
Abstract
The complex interactions between epiphytic bacteria and marine macroalgae are still poorly understood, with limited knowledge about their community structure, interactions, and functions. This study focuses on comparing epiphytic prokaryotes community structure between three seaweed phyla; Chlorophyta, Rhodophyta, and Heterokontophyta in an easternmost rocky intertidal site of the Mediterranean Sea. By taking a snapshot approach and simultaneously collecting seaweed samples from the same habitat, we minimize environmental variations that could affect epiphytic bacterial assembly, thereby emphasizing host specificity. Through 16S rRNA gene amplicon sequencing, we identified that the microbial community composition was more similar within the same seaweed phylum host compared to seaweed host from other phyla. Furthermore, exclusive Amplicon Sequence Variants (ASVs) were identified for each algal phyla despite sharing higher taxonomic classifications across the other phyla. Analysis of niche breadth indices uncovers distinctive affinities and potential specialization among seaweed host phyla, with 39% of all ASVs identified as phylum specialists and 13% as generalists. Using taxonomy function prediction, we observed that the taxonomic variability does not significantly impact functional redundancy, suggesting resilience to disturbance. The study concludes that epiphytic bacteria composition is connected to host taxonomy, possibly influenced by shared morphological and chemical traits among genetically related hosts, implying a potential coevolutionary relationship between specific bacteria and their host seaweeds.
Similar content being viewed by others


Introduction
Marine macroalgae, commonly known as seaweeds, are photosynthetic eukaryotes with key roles in the normal functioning of marine ecosystems1,2. Seaweeds provide food and habitat for a wide range of organisms and are critical components in the biogeochemical cycling of nutrients3. Seaweeds are a polyphyletic group belonging to three phyla; Chlorophyta (green algae), Rhodophyta (red algae), and Heterokontophyta (brown algae from class Phaeophyceae). These phyla are evolutionarily distant from one another and differ in their reproduction strategies, defense mechanisms, pigmentation and other eco-physiological features4,5. Red seaweeds, exhibiting remarkable adaptability, inhabit both shallow and deep marine environments, showcasing their capacity to endure a diverse range of light and temperature conditions6. This diverse group spans from warm tropical waters to frigid polar regions, marking red seaweeds as the most varied among their counterparts7. Green seaweeds thrive abundantly in the shallows, colonizing areas such as rocky shores, tide pools, and coral reefs8. The brown seaweed predominantly occupies the intertidal zone of temperate and cold environments. The resilience of brown algae in low-light conditions is attributed to the distinctive photosynthetic pigment fucoxanthin9. Unlike green and red algae which store energy as true starch, brown algae utilize laminaran as their primary carbohydrate reserve10.
The microbiota in aquatic environments can be found within the water column or attached to natural or artificially introduced substrates11,12,13,14,15. When attached, the bacterial cells generate an extracellular polymeric substance (EPS) matrix, initiating the development of a biofilm14,16. Many marine eukaryotes have strong and consistent relationships with specific bacteria, which play a critical role in the hosts' growth, development, and survival by providing essential nutrients and protecting them from harmful epiphytic colonization and predation13,17,18.
The thallus surface in seaweeds provides a habitat rich in oxygen and organic material, resulting from photosynthetic activity and the release of mucilage and other compounds19. Owing to the generally complex and highly branched structure, the extensive surface area of seaweeds facilitates the settlement and proliferation of opportunistic bacterial colonizers20,21. The relationship between seaweed and their associated bacteria is a rising research field with immense interest in ecology, biotechnology, and aquaculture22. The bacterial interactions with their seaweed hosts may alter the functionality of the seaweeds, for example, the release of carpospores in red seaweeds or the attachment of zoospores in green seaweeds23,24. Epiphytic microbes play a role in preserving the well-being and shaping the morphology of green and brown macroalgae, including genera like Ulva, Monostroma, and Ectocarpus25,26,27,28,29,30. Certain species of red algae ‘garden’ their associated microbes by using their surface-associated chemical compounds to mitigate the attachment of pathogenic bacteria and to attract bacterial strains that confer protection against such pathogens31. In turn, some interactions with bacteria are detrimental to the host seaweed. For example, various lineages within Gammaproteobacteria, Cyanobacteria, Actinobacteria, and the Cytophaga-Flexibacter-Bacteroides group have been linked to macroalgal diseases32,33,34,35.
It has been proposed that the relationship between hosts and their microbiome should be studied as a unified entity, referred to as a "holobiont" or “meta-organism” to better understand the ecology and evolution of eukaryotic hosts36,37,38. This holistic approach can be used to create management strategies that improve the resilience and adaptability of hosts. A crucial first step in this process is determining the factors that shape microbial communities. The establishment and dynamics of seaweed-bacteria relationships are profoundly influenced by environmental factors37,39. The availability of inorganic nutrients and organic matter in the surrounding environment plays a crucial role in shaping these associations40,41. Other water quality parameters like pH, temperature, salinity, and turbidity can affect how seaweed interacts with its associated microbial communities. The microbial communities are also significantly influenced by strong selective forces arising from the macroalgae host physiology and anatomy, including the host structure, chemical composition, specific polysaccharides composition, and other secreted molecules42,43,44,45. In particular, the secreted polysaccharides are often specific to algal clades46, likely playing a key role in microbiome selection47. Given the physiological differences and similarities among various seaweeds, community assembly can vary based on host taxonomy, often at the phylum level33,48,49.
