
Abstract
LINE-1 and Alu retrotransposons are components of the human genome and have been implicated in many human diseases. These elements can influence human transcriptome plasticity in various mechanisms. Chimeric transcripts derived from LINE-1 and Alu can also impact the human transcriptome, such as exonization and post-transcriptional modification. However, its specific role in ASD neuropathology remains unclear, particularly in the cerebellum tissues. We performed RNA-sequencing of post-mortem cerebellum tissues from ASD and unaffected individuals for transposable elements profiling and chimeric transcript identification. The majority of free transcripts of transposable elements were not changed in the cerebellum tissues of ASD compared with unaffected individuals. Nevertheless, we observed that chimeric transcripts derived from LINE-1 and Alu were embedded in the transcripts of differentially expressed genes in the cerebellum of ASD, and these genes were related to developments and abnormalities of the cerebellum. In addition, the expression levels of these genes were correlated with the significantly decreased thickness of the molecular layer in the cerebellum of ASD. We also found that global methylation and expression of LINE-1 and Alu elements were not changed in ASD, but observed in the ASD sub-phenotypes. Our findings showed associations between transposable elements and cerebellar abnormalities in ASD, particularly in distinct phenotypic subgroups. Further investigations using appropriate models are warranted to elucidate the structural and functional implications of LINE-1 and Alu elements in ASD neuropathology.
Similar content being viewed by others


Introduction
Autism spectrum disorder (ASD) is a complex neurodevelopmental disorder that is characterized by two behavioral impairments, including significant (i) deficits in social interactions and communication and (ii) restricted interests and repetitive behaviors according to the diagnostic and statistical manual of mental disorders, Fifth Edition (DSM-5) criteria1. Over the past decade, the ASD prevalence has dramatically increased. The Centers for Disease Control and Prevention (CDC) has estimated the prevalence of ASD in 2020 is approximately 1 in 36 children in the United States2. ASD is known as a multifactorial disorder, and the exact causes of ASD have not been fully identified. Currently, several studies have demonstrated that a combination of genetic and environmental factors affects the heterogeneity of ASD clinical phenotypes3,4,5. This evidence has led to the debate on nongenetic factors or epigenetics such as DNA methylation, which is an intermediate between environmental and genetic factors5,6,7,8,9. DNA methylation mechanism is a fundamental contributor to brain development and pathogenesis of ASD and also related to ASD10.
Transposable elements (TEs) can be classified into two types based on their structure and the requirement of reverse transcription for transposition, including DNA transposons and retro (RNA)-transposons11. There are two main types of retrotransposons: long terminal repeats (LTRs: ERV) and non-long terminal repeats (non-LTRs)11,12. The most common non-LTR retrotransposons are long interspersed nuclear element 1 (LINE-1 or L1) and short interspersed nuclear element (SINE: Alu and MIR) retrotransposons, which remain active and comprise approximately 30% at a copy number of over 1 million elements in the human genome11,12. Retrotransposons are known as mobile genetic material that can create a new copy into a new genomic location. LINE-1 and Alu elements are most of the retrotransposons that remain active in the human genome. These elements can affect the human genome functions in several mechanisms such as insertion, recombination, and transcriptome through introducing alternative promoters or enhancers, novel splicing sites, and epigenetic alteration11. These processes lead to the production of embedded transcripts or chimeric transcripts of LINE-1 and Alu elements, which can impact the human transcriptome by exonization and post-transcriptional modification11. LINE-1 is 6 kb long and consists of four compartments. At 5’UTR region of LINE-1 contains an internal promoter for RNA polymerase II that controls LINE-1 expression. LINE-1 also contains two open reading frames, including ORF1 and ORF2, which encode for proteins that require for retrotransposition mechanism. ORF1 protein has RNA binding and nucleic acid chaperone activity, while ORF2p protein has endonuclease and reverse transcriptase activities13.
Current evidence suggests that aberrant DNA methylation of LINE-1 element is associated with several human brain disorders, especially in individuals with schizophrenia and ASD individuals. LINE-1 methylation and expression were altered in several tissues of individuals with ASD including blood, lymphoblastoid cell lines, and post-mortem brain tissues. We have previously reported alterations in global DNA methylation of LINE-1 and Alu retrotransposons in lymphoblastoid cell lines (LCLs) of ASD subtypes, and such alterations were specific to the ASD case subgroup based on clinical phenotypes14,15. Our previous study also demonstrated that Alu elements were differentially expressed and associated with the expression of host genes in the post-mortem prefrontal cortex of ASD individuals16. Furthermore, whole-genome sequencing analysis showed that insertion of LINE-1 and Alu retrotransposons occurred exclusively in the brain tissues, especially LINE-1 that have a higher number of novel insertions in the ASD brain tissues than observed in normal brain tissues17. The study using whole genome sequencing data of 2288 families with ASD from the Simons Simplex Collection found that there were de novo insertions of LINE-1 in ASD-associated genes18. Following Shpyleva’s study, the binding of MeCp2 repressive protein to LINE-1 promoter was significantly decreased and correlated with overexpression of LINE-1 in the post-mortem cerebellum tissues of ASD individuals19. Another interesting study also found that LINE-1 was upregulated in the ASD subset and showed a potential impact on the expression of ASD-relevant genes20. These findings support that LINE-1 and Alu retrotransposons were changed in ASD brain tissues. However, the specific role of transposable elements and their chimeric transcripts in ASD neuropathology remains unclear, particularly in the cerebellum tissues.
In this study, we performed RNA-sequencing of post-mortem cerebellum tissues for the identification of chimeric transcripts derived from LINE-1 and Alu elements using the ChimeraTE tool. Transposable element profiling was also analyzed for the cerebellum tissues and the Purkinje cells dissected from post-mortem cerebellum tissues using three different methods including TEtranscripts, Teloscope, ERV methods. We compared gene expression profile with a list of genes that produced chimeric transcripts in the cerebellum of ASD individuals and predicted biological function using ingenuity pathway analysis (IPA). Finally, we sought to determine whether transposable elements are associated with neuropathology found in the cerebellum tissues of individuals with ASD. Immunohistochemistry was performed to determine the neuropathology in the cerebellum of ASD, including Purkinje cells (PCs) density, PCs soma size, and the thickness of the cerebellum layer. We then conducted correlation analyses between the expression of genes that produced chimeric transcripts and neuropathology in the cerebellum tissue of ASD individuals. In this study we also determined DNA methylation levels of LINE-1 and Alu retrotransposons in the cerebellum tissues using Combined Bisulfite Restriction Analysis (COBRA) for investigating DNA methylation levels and patterns in the promoters of these elements. To determine the consequences of the DNA methylation on LINE-1 and Alu elements, we conducted quantitative real-time PCR for the relative copy number and expression levels of LINE-1 and Alu elements. This study provided a better understanding of the relationship between retrotransposons and neuropathology in the cerebellum tissues of ASD individuals.
Results
Identification of chimeric transcripts which derived from genes and transposable elements in the post-mortem cerebellum tissues
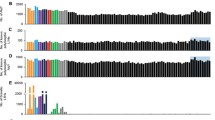
To identify chimeric transcripts derived from genes and retrotransposons (LINE-1, Alu, and MIR), we performed paired-end RNA-sequencing of post-mortem cerebellum tissues from ASD and unaffected individuals. Firstly, there were no significant differences in age, sex, or post-mortem interval (PMI) between the ASD group and the unaffected group (CTRL). The ChimeraTEs package was used to identify chimeric reads that were aligned to both genes and transposable elements (TEs) in different types, including exon embedded, intronic, overlapped exon and intron, initiated, and terminated types. The analyses were performed separately for LINE-1, Alu elements, and MIR, number of chimeric transcripts and genes that produced chimeric transcripts are shown in Table 1 and Fig. 1A,B. We discovered a total of 21,628 and 19,261 chimeric transcripts that were detected more than two reads in at least 70% of the cerebellum tissues from ASD and CTRL (unaffected individuals), respectively (see Supplementary Table S1, S2, and S3). These chimeric transcripts were derived from a total of 12,601 and 11,345 genes from a total unique 70,711 gene models of the GENDOCE annotation (including long non-coding RNAs). Although the total number of chimeric transcripts between ASD and CTRL was not different, we observed that the number of unique chimeric transcripts and genes that produced chimeric transcripts exhibited differences. The number (reads) of chimeric transcripts derived from LINE-1, Alu elements, and MIR in the cerebellum of ASD and unaffected CTRL were shown as heatmaps in Fig. 1C,D,E respectively.

