebral organoids vs isogenic controls. Elife https://doi.org/10.7554/eLife.50333 (2019).
Oh, J. et al. Profound degeneration of wake-promoting neurons in Alzheimer’s disease. Alzheimers Dement 15, 1253–1263 (2019).
Miller, M. B. et al. Somatic genomic changes in single Alzheimer’s disease neurons. Nature 604, 714–722 (2022).
Niikura, T., Tajima, H. & Kita, Y. Neuronal cell death in Alzheimer’s disease and a neuroprotective factor, humanin. Curr. Neuropharmacol. 4, 139–147 (2006).
Hoffman, G. E. & Schadt, E. E. variancePartition: interpreting drivers of variation in complex gene expression studies. BMC Bioinforma. 17, 483 (2016).
Higginbotham, L. et al. Integrated proteomics reveals brain-based cerebrospinal fluid biomarkers in asymptomatic and symptomatic Alzheimer’s disease. Sci Adv. https://doi.org/10.1126/sciadv.aaz9360 (2020).
Minett, T. et al. Microglial immunophenotype in dementia with Alzheimer’s pathology. J. Neuroinflammation 13, 135 (2016).
Shir, D. et al. Association of plasma glial fibrillary acidic protein (GFAP) with neuroimaging of Alzheimer’s disease and vascular pathology. Alzheimers Dement (Amst.) 14, e12291 (2022).
Custodia, A. et al. Endothelial progenitor cells and vascular alterations in Alzheimer’s disease. Front. Aging Neurosci. 13, 811210 (2021).
Tognatta, R. & Miller, R. H. Contribution of the oligodendrocyte lineage to CNS repair and neurodegenerative pathologies. Neuropharmacology 110, 539–547 (2016).
Zhou, Y. et al. Human and mouse single-nucleus transcriptomics reveal TREM2-dependent and TREM2-independent cellular responses in Alzheimer’s disease. Nat. Med. 26, 131–142 (2020).
Quintela-Lopez, T. et al. Abeta oligomers promote oligodendrocyte differentiation and maturation via integrin beta1 and Fyn kinase signaling. Cell Death Dis. 10, 445 (2019).
Pentchev, K., Ono, K., Herwig, R., Ideker, T. & Kamburov, A. Evidence mining and novelty assessment of protein-protein interactions with the ConsensusPathDB plugin for Cytoscape. Bioinformatics 26, 2796–2797 (2010).
Kamburov, A., Wierling, C., Lehrach, H. & Herwig, R. ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res. 37, D623–D628 (2009).
Kamburov, A., Stelzl, U., Lehrach, H. & Herwig, R. The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 41, D793–D800 (2013).
Kamburov, A. et al. ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res. 39, D712–D717 (2011).
Herwig, R., Hardt, C., Lienhard, M. & Kamburov, A. Analyzing and interpreting genome data at the network level with ConsensusPathDB. Nat. Protoc. 11, 1889–1907 (2016).
Florian, M. C. et al. Inhibition of Cdc42 activity extends lifespan and decreases circulating inflammatory cytokines in aged female C57BL/6 mice. Aging Cell 19, e13208 (2020).
Aguilar, B. J., Zhu, Y. & Lu, Q. Rho GTPases as therapeutic targets in Alzheimer’s disease. Alzheimers Res. Ther. 9, 97 (2017).
Saraceno, C. et al. Altered expression of circulating Cdc42 in frontotemporal lobar degeneration. J. Alzheimers Dis. 61, 1477–1483 (2018).
Cui, J. G., Li, Y. Y., Zhao, Y., Bhattacharjee, S. & Lukiw, W. J. Differential regulation of interleukin-1 receptor-associated kinase-1 (IRAK-1) and IRAK-2 by microRNA-146a and NF-kappaB in stressed human astroglial cells and in Alzheimer disease. J. Biol. Chem. 285, 38951–38960 (2010).
Jain, A., Kaczanowska, S. & Davila, E. IL-1 receptor-associated kinase signaling and its role in inflammation, cancer progression, and therapy resistance. Front Immunol. 5, 553 (2014).