We aimed to explore the diversity and specificity of seaweed-associated prokaryotes in the Eastern Mediterranean Sea, a region known for its remarkable biodiversity. For this purpose, we gathered a substantial number of host specimens from the same location simultaneously. This snapshot approach contrasts with previous studies, which involved species from diverse locations and time periods and concentrated on a restricted number of species49,50,51,52. We thus assessed algal microbiome variation in light of limited confounding variables linked to environmental and temporal variation.
Materials and methods
Seaweeds and microbial community sampling
A total of 32 algal specimens representing the three seaweed phyla were collected during morning hours in May 2022 from a flat, rocky intertidal platform in Achziv (33° 03′ 21.9′′ N 35° 06′ 06.9′′ E), Israel (Fig. 1). This sampling approach represents a snapshot of intertidal seaweeds thriving during springtime for this specific geographical location53. For microbial collection, sterile cotton swabs were gently rubbed against the seaweed tissue immediately after harvest and quickly placed into 1 ml of a lysis buffer (40 mM EDTA, 50 mM Tris pH 8.3, and 0.75 M sucrose) transported to the lab and stored at – 20 °C until microbial DNA extractions. From each seaweed sample, about 3 g fresh weight was immersed into 1 ml of the lysis buffer for seaweed molecular taxonomy identification (i.e. barcode). As a background control, 50 ml of seawater from the same collection site was filtered through a Supor 0.2 µm, 25 mm filter (Pall Corporation, 2024). The DNA was then extracted and analyzed in the same manner as the swab samples.

The geographic location of Achziv site in the Israeli Mediterranean Sea (a) and overview of the sampling site with semi-exposed intertidal platforms (b).
DNA extraction for seaweed barcoding
About 100 mg of fresh macroalgae biomass was placed in 1 ml lysis buffer solution (ISOLATE II Plant DNA Kit, Bioline) and homogenized using a small plastic pestle. Genomic DNA extracted as described in the ISOLATE II Plant DNA Kit. Quantity and quality of DNA were verified using a nanodrop (NANODROP 2000c Spectrophotometer, Thermo Scientific, USA). For species identification, the ‘D1,’ ‘D2’ and ‘D3’ hyper-variable domains of the large subunit (LSU) rRNA gene54,55 were amplified using the D1R (5′-ACCCGCTGAATTTAAGCATA-3′) and DC3a (5′-ACGAACGATTTGCACGTCAG-3′) primers55,56. All PCR reactions were run in 30 µL containing 0.75 µL each of forward and reverse primers (10 µM), 14.5 µL of ready Mix (Bioline Meridian Life Science Inc.), 1 µL template DNA, 11 µL of PCR Grade H2O and 2 µL of bovine serum albumin (BSA). The PCR fragments were sequenced using Sanger sequencing by Macrogen Europe (Macrogen Europe BV, Amsterdam, Netherlands). Taxonomy was determined based on BLAST analysis against the NCBI GenBank database57 (http://www.ncbi.nlm.nih.gov), supplemented by microscopic morphological confirmation. We primarily adopted seaweed taxonomy descriptions down to the genus level due to the uncertainty in BLAST resolution for species level identification, and because molecular identification relied on a single marker.
DNA extraction and amplicon sequencing of the 16S rRNA gene
DNA was extracted from the swabs using the phenol–chloroform protocol58. Quantity and quality of DNA were examined using a nanodrop (NANODROP 2000c Spectrophotometer, Thermo Scientific, USA) and DNA concentration was diluted to 0.5 ng/µl. The 16S rRNA V4-V5 primers 515F (5′- GTGYCAGCMGCCGCGGTAA-3′) and 926R (5′-CCGYCAATTYMTTTRAGTTT-3′) amended with CS1 and CS2 linkers were used59,60. PCR reactions were run in 50 µL containing forward and reverse primers (10 µM), ReadyMix (Bioline Meridian Life Science Inc.), template DNA, and PCR-grade water. Library preparation and sequencing were performed at the Genome Research Core (GRC), Research Resources Center (RRC), University of Illinois at Chicago (UIC) according to a standard protocol as described by 61. Out of the 32 samples collected, 29 yielded enough quality reads for microbial taxonomic analysis (1,596,308 reads). Given that archaea represent a minor fraction (5 ASVs, each found in only one sample), our focus will be only on bacteria.