Identification of chimeric transcripts derived from genes and retrotransposons (LINE-1, Alu, and MIR) in the post-mortem cerebellum tissues of ASD and unaffected individuals. (A) Bar chart of the number of unique chimeric transcripts in the cerebellum of CTRL and ASD individuals (B) Bar chart of the number of unique genes that produced chimeric transcripts in the cerebellum of CTRL and ASD individuals. (C) Heatmap of chimeric transcripts derived from LINE-1 family in the cerebellum of CTRL and ASD individuals. (D) Heatmap of chimeric transcripts derived from the Alu family in the cerebellum of CTRL and ASD individuals. (E) Heatmap of chimeric transcripts derived from MIR family in the cerebellum of CTRL and ASD individuals.
Moreover, we observed that most of the chimeric transcripts derived from these elements were embedded in the exons of transcripts. These results indicated that there were unique chimeric transcripts that were produced from genes and LINE-1, Alu elements, and MIR in the post-mortem cerebellum tissues of ASD and CTRL. These transcripts, which embedded transcriptome profiles may contribute to transcriptome plasticity or diversity related to ASD.
Transposable elements profiling in the post-mortem cerebellum tissues and Purkinje cells of ASD and unaffected individuals
We also investigated individual transcripts derived from TEs (free transcripts) in the cerebellum tissues. We performed expression profiling for TEs transcripts using three different methods including TEtranscripts, Teloscope, and ERVmap in our post-mortem cerebellum tissues and GSE211154 data, which were sequenced from Purkinje cells dissected from the cerebellum tissues of ASD and CTRL. Firstly, we found that there were no significant differences in TEs expression levels when combining individual transcripts into family (LINE-1, Alu, MIR) as well as when combined into subfamily (L1H, L1M, L1P, AluJ, AluS, and AluY) (Fig. 2A,B,C). When differential expression was performed for individual transcripts, there were significant TEs transcripts in ASD sub-phenotypes based on ADI-R scores including non-verbal ASD (NV-ASD) and verbal ASD (V-ASD) (Table 2). For all ASD compared with sex- and age-matched unaffected CTRL, we found only 1–10 significantly differentially expressed transposable elements (DETEs) in the Purkinje cells and cerebellum tissues at statistical threshold adjusted P-value less than 0.05. In ASD sub-phenotypes, we found more DETEs in the cerebellum tissues, especially in the NV-ASD (Table 2) (Purkinje cells were not classified due to unavailable phenotypic data). The Teloscope method showed 10, 10, and 38 DETEs in the cerebellum tissues of all ASD, NV-ASD, and V-ASD when compared with sex- and age-matched unaffected individuals (Table 2)(Fig. 2D, E,F). The list of DETEs from Teloscope method at a P-value less than 0.05 are in Supplementary Table S4. Furthermore, the heatmap of the expression level of DETEs in all ASD (Fig. 2G), NV-ASD (Fig. 2H), and NV-ASD (Fig. 2I) exhibited different expression patterns when compared to sex- and age-matched unaffected individuals. However, only a small number of differentially expressed TEs were found out of over a million TEs transcripts, indicating that the majority of free TEs transcripts were not differentially expressed in ASD. These findings indicated that the expression of free TEs transcripts in the cerebellum tissues and Purkinje cells of ASD were not significantly different when compared with sex- and age-matched unaffected individuals.

Expression profiles of transposable elements in the post-mortem cerebellum tissues of ASD and unaffected individuals using Teloscope method. (A) Box plot of expression profile of TEs family in the cerebellum tissues (B) Box plot of expression profile of LINE-1 subfamily in the cerebellum tissues (C) Box plot of expression profile of Alu subfamily in the cerebellum tissues (D) Volcano plot of transposable elements profiling of all ASD versus unaffected CTRL (E) Volcano plot of transposable elements profiling of NV-ASD versus sex- and age-matched unaffected CTRL (F) Volcano plot of transposable elements profiling of V-ASD versus sex- and age-matched unaffected CTRL. (G) Heatmap of expression levels of DETEs in the cerebellum tissue of all ASD and unaffected CTRL. (H) Heatmap of expression levels of DETEs in the cerebellum tissue of NV-ASD and sex- and age-matched unaffected CTRL. (I) Heatmap of expression levels of DETEs in the cerebellum tissue of V-ASD and sex- and age-matched unaffected CTRL.
Differentially expressed transcripts of genes that produced chimeric transcripts were associated with developments and abnormalities of the cerebellum.
Consequently, we performed gene expression profiling and intersected with the list of genes that produced chimeric transcripts in the ASD individuals to figure out whether chimeric transcripts derived from genes and transposable elements (embedded TEs-transcripts) are associated with developments or abnormalities of the cerebellum. Firstly, gene expression profiling showed that only three genes were significantly differentially expressed in the cerebellum of ASD compared with CTRL (Fig. 3A). We also performed an expression profile for transcript level (isoforms level) and found that there are 198 differentially expressed transcripts (DETs) with FDR less than 0.05 in the cerebellum tissues of ASD compared with sex- and age-matched CTRL (Fig. 3B) (see Supplementary Table S5). To determine the biological functions of differentially expressed genes that produced chimeric transcripts, we overlapped the list of DETs and genes that produced chimeric transcripts in ASD. The results show that 76 genes out of 198 DETs were genes that produced chimeric transcripts in the cerebellum of ASD individuals, which corresponded to 209 chimeric transcripts of LINE-1, Alu, MIR family (Fig. 3C). The distribution of chimeric transcripts based on TEs family and type of chimeric showed that most chimeric transcripts belonging to exon-embedded transcripts that derived from Alu and LINE-1 family (Fig. 3D) (see Supplementary Table S6). This finding raised the possibility that altered interactome profiles in the cerebellum of ASD individuals may be linked to LINE-1 and Alu elements embedded in gene exon transcripts.

Chimeric transcript derived from genes and transposable elements using ChimeraTE mode 1 genome-guide method. (A) Volcano plot of gene expression profiling of all ASD versus unaffected CTRL. (B) Volcano plot of mRNA expression profiling (isoform-level) of all ASD versus unaffected CTRL. (C) Venn diagram of intersection between DETs and genes that produced chimeric transcripts in the cerebellum of ASD. (D) Pie chart of distribution of chimeric transcripts derived from LINE-1, Alu elements, and MIR in the cerebellum tissues. (E) Gene regulatory network of DETs in the cerebellum tissues of ASD predicted by IPA.
Next, we then performed an ingenuity pathway analysis (IPA) for 198 DETs in the cerebellum of ASD. We found interesting developmental disorders and neurological diseases predicted by IPA for these genes including autism spectrum disorders, medical comorbidities (seizure disorder, epilepsy, pervasive developmental disorder, movement disorders), and behavioral comorbidities (hyperactive behavior) (Table 3). Moreover, the gene regulatory network that related to cellular development predicted by IPA also showed that some of the genes in this interactome were associated with cerebellar developments and abnormalities including responsiveness of Purkinje cells, development of Purkinje and granule cells, the thickness of molecular layer of the cerebellum, abnormal morphology of cerebellar cortex (Fig. 3E). These findings indicated that differentially expressed genes that produce chimeric transcripts in the ASD were associated with developments and abnormalities of the cerebellum.