Liu, X. et al. BAP31 regulates IRAK1-dependent neuroinflammation in microglia. J. Neuroinflammation 16, 281 (2019).
Wang, X. T., McCullough, K. D., Wang, X. J., Carpenter, G. & Holbrook, N. J. Oxidative stress-induced phospholipase C-gamma 1 activation enhances cell survival. J. Biol. Chem. 276, 28364–28371 (2001).
De Biase, D. & Pennacchietti, E. Glutamate decarboxylase-dependent acid resistance in orally acquired bacteria: function, distribution and biomedical implications of the gadBC operon. Mol. Microbiol. 86, 770–786 (2012).
Mueller, K. A. et al. Hippo signaling pathway dysregulation in human huntington’s disease brain and neuronal stem cells. Sci. Rep. 8, 11355 (2018).
Wu, H., Dunnett, S., Ho, Y. S. & Chang, R. C. The role of sleep deprivation and circadian rhythm disruption as risk factors of Alzheimer’s disease. Front Neuroendocrinol. 54, 100764 (2019).
Musiek, E. S., Xiong, D. D. & Holtzman, D. M. Sleep, circadian rhythms, and the pathogenesis of Alzheimer disease. Exp. Mol. Med. 47, e148 (2015).
Shih, R. H., Wang, C. Y. & Yang, C. M. NF-kappaB signaling pathways in neurological inflammation: a mini review. Front Mol. Neurosci. 8, 77 (2015).
Ju Hwang, C., Choi, D. Y., Park, M. H. & Hong, J. T. NF-kappaB as a key mediator of brain inflammation in Alzheimer’s disease. CNS Neurol. Disord. Drug Targets 18, 3–10 (2019).
Ando, K. et al. N-cadherin regulates p38 MAPK signaling via association with JNK-associated leucine zipper protein: implications for neurodegeneration in Alzheimer disease. J. Biol. Chem. 286, 7619–7628 (2011).
Andreyeva, A. et al. C-terminal fragment of N-cadherin accelerates synapse destabilization by amyloid-beta. Brain 135, 2140–2154 (2012).
Mathys, H. et al. Single-cell transcriptomic analysis of Alzheimer’s disease. Nature 570, 332–337 (2019).
Palop, J. J. et al. Aberrant excitatory neuronal activity and compensatory remodeling of inhibitory hippocampal circuits in mouse models of Alzheimer’s disease. Neuron 55, 697–711 (2007).
Schadt, E. E. et al. Genetics of gene expression surveyed in maize, mouse and man. Nature 422, 297–302 (2003).
Mehrabian, M. et al. Integrating genotypic and expression data in a segregating mouse population to identify 5-lipoxygenase as a susceptibility gene for obesity and bone traits. Nat. Genet 37, 1224–1233 (2005).
Ghazalpour, A. et al. Integrating genetic and network analysis to characterize genes related to mouse weight. PLoS Genet 2, e130 (2006).
Yang, X. et al. Tissue-specific expression and regulation of sexually dimorphic genes in mice. Genome Res. 16, 995–1004 (2006).
Emilsson, V. et al. Genetics of gene expression and its effect on disease. Nature 452, 423–428 (2008).
Dobrin, R. et al. Multi-tissue coexpression networks reveal unexpected subnetworks associated with disease. Genome Biol. 10, R55 (2009).
Zhang, W., Zhu, J., Schadt, E. E. & Liu, J. S. A Bayesian partition method for detecting pleiotropic and epistatic eQTL modules. PLoS Comput Biol. 6, e1000642 (2010).
Zhong, H. et al. Liver and adipose expression associated SNPs are enriched for association to type 2 diabetes. PLoS Genet. 6, e1000932 (2010).
Zhong, H., Yang, X., Kaplan, L. M., Molony, C. & Schadt, E. E. Integrating pathway analysis and genetics of gene expression for genome-wide association studies. Am. J. Hum. Genet. 86, 581–591 (2010).
Millstein, J. et al. Identification of causal genes, networks, and transcriptional regulators of REM sleep and wake. Sleep 34, 1469–1477 (2011).