Bioinformatics and statistical analyses
All sequences were imported into R 62 and analyzed with the DADA2 v 1.8 package (Callahanet al., 2016). Reads were truncated based on quality plots, checked for chimeras, merged, and grouped into amplicon sequence variants (ASVs). Taxonomy was assigned based on the SILVA v138.1 reference database. To determine the ratio of specialist and generalist bacterial taxa, we employed Levins' approach63 as outlined by64. To quantify bacterial habitat specialization within the different phyla (niche breadth), we applied the following equation:
where Bj is the niche breadth and Pij is the proportion of the individuals of species j in phylum i. ASVs that are unique to a single sample were excluded from this calculation, as they may not represent phylum associations but rather specific associations with individual seaweed samples or species. ASVs that are present, and more evenly distributed, along a wider range of habitats will have a higher B-value and can be considered habitat generalists64. Similarly, ASVs with a lower B-value can be regarded as habitat specialists. We considered the three different phyla as habitats. Niche breadth ranged between 1 and 2.99 (with a theoretical maximum of 3). ASVs with B > 2 were arbitrarily defined as generalists, whereas those with B < 1.2 were regarded as specialists.
The microbiome data underwent scaling and normalization using Cumulative Sum Scaling (CSS) and was subsequently clustered based on Bray–Curtis dissimilarity. Dendrograms were constructed using functions from the vegan and ape packages in R. The algal consensus LSU (large subunit ribosomal DNA) sequences were aligned and plotted on a tree using the SILVA ACT online tool65,66.
Alpha-diversity estimates (Shannon index) were computed using even-rarified ASV-level to library size of 2900 and the most prevalent order-level abundance matrices using the MicrobiomeAnalyst web tool67. Beta diversity analysis was performed using the phyloseq package68. The statistical significance of the clustering pattern in the ordination plots was evaluated using Permutational ANOVA (PERMANOVA). For differential abundance, we used the linear discriminant analysis effect size (LEfSe) as implemented in MicrobiomeAnalyst67. Kruskal–Wallis test by ranks was used to identify ASVs with significant abundance. Linear Discriminant Analysis (LDA) was then applied to measure the impact of each differentially abundant ASV. ASVs with a significant impact were defined as those with an LDA score (log10) greater than 2 (p-value < 0.05 for both tests). The metabolic functions and pathways of bacterial communities were predicted using Tax4Fun269. Selected Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways70,71,72 were chosen based on their relevance to the scope of this study. Broad categories such as Cellular Processes and Genetic Information Processing were excluded due to their general nature. Similarly, KEGG orthologs (KOs) unrelated to the focus of this investigation, such as Human Diseases, were also excluded from analysis. Some cellular processes that hold significance within the context of this study, such as Quorum Sensing, Biofilm Formation, and Bacterial Chemotaxis, were retained for further analysis. Furthermore, KOs characterized by low read counts were omitted from the subsequent analysis. The analysis excluded the broad metabolic pathway ko01100. We evaluated the contribution of the KOs to the variation between the different algae using principal coordinates analysis (PCoA) in PAST V473. The core functions were defined as those present in at least 80% of the samples within a group, with an abundance of more than 1% in each sample.
Results
The taxonomic classification of the collected seaweed resulted in 17 representatives of the red seaweeds, 7 of the brown seaweeds, and 5 of the green seaweeds (Supplementary Table S1). After removing sequences of low quality and chimeric nature, a total of 611,824 high-quality reads were extracted from the 29 distinct seaweed samples, which were then used for downstream analysis (Supplementary Table S2). In total, 4582 unique ASVs were identified across the entire sample set after excluding ASVs present in only one sample and those found exclusively in the seawater samples. Within each phylum, ASVs that are present in only one sample contribute to 69% of the ASVs found in brown algae, 65% in green algae, and 71% in red algae. Several ASVs that were found in high abundance in the seawater were also present in the algae samples and exhibited an affinity for specific phylum, particularly to the green phylum. We observed a nearly balanced distribution of ASVs per sample when considering the average for each phylum (averaging 355 ± 140, 355 ± 77, and 350 ± 63 ASVs per sample for red, green, and brown algae hosts, respectively). We note that the high standard deviation in the red phylum is likely influenced by two samples, both belonging to the Nemalion lineage. These samples significantly diverged in their microbial community assembly and exhibited relatively lower ASV counts (96 and 100). The examination of the Shannon Diversity Index, a metric less sensitive to variations in sequencing depth, revealed no statistically significant differences among the three distinct phyla (Fig. S1, p = 0.438). This finding underscores the stability and consistency of alpha diversity across the diverse seaweed host types.
A total of 352 ASVs (7.5%) were shared among the three phyla (Fig. 2). The brown and red phylum exhibited a higher degree of ASV sharing compared to the green and brown, and red and green, with 493 ASVs (10%), 33 ASVs (0.7%), and 255 ASVs (5.5%), respectively. Using the niche breadth index, we determined that 39% of the microbial lineages were likely specialists (B < 1.2), and only 13% were generalists (B > 2), (Supplementary Tables S3).

Venn diagrams showing the unique and shared ASV’s between green seaweed, red seaweed, and brown seaweed.
We observed mostly consistent patterns between the structure of the bacterial community and their algal host phylogeny, with a few exceptions (Fig. 3). The key exceptions included (i) the aberrant Nemalion microbiome and (ii) the remarkably similar communities on the brown seaweed Padina and the red seaweed Thuamtella. The beta diversity analysis further affirmed the consistent patterns between bacterial community structure and algal host phylum, revealing distinct microbiome clustering based on host taxonomy (FDR 0.002) (Fig. 4). When applying pairwise PERMANOVA analysis based on the PCoA coordinates, significant relationships between algal groups were observed. The red and brown algae, as well as the red and green algae, exhibited closer associations with each other (F-values 2.10 and 2.41, respectively), compared to the green and brown algae which displayed greater dissimilarity (F-value 3.96).