Relationship between genes that produced chimeric transcripts and neuropathology in the cerebellum tissues of ASD individuals
To investigate neuropathology in the cerebellum of ASD individuals, we performed immunohistochemistry using paraffin-embedded post-mortem brain tissues (n; ASD = 6, control = 7) stained with anti-calbindin antibody. The linear density of Purkinje cells (PCs) was determined by counting the number of PCs per millimeter length of monolayer (PCs/mm). We then measured PCs soma size, molecular layer thickness, and molecular layer thickness at the folium surface (FS) (Fig. 4A). The result showed that PCs number and soma area were not significant differences in the ASD individuals compared to unaffected CTRL (P-value = 0.771 and 0.362, respectively) (Fig. 4B,C). Nevertheless, the molecular thickness and thickness at the folium surface exhibited significantly decreased in the ASD than unaffected CTRL (Δ thickness = 83.63 and 138.34 \(\upmu\) m, P-value = 0.023 and 0.018, respectively) (Fig. 4D,E). The summarized statistics are provided in Supplementary Table S7.

Immunohistochemical staining of cerebellar Purkinje cells with anti-calbindin antibody in ASD and unaffected CTRL. (A) The paraffin-embedded cerebellum tissue sections were stained with anti-calbindin (DAB-positive cells) and counterstained with hematoxylin. The images were photographed at 20X and 10X, respectively. (B–E) Boxplot of Purkinje cells density (PCs/mm), Purkinje cell body size (PCs soma, mm2), molecular layer thickness (Mol layer length), molecular layer thickness at folium surface (Mol layer length; FS), respectively. (F) Correlation matrix between neuropathology in the cerebellum of ASD and genes that produced chimeric transcripts and related to developments and abnormalities of the cerebellum. (NS = not significant, * = significant at the P-value less than 0.05).
We then selected differentially expressed genes that produced chimeric transcripts from the gene regulatory network that related to developments and abnormalities of the cerebellum for correlation with neuropathology in the cerebellum of ASD, including ENTPD6, HDAC7, JADE1, NEU1, PTAR1, PTPDC1, VPS36, and ZNF423 genes (Fig. 3E). The correlation matrix showed that expression of these genes exhibited correlation with the PCs number, PC soma area, molecular layer thickness, and molecular layer thickness at folium surface (FS) (Fig. 4F). Especially, ENTPD6, JADE1, and ZNF423 genes showed moderate correlation coefficients with the decreasing of molecular layer thickness and molecular layer thickness at FS in the ASD (R-value = 0.40–0.60). While HDAC7, JADE1, and PTAR1 genes showed moderate to high correlation with the number and soma size of PCs which were not change in ASD individuals. These findings indicated that genes that produced chimeric transcripts of transposable elements were related to the neuropathology in the cerebellum of ASD.
Global LINE-1 methylation levels were altered in the cerebellum tissues of ASD with non-verbal phenotype, but Alu elements were unchanged
Because LINE-1 and Alu elements contribute a large number of methylation loci in the genome, we investigated global methylation by measuring their methylation levels. We used combined bisulfite restriction analysis (COBRA) to determine total LINE-1 and Alu methylation levels at two CpG sites in the promoters of LINE-1 and AluSx elements. The percentage of total methylation (%mC) and methylation patterns (%mCmC, %mCuC, %uCmC, %uCuC) were investigated according to our previous study.
The COBRA analysis for LINE-1 methylation showed no significant difference in total LINE-1 methylation (%mC) level and patterns between all ASD compared with their sex- and age-unaffected CTRL (Supplementary Table S8, Fig. 5A). We thus analyzed the COBRA results for ASD sub-phenotypes based on ADI-R scores, which classified ASD individuals into non-verbal ASD (NV-ASD) and verbal ASD (V-ASD). Then we compared each ASD sub-phenotype with their sex- and age-matched unaffected CTRL. We found changes in methylation levels and patterns between the NV-ASD and V-ASD subgroups. The total LINE-1 methylation (∆%mC = 0.621, P-value = 0.018) and hypomethylated pattern (∆%mCmC = -0.578, P-value = 0.0001) were significantly increased and decreased, respectively, in the NV-ASD (Supplementary Table S8, Fig. 5B). In addition, we also found a significantly decreased partially methylated pattern (∆%mCuC = -2.143, P-value = 0.02) in the V-ASD compared with sex- and age-matched individuals (Supplementary Table S8, Fig. 5C). This result suggested LINE-1 methylations were changed in some ASD individuals and specific to clinical phenotype (ADI-R score). Finally, we did not observe different Alu methylation in the cerebellum tissues of ASD compared with unaffected CTRL (Supplementary Figure S1 and Supplementary Table S9). These findings indicated that LINE-1 methylation levels and patterns were altered in the cerebellum of ASD sub-phenotypes not all ASD individuals, while Alu elements were not altered.

Box plots of the COBRA results of LINE-1 methylation levels and patterns in the cerebellum tissues of ASD and unaffected CTRL. (A) Box plot of LINE-1 methylation levels and patterns in the ASD and unaffected CTRL. (B) Box plot of LINE-1 methylation levels and patterns in the non-verbal ASD (NV-ASD) and matched unaffected CTRL. (C) Box plot of LINE-1 methylation levels and patterns in the verbal ASD (V-ASD) and matched unaffected CTRL. (NS = not significant, * = significant at the P-value less than 0.05 level).
LINE-1 expression and relative copy number were altered in the post-mortem cerebellum tissues of ASD sub-phenotypes
Because of the variations in DNA methylation levels observed in the LINE-1 promoter, a mechanism controlling the activity of transposable elements. A dysregulation of LINE-1 in the brain tissues of people with ASD may result from modifications in the DNA methylation of LINE-1 in the cerebellum tissues. Consequently, we determined the relative copy number and expression levels of LINE-1 and Alu retrotransposons in the post-mortem brain tissues of people with ASD and unaffected CTRL.
Because LINE-1 has a long sequence and is independently located around the human genome, so, LINE-1 in the human genome was found in different structures including full-length, 5’UTR truncated, ORF1 truncated, and ORF2 truncated. In this study, we conducted qPCR analysis for the expression level of these LINE-1 including L1UTR, ORF1, and ORF2. The result showed that L1UTR expression was significantly reduced in the cerebellum of ASD compared with unaffected CTRL (log2FC = − 0.87, P-value = 0.044) (Fig. 6A). In contrast, ORF1 and ORF2 were not different between ASD and unaffected individuals. Next, we determined the relative copy number of LINE-1 including L1UTR and ORF2, but we did not observe the difference between ASD and unaffected CTRL (Fig. 6A). When the analysis was performed for the ASD subgroup based on the ADI-R score of ASD individuals, we found that ORF1 expression level was significantly decreased (log2FC = −1.04, P-value = 0.045) in the NV-ASD compared with sex- and age-matched unaffected CTRL, but not in the V-ASD individuals (Fig. 6B). Whereas the relative copy number of LINE-1 exhibited higher in V-ASD individuals not in the NV-ASD (L1UTR; log2FC = 0.43, P-value = 0.020, and ORF2; log2FC = 0.496, P-value = 0.013) (Fig. 6C). The summarized statistics are provided in Supplementary Table S8.

Box plot of the expression levels and relative copy number of LINE-1 in the cerebellum tissues of ASD and unaffected CTRL. (A) Box plot of LINE-1 expression levels and relative copy number in the ASD and unaffected CTRL. (B) Box plot of LINE-1 expression and relative copy number in the non-verbal ASD (NV-ASD) and matched unaffected CTRL. (C) Box plot of LINE-1 expression and relative copy number in the verbal ASD (V-ASD) and matched unaffected CTRL. (NS = not significant, * = significant at the P-value less than 0.05 level).