Schadt, E. E., Woo, S. & Hao, K. Bayesian method to predict individual SNP genotypes from gene expression data. Nat. Genet. 44, 603–608 (2012).
Tu, Z. et al. Integrative analysis of a cross-loci regulation network identifies App as a gene regulating insulin secretion from pancreatic islets. PLoS Genet. 8, e1003107 (2012).
Roussos, P. et al. A role for noncoding variation in schizophrenia. Cell Rep. 9, 1417–1429 (2014).
Miller, C. L. et al. Integrative functional genomics identifies regulatory mechanisms at coronary artery disease loci. Nat. Commun. 7, 12092 (2016).
Myers, A. J. The age of the “ome”: genome, transcriptome and proteome data set collection and analysis. Brain Res. Bull. 88, 294–301 (2012).
Myers, A. J. AD gene 3-D: moving past single layer genetic information to map novel loci involved in Alzheimer’s disease. J. Alzheimer’s Dis.: JAD 33, S15–S22 (2013).
Myers, A. J. et al. A survey of genetic human cortical gene expression. Nat. Genet. 39, 1494–1499 (2007).
Sage Bionetworks. NIH/NIA. https://agora.ampadportal.org/genes (2018).
Srikanth, P. et al. Convergence of independent DISC1 mutations on impaired neurite growth via decreased UNC5D expression. Transl. Psychiatry 8, 245 (2018).
Sullivan, S. E. et al. Candidate-based screening via gene modulation in human neurons and astrocytes implicates FERMT2 in Abeta and TAU proteostasis. Hum. Mol. Genet 28, 718–735 (2019).
Johnson, R. D. et al. Single-molecule imaging reveals abeta42:abeta40 ratio-dependent oligomer growth on neuronal processes. Biophys. J. 104, 894–903 (2013).
Kuperstein, I. et al. Neurotoxicity of Alzheimer’s disease Abeta peptides is induced by small changes in the Abeta42 to Abeta40 ratio. EMBO J. 29, 3408–3420 (2010).
Pajarillo, E., Rizor, A., Son, D. S., Aschner, M. & Lee, E. The transcription factor REST up-regulates tyrosine hydroxylase and antiapoptotic genes and protects dopaminergic neurons against manganese toxicity. J. Biol. Chem. 295, 3040–3054 (2020).
Hwang, J. Y. & Zukin, R. S. REST, a master transcriptional regulator in neurodegenerative disease. Curr. Opin. Neurobiol. 48, 193–200 (2018).
Alzheimer’s Association. 2020 Alzheimer’s disease facts and figures. Alzheimers Dement https://doi.org/10.1002/alz.12068 (2020).
Villa, C. et al. Genetics and expression analysis of the specificity protein 4 gene (SP4) in patients with Alzheimer’s disease and frontotemporal lobar degeneration. J. Alzheimers Dis. 31, 537–542 (2012).
Soulie, C., Nicole, A., Delacourte, A. & Ceballos-Picot, I. Examination of stress-related genes in human temporal versus occipital cortex in the course of neurodegeneration: involvement of 14-3-3 zeta in this dynamic process. Neurosci. Lett. 365, 1–5 (2004).
Umahara, T. et al. 14-3-3 proteins and zeta isoform containing neurofibrillary tangles in patients with Alzheimer’s disease. Acta Neuropathol. 108, 279–286 (2004).
Hernandez, F., Cuadros, R. & Avila, J. Zeta 14-3-3 protein favours the formation of human tau fibrillar polymers. Neurosci. Lett. 357, 143–146 (2004).
Mateo, I. et al. 14-3-3 zeta and tau genes interactively decrease Alzheimer’s disease risk. Dement Geriatr. Cogn. Disord. 25, 317–320 (2008).
Hardy, J. & Selkoe, D. J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science 297, 353–356 (2002).
Johansson, C. et al. The roles of Jumonji-type oxygenases in human disease. Epigenomics 6, 89–120 (2014).
Kwok, J., O’Shea, M., Hume, D. A. & Lengeling, A. Jmjd6, a JmjC dioxygenase with many interaction partners and pleiotropic functions. Front Genet. 8, 32 (2017).