Phylogenetic tree based on the LSU marker of the seaweeds (left) plotted against the dendrogram tree of clustered bacteria assembly of the different samples using the BC dissimilarity index (right). Samples are colored by their taxonomic classification: green, red, or brown.

PCoA plot using bray distance to express the differences in epiphytic bacterial microbiome assembly between the three algae phyla. The explained variances are shown in brackets. (F-value: 2.2; R-squared: 0.15; p-value: 0.001).
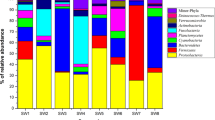
We identified several key bacteria clades in association with three host phyla. These clades (Class level) comprised Alphaproteobacteria (28%, 30%, 36%), Bacteroidia (27%, 37%, 35%), Gammaproteobacteria (10%, 4%, 9%), Verrucomicrobiae (12%, 9%, 6%), and Cyanobacteriia (10%, 9%, 4%), in red, green, and brown algae, respectively. The bacterial families Flavobacteriaceae (17%, 9% and 7% in brown, green and red algae, respectively) and Saprospiraceae (14%, 17% and 24% in brown, green and red algae, respectively) were prominent across all algal groups. The genus Lewinella within the Saprospiraceae family emerged as one of the most abundant genera across all algal groups, representing 3.07% of the total reads within the green phylum and 1.24% and 0.95% within the red and brown phyla, respectively. Several genera from the most abundant families, such as Rubidimonas (Saprospiraceae) and Tenacibaculum (Flavobacteriaceae), were found to be shared among all algal phyla, with each phylum having its own distinct ASVs. A list of distinct ASVs identified in more than two samples within a phylum, and exclusive to that phylum, is provided in the Supplementary Tables S4, S5, and S6.
Using the LEfSe we found 11 differentially abundant ASVs with an LDA score higher than two and a p-value cutoff of 0.05 (Fig. 5). Flavobacteriaceae (ASV38), Saprospiraceae (ASV114), Trueperaceae (ASV 86), Sphingomonadaceae (ASV19), Rhodobacteraceae (ASV50), Hyphomonadaceae (ASV37) and Cryomorphaceae (ASV113) were characteristic of the green algae bacterial community. A single Flavobacteriaceae from the Tenacibaculum genus (ASV64) was enriched in brown algae. A single Granulosicoccales )ASV91( was predominant in red algae. Most of the taxa found on both green and brown algae were also found on red algae but in lower abundance.

LDA (Linear Discriminant Analysis) scores of differentially abundant ASVs among red, brown, and green algae groups. Taxa with LDA scores > 2 and P < 0.05 are considered significant. The mini heatmap indicates relative abundance (red: higher, blue: lower) in each group.
The Trueperaceae family exhibited a higher abundance in the green algae phylum compared to the other phyla. Specifically, the relative abundance of Trueperaceae ASVs was 0.8% in the brown phylum, 0.5% in the red phylum, and 2.3% in the green phylum. Especially the Truepera genus with a notable presence across all green samples, amounting to a total of 6751 reads across 18 different ASVs (p-value = 0.01, Fig. 6). In turn, the Olleya genus (Flavobacteriaceae) was significantly enriched in brown algae (p-value = 0.001, Fig. 6).

Box plots of the bacterial abundance within the three algae groups. (a) Truepera genus. (b) Olleya genus. *** represent P < 0.05.
Taxonomy-based functional predictions hinted at no significant differences in the microbial functions among the different algae groups sampled (Figs. 7 and 8). There was little variation in the predicted functional core across all three groups, considering the prevalence and relative abundance of the function distribution. We estimated a total of 316 KEGG orthologs (KOs) while 183 KOs were associated with prokaryotic functions and considered relevant for our analysis. The most common predicted pathways (more than 1% of the reads in every sample), listed in descending order, were biosynthesis of antibiotics (ko01130), biosynthesis of secondary metabolites (ko01110), carbon metabolism (ko01200), biosynthesis of amino acids (ko01230), purine metabolism (ko00230), and valine, leucine, and isoleucine degradation (ko00280). Other prominent functional categories were bacterial ABC transporters (ko02010), two-component systems (ko02020), and quorum sensing (ko02024) (Supplementary Table S7).

PCoA plot using bray distance to express the differences of epiphytic bacterial KEGG Ortholog (KO) Functions between the three algae phyla.

Average relative abundance of estimated core KEGG Ortholog (KO) functions with standard deviation (STD) among the three phyla of seaweed.