Relationship between LINE-1 and neuropathology in the cerebellum tissues of ASD
We further performed correlation analysis between global LINE-1 methylation, expression, relative copy number, and neuropathology in the cerebellum of ASD and unaffected CTRL. Interestingly, we found several histological parameters were significantly correlated with LINE-1 methylation. Total methylation (%mC) showed a negative correlation with PCs soma area (R-value = -0.742) as well as a hypermethylated pattern (%mCmC) (Fig. 7). While partially methylated patterns (%mCuC and %uCmC) had a positive correlation with molecular layer thickness and a negative correlation with PCs density, respectively. These findings indicated that neuropathology in the cerebellum tissues was related to global LINE-1 methylation levels in the ASD, especially differentially methylated pattern (%mCuC) that was strongly correlated to molecular layer thickness (R = 0.75–0.79). Moreover, neuropathology was also related to the relative copy number and expression levels of LINE-1 in the cerebellum tissues of ASD individuals. This finding indicated that LINE-1 in the cerebellum of ASD individuals may related to the altered neuropathology of ASD.

Scatter plot matrix of correlation between neuropathology and relative copy number, expression, and DNA methylation of LINE-1 in the cerebellum tissues of ASD and unaffected CTRL. The LINE-1 relative copy number, expression, methylation levels, and patterns were correlated with Purkinje cells density (PCs/mm), molecular layer thickness (Mol layer length), molecular layer thickness at folium surface (Mol layer length; FS), and Purkinje cell body size (PCs soma, mm2).
Correlation analysis between ADI-R scores and LINE-1 in the ASD cerebellum tissues
To determine whether LINE-1 was related to ASD severity based on AID-R scores, we correlated the DNA methylation, expression, and relative copy number of LINE-1 with ADI-R scores of ASD individuals. We found that the decreasing LINE-1 methylation (total %mC) was correlated with worse ADI-R scores, especially in the social interaction domain (ADI-R A) and communication domain for verbal (ADI-R B V). While the relative copy number showed negative correlations, the LINE-1 expression levels were positively correlated with worse ADI-R scores of the repetitive behavior domain (ADI-R C) and the communication domain for nonverbal (ADI-R B NV) (Fig. 8). These findings indicated that LINE-1 related to the ASD severity based on ADI-R scores and deserved further study using animal models and behavioral examination.

Correlation matrix between ADI-R scores and DNA methylation, expression, and relative copy number of LINE-1 in the cerebellum tissues of ASD individuals.
Discussion
Both LINE-1 and Alu retrotransposons were linked to ASD, according to a number of studies by different investigators. These studies focused on the insertion or retrotransposition of these elements and how it affected the expression of genes linked to ASD14,15,16,17,18,19,20,21. However, chimeric transcripts in the cerebellum tissues of ASD individuals have not been identified. In this study, we have identified the chimeric transcripts derived from genes and LINE-1/Alu retrotransposons in the cerebellum tissues of ASD and unaffected individuals. As well as several genes that produced these chimeric transcripts were also identified in our study. These findings suggested that LINE-1 or Alu embedded in the transcripts may be involved with the transcriptome diversity of the cerebellum tissues. Transposable elements embedded in the transcripts or genes can drive transcriptome diversity and post-transcriptional regulation in the human brain development by contributing regulatory sequence, alternative promoters, source of miRNA, and post-transcriptional modification such as RNA editing11,22,23,24,25,26,27. We observed differences in the number of unique chimeric transcripts and genes that produced chimeric transcripts between ASD and unaffected CTRL, especially LINE-1 and Alu retrotransposons embedded in the exon of differentially expressed transcripts (Table 1). According to our transcriptome analysis, we found there were no significantly differentially expressed genes in the cerebellum tissues of ASD compared to sex- and age-matched unaffected individuals. This finding is consistent with several transcriptome studies of both post-mortem cerebellum tissues and Purkinje cells from ASD individuals compared to unaffected individuals28,29,30. Nevertheless, some studies have reported that long noncoding RNAs associated with ASD were dysregulated in the post-mortem frontal, temporal, and cerebellum tissues of ASD individuals29,31. In this study, we also performed an analysis for isoform levels in the cerebellum and found the differentially expressed transcripts when compared to unaffected individuals. IPA results showed that functions of these transcripts were associated with ASD and developments and abnormalities of cerebellum tissues.
For neuropathology in the cerebellum of ASD, several studies have reported the reduction of Purkinje cell number in the ASD32,33,34. However, there is an inconsistency among studies as well as neuronal cell numbers in the prefrontal cortex34,35. The heterogeneity within the ASD population might cause this inconsistency, subtyping ASD individuals based on clinical phenotypes using larger samples may be helpful. In this study, we did not observe a change in Purkinje cell number and soma size in the ASD, only cerebellar molecular thickness was significantly reduced in the ASD. Although the reduction of cerebellar molecular thickness has not been reported in ASD, abnormalities in the cerebellar lobules have been observed in individuals with ASD, with lower cortical thickness36. The structural changes in the cerebellum may influence the neuropathogenesis of ASD, highlighting the importance of understanding cerebellar mechanisms in ASD. At the molecular layer thickness, there are molecular interneurons that play an essential role in cerebellar processing by regulating Purkinje cell activity37. Molecular interneurons consist of basket cells and stellate cells, which function as inhibitory synaptic transmission regulators for controlling excitatory circuits37,38. Abnormalities of interneurons in ASD have been widely reported across brain regions as well as in the cerebellum tissues39. According to Whitney et al., the density of both interneurons in the cerebellum did not differ from that of unaffected individuals; however, the expression of parvalbumin, an interneuron marker, in the cerebellum was considerably lower in ASD40,41.
We thus performed a correlation analysis between neuropathology in the cerebellum of ASD and chimeric genes selected from the interactome related to developments and abnormalities of cerebellum predicted by IPA including ENTPD6, JADE1, HDAC7, PTAR1, and ZNF423 genes. Interestingly, ZNF423 was related to abnormal morphology of the cerebellar cortex, developments, and quantity of Purkinje cells, and proliferation of granule cells in the cerebellum (Fig. 3E). The expression of the ZNF423 gene exhibited a moderate correlation with the molecular layer thickness of cerebellum tissues. ZNF423 belongs to the family of Kruppel-like C2H2 zinc finger proteins which functions as a DNA-binding transcription factor involved in various signaling pathways related to brain development42. ZNF423 regulated the cell cycle and division of Purkinje neuron progenitors, and mutation of this gene was linked to cerebellar malformations like Joubert syndrome43. However, the role of ZNF423 and other correlated genes (ENTPD6, JADE1, HDAC7, PTAR1) was not thoroughly studied. Their potential impact on the molecular layer thickness of the cerebellum and ASD neuropathology requires further investigation.