Uhlen, M. et al. Proteomics. Tissue-based map of the human proteome. Science 347, 1260419 (2015).
Meyer, K. et al. REST and neural gene network dysregulation in iPSC models of Alzheimer’s disease. Cell Rep. 26, 1112–1127.e1119 (2019).
Lu, T. et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 507, 448–454 (2014).
Esposito, M. & Sherr, G. L. Epigenetic modifications in Alzheimer’s neuropathology and therapeutics. Front. Neurosci. 13, 476 (2019).
Wen, K. X. et al. The role of DNA methylation and histone modifications in neurodegenerative diseases: a systematic review. PLoS One 11, e0167201 (2016).
Day, J. J. & Sweatt, J. D. Epigenetic mechanisms in cognition. Neuron 70, 813–829 (2011).
Butler, A. A., Webb, W. M. & Lubin, F. D. Regulatory RNAs and control of epigenetic mechanisms: expectations for cognition and cognitive dysfunction. Epigenomics 8, 135–151 (2016).
Xu, X. DNA methylation and cognitive aging. Oncotarget 6, 13922–13932 (2015).
Torres, R. F., Kouro, R. & Kerr, B. Writers and readers of DNA methylation/Hydroxymethylation in physiological aging and its impact on cognitive function. Neural Plast. 2019, 5982625 (2019).
Maddock, J. et al. DNA methylation age and physical and cognitive aging. J. Gerontol. A Biol. Sci. Med. Sci. 75, 504–511 (2020).
Ianov, L., Riva, A., Kumar, A. & Foster, T. C. DNA methylation of synaptic genes in the prefrontal cortex is associated with aging and age-related cognitive impairment. Front Aging Neurosci. 9, 249 (2017).
Haberman, R. P., Quigley, C. K. & Gallagher, M. Characterization of CpG island DNA methylation of impairment-related genes in a rat model of cognitive aging. Epigenetics 7, 1008–1019 (2012).
Chouliaras, L. et al. Peripheral DNA methylation, cognitive decline and brain aging: pilot findings from the Whitehall II imaging study. Epigenomics 10, 585–595 (2018).
Hwang, J. Y., Aromolaran, K. A. & Zukin, R. S. The emerging field of epigenetics in neurodegeneration and neuroprotection. Nat. Rev. Neurosci. 18, 347–361 (2017).
Furia, M. Vol. R Package Version 1.11-1. http://www.sagebase.org (2015).
Conway, O. J. et al. ABI3 and PLCG2 missense variants as risk factors for neurodegenerative diseases in Caucasians and African Americans. Mol. Neurodegener. 13, 53 (2018).
Bennett, D. A. et al. Religious orders study and rush memory and aging project. J. Alzheimers Dis. 64, S161–S189 (2018).
Allen, M. et al. Gene expression, methylation and neuropathology correlations at progressive supranuclear palsy risk loci. Acta Neuropathol. 132, 197–211 (2016).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Langmead, B., Trapnell, C., Pop, M. & Salzberg, S. L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25 (2009).
Anders, S., Pyl, P. T. & Huber, W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015).
Babraham Bioinformatics. FastQC. http://www.bioinformatics.babraham.ac.uk/projects/fastqc (2010).
Wang, L., Wang, S. & Li, W. RSeQC: quality control of RNA-seq experiments. Bioinformatics 28, 2184–2185 (2012).
Hansen, K. D., Irizarry, R. A. & Wu, Z. Removing technical variability in RNA-seq data using conditional quantile normalization. Biostatistics 13, 204–216 (2012).
Statham, A. L. et al. Repitools: an R package for the analysis of enrichment-based epigenomic data. Bioinformatics 26, 1662–1663 (2010).
Li, H. et al. The sequence alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Institute, B. A Set of Command Line Tools (in Java) for Manipulating High-Throughput Sequencing (HTS) Data And Formats Such as SAM/BAM/CRAM and VCF. http://broadinstitute.github.io/picard (2019).
Hardik Shah, Y.-C. W., R. Castellanos, C. & Pandya, Z. The 65th Annual Meeting of The American Society of Human Genetics (Hunter Medical Research Institute, 2015).