Discussion
This study investigated epiphytic bacterial communities hosted by seaweeds in the rocky intertidal zone of the Eastern Mediterranean Sea. Our findings demonstrate that the epiphytic communities of seaweeds from the rocky intertidal platform at Achziv exhibit a stronger association with genetically similar host seaweeds, thereby linking the epiphytic microbiome to host taxonomy (Figs. 3, 4). Even in close spatial proximity, the community structure was linked to host taxonomy, extending to the phylum level, indicating that the selection of these communities can be linked to host traits, which are likely most conserved among related seaweed taxa. We observed some discrepancies in the clustering of bacterial assemblages that transcend the host taxonomy. For example, the bacterial community associated with the brown alga Padina sp. was found to be more similar to that of certain red algae rather than other brown algae. The complexity and structure of the thallus can also influence the epiphytic microbiome associated with the seaweed host21. Padina sp., one of the only two known calcified brown algae, exhibits a unique epiphytic microbiome that differs from other brown algae. This distinction can be attributed to its calcification trait, a characteristic it shares with Jania sp., as well as other red algae. The calcified nature of Padina sp. may foster a microenvironment favorable to a specific microbiome, which could explain its closer association with other red algae, including Jania sp., rather than with other brown algae. ASVs-level variations among host-specific bacteria indicate that host specificity varies at bacterial species or sub-species levels, and host-specific specialists are widespread among various bacterial phyla.
Our data reveal substantial distinctions in the epiphytic bacterial communities among the three phyla of seaweed hosts. Notably, the bacterial communities associated with red and brown algae exhibit a higher degree of similarity to each other compared to those found in green algae (Fig. 4). This pattern may be attributed to the carbohydrate-rich extracellular matrix found in red and brown algae, which is less prevalent in green algae74. The unique composition of this extracellular matrix in red and brown algae likely provides a conducive environment for similar bacterial communities, highlighting the role of host-specific biochemical properties in shaping microbial assemblages. Moreover, specific algal defenses can selectively inhibit the growth of non-native biofilms or repel already attached bacteria, further contributing to shaping the composition of the epiphytic microbial community. For instance, the red algae Delisea pulchra produces secondary metabolites that deter the colonization of biofilms not specific to the alga, as demonstrated by Steinberg et al.75. These defense mechanisms are well-documented for red and brown seaweeds but not for green algae, suggesting potentially lower selectivity in the latter76,77. Another indication of lower selectivity within the green algae phylum in the Achziv site is evident from the analysis of the seawater samples. Most of the ASVs associated with the seawater samples were not found in the algae samples. However, five of the ASVs were consistently found in high abundance across most green algae samples and contributed to some of the observed variations from other phyla, as highlighted by the LDA (Fig. 5).
This observation is consistent with previous studies emphasizing the tendency for epiphytic bacterial communities to cluster within the same algal phylum, showcasing discernible differences from those in other phyla21,49,78. In parallel to our findings, Chen et al.78 reported the similarities in the microbiomes of red and brown seaweeds. However, Selvarajan et al.49 highlighted a marked dissimilarity among specific brown algae when compared to both red and green seaweeds. The substantial dissimilarity between the brown phylum and the other algae phyla could be attributed to the distinct species of brown algae examined in these studies.
While we showed the presence of generalist bacteria that thrive in association with all three host phyla, most taxa (39%) were host-specific, indicating a specialization in microbe-algae associations, possibly to an obligate extent in the development of those seaweeds. These specialized microbes might share common dietary preferences, favoring compounds like polysaccharides linked to specific algal phylum47. They may also exhibit similar colonization preferences based on substrate chemical or mechanical features21. These findings align with prior research that has observed a higher abundance of specialists compared to generalists within certain aquatic niches79,80,81.
Our findings confirm the key role of Flavobacteriaceae and Saprospiraceae in the seaweed holobiont39,50,77,82,83,84. Flavobacteriaceae are the key generalist degraders of macromolecules, such as glycans, in marine environments85,86,87,88. They can play a key role in the degradation of fucoidan, a sulfated polysaccharide found in various species of brown seaweeds89. Some Saprospiraceae are likely pivotal in the decomposition and breakdown of complex organic compounds90. We identified Lewinella (Saprospiraceae) across all three seaweed phyla, whereas some ASVs were phylum-specific. A parallel pattern was observed within the Flavobacteriaceae (e.g. Tenacibaculum genus). Our results hint at a certain degree of host specificity at the phylum level in Saprospiraceae and Flavobacteriaceae. The relationship between the variation in these taxa and the distinct macromolecular arsenal of the hosts, which is known to differ significantly among the three phyla49,91,92, remains to be elucidated. Our findings indicate that seaweed-associated bacteria may carry out functions crucial for the host's well-being, such as nutrient cycling and the synthesis of secondary metabolites that offer protection to chemically vulnerable macroalgae, thereby mitigating the risk of secondary colonization by other microscopic and macroscopic epibiota37,93. These key functions appear to remain conserved among diverse taxa. The functional redundancy among the seaweed-associated microbes likely implies the resilience of the meta-organism during disturbances, while maintaining its essential functions. However, taxonomy-based functional analysis falls short of capturing functional disparities emerging from diverse factors, especially given variation at the ASV level rather than at higher taxonomic levels. For more accurate identification of distinct bacterial functions such as degradation of macromolecules, in different seaweed species, experimental and omics approaches are needed94.