Current evidence links aberrant DNA methylation of LINE-1 and Alu retrotransposons to human brain disorders, especially in individuals with schizophrenia and ASD44. In the post-mortem brain tissues of ASD, whole-genome sequencing analysis shows a higher number of novel insertions of LINE-1 and Alu retrotransposons than found in normal brain tissues17. Analysis using whole genome sequencing data of 2,288 families with ASD from the Simons Simplex Collection found that there was de novo insertion of LINE-1 in ASD-associated genes18. In this study, we also investigated DNA methylation, expression, and relative copy number in individuals with ASD and ASD sub-phenotypes based on ADI-R scores45,46. Reducing the heterogeneity of ASD by classifying ASD based on clinical phenotype may be beneficial, as demonstrated by previous findings from our investigators14,15,45,46,47. Subcategorizing ASD allowed us to observe the molecular signature of ASD individuals. We have previously reported alterations in global DNA methylation of LINE-1 and Alu retrotransposons in lymphoblastoid cell lines (LCLs) of ASD sub-phenotypes, and such alterations were specific to the ASD subset based on clinical phenotypes. Alteration of LINE-1 methylation in LCLs was specific for ASD with severe language impairment, whereas alteration of Alu methylation was specific for ASD with mild symptoms. Furthermore, several genes with LINE-1 and Alu insertions were differentially expressed in peripheral blood and LCLs of ASD. These genes are known to be associated with neurodevelopmental disorders14,15. In this study, we used COBRA to determine global LINE-1 and AluSx methylation levels and patterns in the cerebellum of ASD individuals. We observed the same results as our previous study and other authors that there was no significant difference in the DNA methylation levels and expression of LINE-1 and Alu in the brain tissues of all ASD compared with unaffected individuals16,19,20.
When we classified ASD individuals based on ADI-R score for nonverbal and verbal ASD, we found the difference in LINE-1 methylation, expression, and copy number in the cerebellum tissues of each ASD sub-phenotype, while Alu was not changed. For global LINE-1 methylation, we found that total LINE-1 methylation (%mC) was explicitly altered in the cerebellum of nonverbal ASD. Intriguingly, LINE-1 expression was decreased in the cerebellar tissue while total LINE-1 methylation showed hypermethylation in the nonverbal ASD compared to matched unaffected individuals. Moreover, the decreasing LINE-1 methylation (total %mC) was correlated with worse ADI-R scores and reduced cerebellar molecular layer thickness in the cerebellum of ASD. These findings deserved further study of LINE-1 and Alu retrotransposons in autism-related behaviors and neuropathogenesis in ASD by other mechanisms such as genetic instability, gene fusion, and somatic mosaicism11. It is very important to note that our finding is a preliminary study that highlighted the relationship, especially LINE-1 on neuropathology in the cerebellum of individuals with ASD.
Identification of chimeric transcripts and transposable elements profiling can be affected by several factors, such as low mappability of multiple mapped sequences, RNA library preparation, and mappability of some active young TEs. The use of post-mortem brain tissues and the small sample size resulting from the limited availability of brain tissues were additional limitations. Larger sample sizes would enhance the generalizability of our findings, and future studies with larger cohorts are necessary to validate and extend our results. Our analyses might not accurately represent the overall methylation status in the LINE-1 and Alu retrotransposons since COBRA is predicated on restriction enzyme recognition sites. Pyrosequencing analysis may be used to confirm this limitation in the future. While our study included supporting data, it lacked a functional analysis to establish the presence of LINE-1 and Alu retrotransposons, which are linked to autism spectrum disorders, in the cerebellar tissues of ASD individuals. This study is a pilot study that demonstrated the association between LINE-1 and neuropathology in the cerebellum tissues of ASD.
Our findings should be confirmed in the future using appropriate models to study the role of transposable elements in both structural and functional aspects of the brain. DNA methylation inhibitors (5-azacytidine and 5-aza-2'-deoxycytidine) and synthetic and anti-retroviral drugs can be used in conjunction to investigate the role of LINE-1 in neuronal functions using a neuronal cell model or brain organoids48,49. Moreover, the in-utero electroporation approach can help induce specific brain abnormalities of LINE-1 during developmental stages and follow the behavioral deficits associated with ASD. It is also important to treat or correct LINE-1 epigenetic abnormalities. We can investigate using appropriate techniques in animal models, such as: in vivo locus-specific epigenome editing using the CRISPR-dCAS9 system50. We can moderate transcription or restore the epigenetic status of LINE-1 by combining the CRISPR-dCAS9 system with DNMTs and TET enzymes, which are DNA methylation and reversal-DNA methylation enzymes, respectively.
Conclusion
In this study, we identified chimeric transcripts derived from genes and LINE-1 and Alu retrotransposons in the cerebellum tissues of people with ASD. These transcripts were linked to abnormalities and developments in the cerebellum tissues as well as ASD. Furthermore, we pointed out a relationship between neuropathology in the cerebellum of ASD, LINE-1, and ASD severity (ADI-R scores). Our findings encourage more functional research utilizing suitable models, such as brain organoids or animal models, to examine the function of transposable elements in the structural and functional aspects of the brains of people with ASD.
Methods
Ethics declarations
This research protocols were approved by the research ethics review committee of Chulalongkorn University which all methods were performed in accordance with the relevant guidelines and regulations . Furthermore, the post-mortem cerebellar tissues used in this study were obtained from the Harvard Brain Tissue Resource Center. These tissues were acquired ethically, and informed consent was obtained in accordance with the relevant guidelines and regulations.
Post-mortem brain tissues from ASD and unaffected controls
The post-mortem brain tissues used in this study consist of samples from 15 individuals in total: 8 individuals with ASD and 7 unaffected controls. Snap-frozen post-mortem brain tissues (n; ASD = 8, control = 5) were utilized for DNA and RNA isolations, which were then subjected to molecular analysis. In contrast, paraffin-embedded post-mortem brain tissues (n; ASD = 6, control = 7) were used for immunohistochemistry staining to facilitate neuropathological analysis. It is important to note that not all individuals had both types of tissues available, resulting in some individuals being represented in only one type of analysis. The demographic data of our sample is shown in Supplementary Table S10. For experimental analysis, the ASD samples were analyzed and compared with sex (all male) and age-matched unaffected individuals.
DNA and RNA extraction from post-mortem brain tissues
Total RNA and genomic DNA were isolated from post-mortem brain tissue from ASD and unaffected control using mirVana miRNA Isolation Kit (Ambion, USA) according to the manufacturer’s protocol. Firstly, about 100 mg of tissue was washed with PBS buffer on ice and repeated two times. Then were homogenized using 0.5 mm glass beads in the tube containing the lysis solution. Homogenization was performed by using Disruptor Genie (Scientific Industries, USA) using a high speed at 4 °C. If homogenization is incomplete, repeat the homogenization step. Acid-phenol: chloroform was added to homogenate and centrifuge at 10,000 g 5 min, then clear homogenate (upper) was transferred to a new tube for RNA isolation using mirVana miRNA Isolation Kit according to the manufacturer’s protocol. Briefly, miRNA homogenate additive and Acid-Phenol: Chloroform was added into the homogenate, then vortex for 1 min. The mixed homogenate was centrifuged at 10,000 g for 5 min, then carefully transfer the aqueous phase to a new tube. Ethanol was then added to the aqueous phase and passed through a filter cartridge containing a filter that immobilized the RNA. The total RNA was washed and eluted to a collecting tube. For genomic DNA extraction, 100% ethanol was added to the interphase and the lower phenol–chloroform phase and centrifuge at 2000 g for 5 min. Then, the DNA pellet was resuspended in 0.1 M sodium citrate in 10% ethanol incubated for 30 min and then washed with 75% ethanol. The DNA pellet was air-dried for 5–10 min and resuspended in 8 mM NaOH and HEPES. The quality and quantity of purified RNA and DNA were measured using NanoDrop One (Thermo Fisher Scientific, Inc.).
Data collection
We also obtained a previous publicly available RNA sequencing dataset (GSE211154) in the NCBI Gene Expression Omnibus database (GEO DataSets: https://www.ncbi.nlm.nih.gov/gds)51. Publicly available RNA-seq raw data of GSE211154 provided in the FASTQ files were obtained from the Sequence Read Archive (SRA) database for sequencing data generated by high-throughput sequencing such as RNA sequencing.