Robinson, M. D. & Oshlack, A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25 (2010).
Robinson, M. D., McCarthy, D. J. & Smyth, G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47 (2015).
Purcell, S. et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575 (2007).
Zheng, X. et al. A high-performance computing toolset for relatedness and principal component analysis of SNP data. Bioinformatics 28, 3326–3328 (2012).
Patterson, N., Price, A. L. & Reich, D. Population structure and eigenanalysis. PLoS Genet. 2, e190 (2006).
Chang, C. C. et al. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience 4, 7 (2015).
Genomes Project, C. et al. A global reference for human genetic variation. Nature 526, 68–74 (2015).
Howie, B. N., Donnelly, P. & Marchini, J. A flexible and accurate genotype imputation method for the next generation of genome-wide association studies. PLoS Genet 5, e1000529 (2009).
DeLuca, D. S. et al. RNA-SeQC: RNA-seq metrics for quality control and process optimization. Bioinformatics 28, 1530–1532 (2012).
Shabalin, A. A. Matrix eQTL: ultra fast eQTL analysis via large matrix operations. Bioinformatics 28, 1353–1358 (2012).
Benjamini, Y., Drai, D., Elmer, G., Kafkafi, N. & Golani, I. Controlling the false discovery rate in behavior genetics research. Behav. Brain Res. 125, 279–284 (2001).
Michael Linderman, B. Z. coexpp: Large-Scale Co-Expression Network Creation And Manipulation Using WGCNA. R Package Version 0.1.0. https://bitbucket.org/multiscale/coexpp (2011).
Chang, R. et al. Predictive metabolic networks reveal sex- and APOE genotype-specific metabolic signatures and drivers for precision medicine in Alzheimer’s disease. Alzheimers Dement https://doi.org/10.1002/alz.12675 (2022).
Cadiz, M. P. et al. Culture shock: microglial heterogeneity, activation, and disrupted single-cell microglial networks in vitro. Mol. Neurodegener. 17, 26 (2022).
Tarjan, R. Depth-first search and linear graph algorithms. SIAM J. Comput. 1, 146–160 (1972).
Acknowledgements
The authors thank the following funding resources which supported this study: NIH/NIA 1R56AG062620-01 and NIH/NINDS/Mayo Clinic U54NS110435 subaward to R.C.; NIH/NIA 1RF1AG057457-01 to R.C. and T.L.Y.-P.; and NIH/NIA R01AG055909 and RF1NS117446 to T.L.Y.-P. We also thank the Center of Innovations in Brain Sciences at University of Arizona for seed funding to R.C. R.C. is the founder of INTelico Therapeutics LLC and a co-founder of PATH Biotech LLC. This study is not supported by any funding from INTelico Therapeutics LLC or PATH Biotech LLC.
Author information
Authors and Affiliations
Contributions
T.L.Y.-P. and R.C. conceptualized this study. M.Y.R.H. and N.E.T. pre-processed the MAYO RNAseq and whole genome sequencing data; M.Y.R.H. pre-processed the ROSMAP RNAseq and whole genome sequencing data. K.Z., R.C., M.Y.R.H., and N.E.T. analyzed cell type-specific gene expression. K.Z., B.L., S.L.M., M.A., S.S.A.Z, M.Y.R.H., and R.C. performed network construction and data analysis. J.P.M., R.V.P.II, and T.L.Y.-P. designed and implemented induced neuron shRNA gene knockdown experiments. D.A.B. provided technical assistance. K.Z., J.P.M, and R.C. wrote the manuscript and generated the figures. J.P.M., T.L.Y.-P., and R.C. edited the manuscript and figures.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Communications Biology thanks Devika Agarwal and Zhisong He for their contribution to the peer review of this work. Primary Handling Editors: Alex Nord and George Inglis.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Merchant, J.P., Zhu, K., Henrion, M.Y.R. et al. Predictive network analysis identifies JMJD6 and other potential key drivers in Alzheimer’s disease. Commun Biol 6, 503 (2023). https://doi.org/10.1038/s42003-023-04791-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s42003-023-04791-5
- Springer Nature Limited