Our data hints that the strong UV radiation in the littoral may also play a role in the seaweed microbiome assembly. For example, the prominent Trueperaceae, found in association with all three host phyla, gained recognition for its exceptional resistance to ionizing radiation 95. In this study, all the sampled seaweeds were collected from the intertidal zones, which are characterized by substantial radiation exposure, particularly during low tides. However, the increased prevalence of the Trueperaceae family in the green algae group may be linked to the preference of green algae for shallower waters and, generally, higher levels of radiation exposure6,53.
In this study, host-specialist taxa appeared to have a competitive advantage over generalists. We note that this outcome does not universally apply to all ecological niches, as generalists can outcompete specialists in certain scenarios96,97. The decline of specialists contributes to the phenomenon known as "functional homogenization" within biodiversity. Clavel et al.98 have proposed using community-level specialization as an indicator to gauge the impacts of global changes, including habitat and climate disruptions, on biodiversity. In our study, the prominence of certain specialist species becomes apparent, offering insights into the current state of the ecosystem. By revisiting the same niche in the future and comparing the relative abundance of host-specific strains, we can glean valuable insights into alterations within this ecosystem, providing a nuanced understanding of its health and stability.
Conclusions
Our results indicate that, despite the differences in bacterial taxonomy across various seaweed phyla, the resilience of these communities relies on conserved functionality which is linked to the host taxonomy. Screening epiphytic bacteria that are strongly linked to phylum taxonomy can identify strains with unique biotechnological applications, such as the usage of polysaccharide-degrading enzymes, as well as antioxidants, anticoagulants, anti-inflammatories, and immunostimulants99. This research sets the stage for further exploration of the intricate relationships within the seaweed holobiont, offering new opportunities to harness the ecological and functional potential of epiphytic bacteria for the sustainable management of seaweed ecosystems.
Data availability
The raw 16S amplicon reads have been uploaded to NCBI (www.ncbi.nlm.nih.gov) under the BioProject: PRJNA1074237 with accessions numbers SAMN39854491 to SAMN39854519. Sanger sequences utilized for verifying host species identity have been submitted to NCBI with accessions numbers PP339846-PP339874.
References
Sahoo, D. & Seckbach, J. (eds) The Algae World (Springer Netherlands, 2015).
Wiencke, C. & Bischof, K. Seaweed Biology Novel Insights into Ecophysiology, Ecology and Utilization (Springer, 2012).
Cotas, J., Gomes, L., Pacheco, D. & Pereira, L. Ecosystem services provided by seaweeds. Hydrobiology 2, 75–96 (2023).
Baweja, P. & Sahoo, D. The Algae World. Classification of Algae (Springer, 2015).
El-Manaway, I. M. & Rashedy, S. H. The Ecology and Physiology of Seaweeds: An Overview. Sustainable Global Resources of Seaweeds (Springer, 2022). https://doi.org/10.1007/978-3-030-91955-9_1.
Eggert, A. Seaweed responses to temperature. Seaweed Boil. Novel Insights Ecophysiol. Ecol. utilization. 27, 47–66 (2012).
Wiencke, C. & Bischof, K. Seaweed Biology. Novel Insights into Ecophysiology, Ecology and Utilization (Springer, 2012).
Buchholz, C. M., Krause, G. & Buck, B. H. Seaweed biology. Seaweed Biol. 219, 471–493 (2012).
Kirk, J. T. Light and Photosynthesis in Aquatic Ecosystems (Cambridge University Press, 1994).
Rioux, L. E., Turgeon, S. L. & Beaulieu, M. Structural characterization of laminaran and galactofucan extracted from the brown seaweed Saccharina longicruris. Phytochemistry 71, 1586–1595 (2010).
Costerton, J. W. et al. Microbial biofilms. Ann. Rev. Microbiol. 49, 711–745 (1995).
Stal, L. J. & Cretoiu, E. The Marine Microbiome (Springer, 2016).
Wahl, M., Goecke, F., Labes, A., Dobretsov, S. & Weinberger, F. The second skin: Ecological role of epibiotic biofilms on marine organisms. Front. Microbiol. https://doi.org/10.3389/fmicb.2012.00292 (2012).
Flemming, H. C. Eps—Then and now. Microorganisms 4, 1–18 (2016).
Flemming, H. C. & Wuertz, S. Bacteria and archaea on earth and their abundance in biofilms. Nat. Rev. Microbiol. 17, 247–260 (2019).
Costerton, J. W., Geesey, G. G. & Cheng, K. J. How bacteria stick. Sci. Am. 238, 86–95 (1978).
Egan, S., Thomas, T. & Kjelleberg, S. Unlocking the diversity and biotechnological potential of marine surface associated microbial communities. Curr. Opin Microbiol. 11, 219–225 (2008).
Wilkins, L. G. E. et al. Host-associated microbiomes drive structure and function of marine ecosystems. PLoS Biol. 17, 1–15 (2019).
Hurd, C. L., Harrison, P. J., Bischof, K. & Lobban, C. S. Seaweed thalli and cells. Seaweed Ecol. Physiol. https://doi.org/10.1017/cbo9781139192637.002 (2014).