RNA-sequencing of the post-mortem cerebellum tissues
Total RNA was sent to BGI Hong Kong for RNA-sequencing analysis using the MGI DNBSEQ technology platform. Briefly, total RNA was used to prepare stranded mRNA library construction by mRNA enrichment and was then sequenced by the DNBSEQ-G400 (150 bp paired-end). RNA-sequencing data were qualified by filtering low-quality reads and adaptors using SOAPnuke software developed by BGI. Clean reads were mapped to reference gene sequence (transcriptome) by Bowtie252 and quantified expression level using RSEM (RNA-Seq by Expectation Maximization)53. Read counts were then performed for differential expression analysis using the DESeq2 package, which is a method based on the negative binomial distribution54. The genes or transcripts with an adjusted P-value less than 0.05 were considered statistically significant. The list of significant genes was subjected to IPA software (QIAGEN Inc., Hilden, Germany, https://www.qiagenbioinformatics.com/products/ingenuitypathway-analysis) for the prediction of biological functions, pathways, and gene regulatory networks related to ASD.
Transposable elements profiling in the post-mortem cerebellum tissues
In this study, we used the atena R package to quantify transposable elements profiling in the RNA-sequencing data that include a pipeline from three different methods including TEtranscripts55, ERVmap56, and Teloscope57 in the RStudio58,59. Firstly, cleaned reads (FASTQ) were aligned to the human reference genome using STAR aligner in the Galaxy (https://usegalaxy.org/) which is an open-source, web-based platform for data-intensive biomedical research60. The parameter used for mapping to reference genome was adapted from the Teissandier study that compared the tools for alignment and quantification of transposable elements analysis using high-throughput sequencing data61. Then, transposable elements were quantified by three methods in the atena package and differential expression analysis was performed using the DESeq2 package (adjusted P-value less than 0.05 was considered statistically significant).
Chimeric transcript identification in the post-mortem cerebellum tissues of ASD and unaffected control using ChimeraTE package
Chimeric transcripts derived from genes and LINE-1 or Alu or MIR were identified using the ChimeraTE package with a genome-guided approach which chimeric transcripts were detected by aligning reads against the reference62. In this approach, chimeric reads containing both transposable elements and exon sequences can be classified as initiated transcripts (upstream of the gene), exonized transcripts (TE within introns or embedded in gene exons), and terminated transcripts (downstream of the gene). In this study, we performed ChimeraTE analysis by obtaining gene annotation from GENCODE database (GENCODE human release 45, GRCh38) and transposable elements annotation, RepeatMasker annotation for hg38, from UCSC Table Browser63,64. The RNA-seq data were aligned to the human reference genome (GRCh38), reads were considered as chimeric reads when one mate of the paired reads originated from transposable elements into exon. Chimeric reads with greater or equal to 2 reads in at least 70% of replicates in each group were considered actual chimeric reads derived from genes and transposable elements.
Combined bisulfite restriction analysis (COBRA) for global LINE-1 and Alu methylation
To determine global DNA methylation at the promoter of LINE-1 and Alu retrotransposons, we conducted a COBRA method which is a quantitative technique to determine locus-specific DNA methylation of interesting loci. This technique is based on the pattern of the PCR products digested with an enzyme for which the recognition sequence is based on the methylation state in the original DNA.
Firstly, 1 \(\mu\) g of gDNA from post-mortem cerebellum tissues was treated with sodium bisulfite using EZ DNA Methylation-Gold™ Kit (Zymo, Irving, CA, USA) according to the manufacturer’s protocol. Then, the bisulfite-treated DNA was amplified with PCR (Hot Start Taq DNA polymerase, QIAGEN, USA) using specific primers for LINE-1 and Alu consensus sequences as described in our previous studies14,15,16. Then, the PCR amplicons were digested using a restriction enzyme and incubated at 65 °C for 15 h. Finally, the digested products were electrophoresed on an 8% non-denaturing polyacrylamide gel. The band intensity was measured using GelAnalyzer 19.1 (www.gelanalyzer.com). The intensity of the SH-KN-FI cell line was used as inter-assay normalization using calibration mode. The percentages of Alu elements and LINE-1 methylation levels and patterns were calculated according to the previous studies14,15,16.
Quantitative reverse transcription PCR (qRT-PCR) for expression and relative copy number of LINE-1 and Alu
qRT-PCR was used to quantify expression levels of LINE-1 and Alu retrotransposons in the post-mortem cerebellum tissues of ASD and unaffected individuals. A total of 1 \(\mu\) g RNA was treated with DNaseI enzyme (Thermo Fisher Scientific, USA). The DNase-treated RNA was converted to cDNA using Thermo Scientific™ RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, USA) according to the manufacturer’s protocol. Firstly, 1 \(\mu\) g of total RNA was mixed with 1 \(\mu\) l of random primer in 12 \(\mu\) l final volume. The reactions were incubated at 65 °C for 5 min and placed on ice. For the cDNA synthesis step, 1 \(\mu\) l of reverse transcriptase, 1 \(\mu\) l of RNase inhibitor, 1.5 \(\mu\) l of dNTP mix, and 4 \(\mu\) l of the buffer were added to the reaction. The cDNA synthesis reaction was performed by incubating the reaction at 25 °C for 5 min and 42 °C for 60 min, respectively. Quantification reverse transcription PCR was performed to quantify the expression level of LINE-1 and Alu using RealMOD Real-time PCR Master Mix Kit (iNtRON, Korea) and Bio-Rad CFX Connect Real-Time System (Bio-Rad, USA). A total of 5 ng of cDNA was added to 20 \(\mu\) l reaction in the total volume of RealMOD reagent according to the manufacturer’s protocol, and all samples were performed in triplicate. The PCR condition consisted of an initial step at 95 °C for 15 min followed by 40 cycles of 45 s at 95 °C for denature step and 30 s at 60 °C for annealing/extension followed by melting curve analysis (55 to 94 °C). Finally, the relative expression levels were calculated using the 2−ΔΔCt method using 18S ribosomal RNA as an endogenous control. The specific primers were used in our previous studies15,16.
For the relative copy number of LINE-1 and Alu retrotransposons quantification, we performed direct qPCR to amplify copies from genomic DNA using specific primers as described in previous studies19,65,66. Briefly, 250 pg of genomic DNA was performed directly to qPCR assay consisting of an initial step at 95 °C for 15 min followed by 40 cycles of 45 s at 95 °C for denature step and 30 s at 60 °C for annealing/extension. The LINE-1 and Alu retrotransposons copy number was normalized by SATA elements and calculated using the 2−ΔΔCt method.
Immunohistochemistry
To investigate neuropathology in the cerebellum tissues of ASD individuals, we performed immunohistochemistry (IHC) using Paraffin-embedded cerebellum tissues. Paraffin-embedded post-mortem cerebellum tissues were cut using an IEC microtome, and then the paraffin sections were deparaffinized with xylene and rehydrated via ethanol series. Briefly, a deparaffinized slide section was performed heat-induced antigen retrieval using sodium citrate buffer at 95 °C for 30 min. After cooling down, the slide was placed in PBS for 5 min 3 times and incubated with 1:1000 primary antibody (Recombinant Anti-Calbindin antibody [EP3478] (ab108404), abcam) in a moisture storage box for 18 h. The slide was placed in PBS for 5 min 3 times and then incubated with 1:1000 secondary antibody. Finally, the slide was incubated with ABC solution (ABC-HRP Kit, VECTASTAIN) and developed a signal using DAB (3,3'-Diaminobenzine) solution for 3–7 min, followed by hematoxylin staining. The criteria for Purkinje cell identification included the following: (i) large round or pyramidal-shaped neurons (ii) localized to a monolayer between the granular and molecular layers (iii) anti-calbindin positive cells (DAB +). The PCs were counted at a magnification of 20X. The linear density of PCs was determined by counting the number of PCs per millimeter line length of monolayer (PCs/mm). The PCs soma area was investigated at a magnification of 40X. The overall molecular layer thickness and thickness at the folium surface were measured at a magnification of 20X. Tissue sections were photographed using light microscopy (DM1000; Leica Microsystems, Wetzlar, Germany) and analyzed using ImageJ software67.
Statistical analysis
Statistical analyses for comparison of the means between two groups, the data was firstly performed Shapiro–Wilk Test of Normality. If data are normally distributed, a two-tailed Student’s t-test was used; If not, the Mann–Whitney test or Wilcoxon sign-rank test was used alternatively. A P-value less than 0.05 was considered statistically significant. For transcriptome and transposable elements profiling, the statistical analysis was performed as introduced in the package used in the analysis, and the Benjamini Hochberg correction method was used at FDR 0.05. Graphs were created using the ggplot2 package68.
Data availability
All data generated or analyzed in this study are included in this published article and supplementary information files. The raw RNA-seq data from postmortem cerebellum tissues have been deposited in the GEO database under the accession number GSE269105. This dataset will be accessible via this number following the publication of the article.
References
American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders. 5th edition. 2013.
Maenner, M. J. et al. Prevalence and characteristics of autism spectrum disorder among children aged 8 years—autism and developmental disabilities monitoring network, 11 sites, United States, 2020. MMWR Surveill. Summ. 72(2), 1–14 (2023).
Bolte, S., Girdler, S. & Marschik, P. B. The contribution of environmental exposure to the etiology of autism spectrum disorder. Cell Mol. Life Sci. 76(7), 1275–1297 (2019).
Stamou, M., Streifel, K. M., Goines, P. E. & Lein, P. J. Neuronal connectivity as a convergent target of gene x environment interactions that confer risk for autism spectrum disorders. Neurotoxicol. Teratol. 36, 3–16 (2013).
Tordjman, S. et al. Gene x environment interactions in autism spectrum disorders: role of epigenetic mechanisms. Front. Psychiatry 5, 53 (2014).
Kanlayaprasit, S. et al. Sex-specific impacts of prenatal bisphenol A exposure on genes associated with cortical development, social behaviors, and autism in the offspring’s prefrontal cortex. Biol. Sex Differ. 15(1), 40 (2024).
Thongkorn, S. et al. Investigation of autism-related transcription factors underlying sex differences in the effects of bisphenol A on transcriptome profiles and synaptogenesis in the offspring hippocampus. Biol. Sex Differ. 14(1), 8 (2023).
Kanlayaprasit, S. et al. Autism-related transcription factors underlying the sex-specific effects of prenatal bisphenol a exposure on transcriptome-interactome profiles in the offspring prefrontal cortex. Int. J. Mol. Sci. 22(24), 13201 (2021).
Thongkorn, S. et al. Sex differences in the effects of prenatal bisphenol A exposure on autism-related genes and their relationships with the hippocampus functions. Sci. Rep. 11(1), 1241 (2021).
Tremblay, M. W. & Jiang, Y. H. DNA methylation and susceptibility to autism spectrum disorder. Annu. Rev. Med. 70, 151–166 (2019).
Garcia-Perez, J. L., Widmann, T. J. & Adams, I. R. The impact of transposable elements on mammalian development. Development. 143(22), 4101–4114 (2016).
Lander, E. S. et al. Initial sequencing and analysis of the human genome. Nature. 409(6822), 860–921 (2001).
Moran, J. V. et al. High frequency retrotransposition in cultured mammalian cells. Cell. 87(5), 917–927 (1996).
Saeliw, T. et al. Integrated genome-wide Alu methylation and transcriptome profiling analyses reveal novel epigenetic regulatory networks associated with autism spectrum disorder. Mol. Autism. 9, 27 (2018).
Tangsuwansri, C. et al. Investigation of epigenetic regulatory networks associated with autism spectrum disorder (ASD) by integrated global LINE-1 methylation and gene expression profiling analyses. PLoS One. 13(7), e0201071 (2018).
Saeliw, T. et al. Epigenetic gene-regulatory loci in Alu elements associated with autism susceptibility in the prefrontal cortex of ASD. Int. J. Mol. Sci. 24(8), 7518 (2023).
Jacob-Hirsch, J. et al. Whole-genome sequencing reveals principles of brain retrotransposition in neurodevelopmental disorders. Cell Res. 28(2), 187–203 (2018).
Borges-Monroy, R. et al. Whole-genome analysis reveals the contribution of non-coding de novo transposon insertions to autism spectrum disorder. Mob. DNA. 12(1), 28 (2021).
Shpyleva, S., Melnyk, S., Pavliv, O., Pogribny, I. & Jill, J. S. Overexpression of LINE-1 retrotransposons in autism brain. Mol. Neurobiol. 55(2), 1740–1749 (2018).
Spirito, G. et al. Exploratory analysis of L1 retrotransposons expression in autism. Mol. Autism. 14(1), 22 (2023).
Saeliw, T. et al. LINE-1 and Alu methylation signatures in autism spectrum disorder and their associations with the expression of autism-related genes. Sci. Rep. 12(1), 13970 (2022).
Thomas, C. A., Paquola, A. C. & Muotri, A. R. LINE-1 retrotransposition in the nervous system. Annu. Rev. Cell Dev. Biol. 28, 555–573 (2012).
Thomas, C. A. & Muotri, A. R. LINE-1: creators of neuronal diversity. Front. Biosci. (Elite Ed). 4(5), 1663–1668 (2012).
Chesnokova, E., Beletskiy, A. & Kolosov, P. The role of transposable elements of the human genome in neuronal function and pathology. Int. J. Mol. Sci. 23(10), 5847 (2022).
Playfoot, C. J., Sheppard, S., Planet, E. & Trono, D. Transposable elements contribute to the spatiotemporal microRNA landscape in human brain development. RNA. 28(9), 1157–1171 (2022).
Playfoot, C. J. et al. Transposable elements and their KZFP controllers are drivers of transcriptional innovation in the developing human brain. Genome Res. 31(9), 1531–1545 (2021).
Mustafin, R. N. & Khusnutdinova, E. K. Involvement of transposable elements in neurogenesis. Vavilovskii Zhurnal Genet Selektsii. 24(2), 209–218 (2020).
Voineagu, I. et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 474(7351), 380–384 (2011).
Parikshak, N. N. et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature. 540(7633), 423–427 (2016).
Brandenburg, C. et al. Transcriptomic analysis of isolated and pooled human postmortem cerebellar Purkinje cells in autism spectrum disorders. Front. Genet. 13, 944837 (2022).
Ziats, M. N. & Rennert, O. M. Aberrant expression of long noncoding RNAs in autistic brain. J. Mol. Neurosci. 49(3), 589–593 (2013).
Ritvo, E. R. et al. Lower Purkinje cell counts in the cerebella of four autistic subjects: initial findings of the UCLA-NSAC autopsy research report. Am. J. Psychiatry. 143(7), 862–866 (1986).
Skefos, J. et al. Regional alterations in Purkinje cell density in patients with autism. PLoS One. 9(2), e81255 (2014).
Whitney, E. R., Kemper, T. L., Bauman, M. L., Rosene, D. L. & Blatt, G. J. Cerebellar Purkinje cells are reduced in a subpopulation of autistic brains: A stereological experiment using calbindin-D28k. Cerebellum. 7(3), 406–416 (2008).
Fatemi, S. H. et al. Purkinje cell size is reduced in cerebellum of patients with autism. Cell Mol. Neurobiol. 22(2), 171–175 (2002).
Lu, Q. et al. Cerebellar structural abnormality in autism spectrum disorder: A magnetic resonance imaging study. Psychiatry Investig. 20(4), 334–340 (2023).
Kim, J. & Augustine, G. J. Molecular layer interneurons: Key elements of cerebellar network computation and behavior. Neuroscience. 462, 22–35 (2021).