Armstrong, E., Yan, L., Boyd, K. G., Wright, P. C. & Burgess, J. G. The symbiotic role of marine microbes on living surfaces. Hydrobiologia 461, 37–40 (2001).
Lemay, M. A. et al. Morphological complexity affects the diversity of marine microbiomes. ISME J. 15, 1372–1386 (2021).
Singh, R. P. & Reddy, C. R. K. Seaweed–microbial interactions: Key functions of seaweed-associated bacteria. FEMS Microbiol. Ecol. 88(2), 213–230 (2014).
Weinberger, F. et al. Spore release in Acrochaetium sp. (Rhodophyta) is bacterially controlled. J. Phycol. 43, 235–241 (2007).
Singh, R. P., Shukla, M. K., Mishra, A., Reddy, C. R. K. & Jha, B. Bacterial extracellular polymeric substances and their effect on settlement of zoospore of Ulva fasciata. Colloids Surf. B Biointerfaces 103, 223–230 (2013).
Spoerner, M., Wichard, T., Bachhuber, T., Stratmann, J. & Oertel, W. Growth and thallus morphogenesis of Ulva mutabilis (Chlorophyta) depends on A combination of two bacterial species excreting regulatory factors. J. Phycol. 48, 1433–1447 (2012).
Provasoli, L. & Pintner, I. J. Bacteria induced polymorphism in an axenic laboratory strain of Ulva lactuca (Chlorophyceae). J. Phycol. 16, 196–201 (1980).
Nakanishi, K., Nishijima, M., Nishimura, M., Kuwano, K. & Saga, N. Bacteria that induce morphogenesis in Ulva pertusa (Chlorophyta) grown under axenic conditions. J. Phycol. 32, 479–482 (1996).
Alsufyani, T. et al. Macroalgal-bacterial interactions: Identification and role of thallusin in morphogenesis of the seaweed Ulva (Chlorophyta). J. Exp. Bot. 71, 3340–3349 (2020).
Burgunter-Delamare, B. et al. Metabolic complementarity between a brown alga and associated cultivable bacteria provide indications of beneficial interactions. Front. Mar. Sci. 7, 85 (2020).
Tapia, J. E., González, B., Goulitquer, S., Potin, P. & Correa, J. A. Microbiota influences morphology and reproduction of the brown alga Ectocarpus sp.. Front. Microbiol. 7, 1–14 (2016).
Saha, M. & Weinberger, F. Microbial, “gardening” by a seaweed holobiont: Surface metabolites attract protective and deter pathogenic epibacterial settlement. J. Ecol. 107, 2255–2265 (2019).
Wang, G. et al. Phylogenetic analysis of epiphytic marine bacteria on Hole-Rotten diseased sporophytes of Laminaria japonica. J. Appl. Phycol. 20, 403–409 (2008).
Beleneva, I. A. & Zhukova, N. V. Bacterial communities of some brown and red algae from Peter the Great Bay, the Sea of Japan. Microbiology 75, 348–357 (2006).
Vairappan, C. S. et al. Distribution and symptoms of epiphyte infection in major carrageenophyte-producing farms. J. Appl. Phycol. 20, 477–483 (2008).
Kusuda, R., Kawai, K., Salati, Fu., Kawamura, Y. & Yamashita, Y. Characteristics of Flavobacterium sp. causing ‘suminori’ disease in cultivated. Porphyra Suisanzoshoku 40, 457–461 (1992).
Dittami, S. M. et al. A community perspective on the concept of marine holobionts: Current status, challenges, and future directions. PeerJ 9, 1–35 (2021).
Egan, S. et al. The seaweed holobiont: Understanding seaweed-bacteria interactions. FEMS Microbiol. Rev. 37, 462–476 (2013).
Forest, R., Victor, S., Farooq, A. & Nancy, K. Diversity and distribution of coral-associated bacteria. Mar. Ecol. Prog. Ser. 243, 1–10 (2002).
Juhmani, A. S. et al. Diversity and dynamics of seaweed associated microbial communities inhabiting the lagoon of Venice. Microorganisms 8, 1–23 (2020).
Qu, T. et al. Structure-function covariation of phycospheric microorganisms associated with the typical cross-regional harmful macroalgal bloom. Appl. Environ. Microbiol https://doi.org/10.1128/aem.01815-22 (2023).
Aires, T., Muyzer, G., Serrão, E. A. & Engelen, A. H. Seaweed loads cause stronger bacterial community shifts in coastal lagoon sediments than nutrient loads. Front. Microbiol. 10, 1–18 (2019).
Bauer, M. A., Kainz, K., Carmona-Gutierrez, D. & Madeo, F. Microbial wars: Competition in ecological niches and within the microbiome. Microb. Cell. 5, 215–219 (2018).
Coyte, K. Z., Schluter, J. & Foster, K. R. The ecology of the microbiome: Networks, competition, and stability. Science 1979(350), 663–666 (2015).
Wood, G. et al. Host genetics, phenotype and geography structure the microbiome of a foundational seaweed. Mol. Ecol. 31, 2189–2206 (2022).