Lafarga, M. et al. Cytology and organization of reactive astroglia in human cerebellar cortex with severe loss of granule cells: A study on the ataxic form of Creutzfeldt–Jakob disease. Neuroscience. 40(2), 337–352 (1991).
Fetit, R., Hillary, R. F., Price, D. J. & Lawrie, S. M. The neuropathology of autism: A systematic review of post-mortem studies of autism and related disorders. Neurosci. Biobehav. Rev. 129, 35–62 (2021).
Edmonson, C., Ziats, M. N. & Rennert, O. M. Altered glial marker expression in autistic post-mortem prefrontal cortex and cerebellum. Mol. Autism. 5(1), 3 (2014).
Whitney, E. R., Kemper, T. L., Rosene, D. L., Bauman, M. L. & Blatt, G. J. Density of cerebellar basket and stellate cells in autism: Evidence for a late developmental loss of Purkinje cells. J. Neurosci. Res. 87(10), 2245–2254 (2009).
Al-Naama, N., Mackeh, R. & Kino, T. C(2)H(2)-type zinc finger proteins in brain development, neurodevelopmental, and other neuropsychiatric disorders: Systematic literature-based analysis. Front. Neurol. 11, 32 (2020).
Casoni, F. et al. Zfp423/ZNF423 regulates cell cycle progression, the mode of cell division and the DNA-damage response in Purkinje neuron progenitors. Development. 144(20), 3686–3697 (2017).
Lapp, H. E. & Hunter, R. G. Early life exposures, neurodevelopmental disorders, and transposable elements. Neurobiol. Stress. 11, 100174 (2019).
Hu, V. W. & Steinberg, M. E. Novel clustering of items from the autism diagnostic interview-revised to define phenotypes within autism spectrum disorders. Autism. Res. 2(2), 67–77 (2009).
Hu, V. W. et al. Gene expression profiling differentiates autism case-controls and phenotypic variants of autism spectrum disorders: Evidence for circadian rhythm dysfunction in severe autism. Autism. Res. 2(2), 78–97 (2009).
Lee, E. C. & Hu, V. W. Phenotypic subtyping and re-analysis of existing methylation data from autistic probands in simplex families reveal ASD subtype-associated differentially methylated genes and biological functions. Int. J. Mol. Sci. 21(18), 6877 (2020).
Christman, J. K. 5-Azacytidine and 5-aza-2’-deoxycytidine as inhibitors of DNA methylation: Mechanistic studies and their implications for cancer therapy. Oncogene. 21(35), 5483–5495 (2002).
Banuelos-Sanchez, G. et al. Synthesis and characterization of specific reverse transcriptase inhibitors for Mammalian LINE-1 retrotransposons. Cell Chem. Biol. 26(8), 1095–1109 (2019).
Yim, Y. Y., Teague, C. D. & Nestler, E. J. In vivo locus-specific editing of the neuroepigenome. Nat. Rev. Neurosci. 21(9), 471–484 (2020).
Barrett, T. et al. NCBI GEO: archive for functional genomics data sets–10 years on. Nucleic Acids Res. 39(Database issue), D1005–D1010 (2011).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9(4), 357–359 (2012).
Li, B. & Dewey, C. N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 12, 323 (2011).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15(12), 550 (2014).
Jin, Y., Tam, O. H., Paniagua, E. & Hammell, M. TEtranscripts: A package for including transposable elements in differential expression analysis of RNA-seq datasets. Bioinformatics. 31(22), 3593–3599 (2015).
Tokuyama, M. et al. ERVmap analysis reveals genome-wide transcription of human endogenous retroviruses. Proc. Natl. Acad. Sci. U S A. 115(50), 12565–12572 (2018).
Bendall, M. L. et al. Telescope: Characterization of the retrotranscriptome by accurate estimation of transposable element expression. PLoS Comput. Biol. 15(9), e1006453 (2019).
Team R. RStudio: Integrated development for R (RStudio, PBC, 2023).
Team Rc. R: A language and environment for statistical computing. Vienna, Austria: R Foundation for Statistical Computing; 2023. Available from: https://www.R-project.org/.
Jalili, V. et al. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2020 update. Nucleic Acids Res. 48(W1), W395–W402 (2020).
Teissandier, A., Servant, N., Barillot, E. & Bourc’his, D. Tools and best practices for retrotransposon analysis using high-throughput sequencing data. Mobile DNA. 10(1), 52 (2019).
Oliveira, D. S. et al. ChimeraTE: a pipeline to detect chimeric transcripts derived from genes and transposable elements. Nucleic Acids Res. 51(18), 9764–9784 (2023).
Navarro Gonzalez, J. et al. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 49(D1), D1046–D1057 (2021).
Frankish, A. et al. Gencode 2021. Nucleic Acids Res. 49(D1), D916–D923 (2021).
Coufal, N. G. et al. L1 retrotransposition in human neural progenitor cells. Nature. 460(7259), 1127–1131 (2009).
Gualtieri, A. et al. Increased expression and copy number amplification of LINE-1 and SINE B1 retrotransposable elements in murine mammary carcinoma progression. Oncotarget. 4(11), 1882–1893 (2013).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods. 9(7), 671–675 (2012).
Wickham, H. ggplot2: Elegant graphics for data analysis (Springer, 2016).
Acknowledgements
We gratefully acknowledge the contribution of brain tissue donors as well as those involved in every process and the Research Affairs, Faculty of Medicine, Chulalongkorn University, for assistance regarding microscopic analysis and software. We would like to thank Mrs. Wanida Buasorn, Department of Anatomy, Faculty of Medicine, Chulalongkorn University, for her assistance in training TS (Thanit Saeliw) to prepare paraffin-embedded tissue sections.
Funding
This study was supported by the Program Management Unit for Human Resources and Institutional Development, Research and Innovation (PMU-B; B36G660008), the Thailand Science Research and Innovation Fund Chulalongkorn University (HEA663700091 and HEAF67370092), and the Ratchadapisek Somphot Fund for Supporting Center of Excellence, Chulalongkorn University (GRU 6300437001-1, GRU_64_033_37_004, GRU 6506537004-1, and CE66_046_3700_003) to T.S. (Tewarit Sarachana). T.S. (Thanit Saeliw) and S.K. were supported by the Second Century Fund (C2F), Chulalongkorn University. The Scholarship from the Graduate School, Chulalongkorn University, to commemorate the 72nd anniversary of His Majesty King Bhumibol Adulyadej, is gratefully acknowledged for financially supporting K.S.
Author information
Authors and Affiliations
Contributions
Conceptualization, TS (Tewarit Sarachana); data curation, TS (Thanit Saeliw) and TS (Tewarit Sarachana); formal analysis, TS (Thanit Saeliw) and TS (Tewarit Sarachana); funding acquisition, TS (Tewarit Sarachana); investigation, TS (Thanit Saeliw) and TS (Tewarit Sarachana); methodology, TS (Thanit Saeliw), SK, ST, KS, BS, DJ, VWH, and TS (Tewarit Sarachana); project administration, TS (Tewarit Sarachana); resources, TS (Tewarit Sarachana); supervision, BS, DJ, and TS (Tewarit Sarachana); validation, TS (Thanit Saeliw) and TS (Tewarit Sarachana); writing original draft, TS (Thanit Saeliw) and TS (Tewarit Sarachana); writing review and editing, TS (Thanit Saeliw), VWH, and TS (Tewarit Sarachana). All authors have read and agreed to the published version of the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Saeliw, T., Kanlayaprasit, S., Thongkorn, S. et al. Investigation of chimeric transcripts derived from LINE-1 and Alu retrotransposons in cerebellar tissues of individuals with autism spectrum disorder (ASD). Sci Rep 14, 21889 (2024). https://doi.org/10.1038/s41598-024-72334-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-72334-x
- Springer Nature Limited