Sneed, J. & Puglisi, M. P. The role of natural products in structuring microbial communities of marine algae. In Chemical Ecology 221–235 (CRC Press, 2018).
Kloareg, B. & Quatrano, R. S. Structure of the cell walls of marine algae and ecophysiological functions of the matrix polysaccharides. Oceanogr. Mar. Biol. Ann. Rev. 26, 259–315 (1988).
Malik, A. A. S. et al. Defence on Surface: Macroalgae and Their Surface-Associated Microbiome. Advances in Botanical Research Vol. 95 (Elsevier, 2020).
Wang, Z., Xiao, T., Pang, S., Liu, M. & Yue, H. Isolation and identification of bacteria associated with the surfaces of several algal species. Chin. J. Oceanol. Limnol. 27, 487–492 (2009).
Selvarajan, R. et al. Distribution, interaction and functional profiles of epiphytic bacterial communities from the rocky intertidal seaweeds, South Africa. Sci. Rep. 9, 1–13 (2019).
Korlević, M., Markovski, M., Zhao, Z., Herndl, G. J. & Najdek, M. Seasonal dynamics of epiphytic microbial communities on marine macrophyte aurfaces. Front. Microbiol. 12, 1–17 (2021).
Pei, P. et al. Environmental factors shape the epiphytic bacterial communities of Gracilariopsis lemaneiformis. Sci. Rep. 11, 1–16 (2021).
Bondoso, J. et al. Epiphytic Planctomycetes communities associated with three main groups of macroalgae. FEMS Microbiol. Ecol. https://doi.org/10.1093/femsec/fiw255 (2017).
Israel, A., Golberg, A. & Neori, A. The seaweed resources of Israel in the Eastern Mediterranean Sea. Botanica Mar. 63, 85–95 (2019).
Michot, B. & Bachellerie, J. P. Comparisons of large subunit rRNAs reveal some eukaryote-specific elements of secondary structure. Biochimie 69, 11–23. https://doi.org/10.1016/0300-9084(87)90267-7 (1987).
Lenaers, G., Maroteaux, L., Michot, B. & Herzog, M. Dinoflagellates in evolution. A molecular phylogenetic analysis of large subunit ribosomal RNA. J. Mol. Evol. 29(1), 40–51. https://doi.org/10.1007/BF02106180 (1989).
Orsini, L. et al. Toxic Pseudo-nitzschia multistriata (Bacillariophyceae) from the Gulf of Naples: Morphology, toxin analysis and phylogenetic relationships with other Pseudo-nitzschia species. Eur. J. Phycol. 37, 247–257 (2002).
Benson, D. A. et al. GenBank. Nucl. Acids Res. 41, 36–42 (2013).
Neumann, B., Pospiech, A. & Schairer, H. Rapid isolation of genomic DNA from gram-negative bacteria. Trends Genet. 8, 332–333 (1992).
Parada, A. E., Needham, D. M. & Fuhrman, J. A. Every base matters: Assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ. Microbiol. 18, 1403–1414 (2016).
Caporaso, J. G. et al. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U.S.A. 108, 4516–4522 (2011).
Deutsch, Y., Ofek-Lalzar, M., Borenstein, M., Berman-Frank, I. & Ezra, D. Re-introduction of a bioactive bacterial endophyte back to its seaweed (Ulva sp.) host, influences the host’s microbiome. Front. Mar. Sci. 10, 1–12 (2023).
R Team Core. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. (2023).
Levins, R. Evolution in Changing Environments: Some Theoretical Explorations (Princeton University Press, 1968).
Pandit, S. N., Kolasa, J. & Cottenie, K. Contrasts between habitat generalists and specialists: An empirical extension to the basic metacommunity framework. Ecology 90, 2253–2262 (2009).
Pruesse, E., Peplies, J. & Glöckner, F. O. SINA: Accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28, 1823–1829 (2012).
Quast, C. et al. The SILVA ribosomal RNA gene database project: Improved data processing and web-based tools. Nucl. Acids Res. 41, 590–596 (2013).
Chong, J., Liu, P., Zhou, G. & Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 15, 799–821 (2020).
McMurdie, P. J. & Holmes, S. Phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217 (2013).
Wemheuer, F. et al. Tax4Fun2: A R-based tool for the rapid prediction of habitat-specific functional profiles and functional redundancy based on 16S rRNA gene marker gene sequences. BioRxiv 490037 (2018).
Kanehisa, M., Furumichi, M., Sato, Y., Kawashima, M. & Ishiguro-Watanabe, M. KEGG for taxonomy-based analysis of pathways and genomes. Nucl. Acids Res. 51, D587–D592 (2023).
Kanehisa, M. Toward understanding the origin and evolution of cellular organisms. Protein Sci. 28, 1947–1951. https://doi.org/10.1002/pro.3715 (2019).
Kanehisa, M. & Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Research vol. 28 http://www.genome.ad.jp/kegg/ (2000).
Hammer, Ø., Harper, D. A. T. & Pau, D. R. Past: Paleontological statistics software package for education and data analysis. Palaeontol. Electron. 4, 1–9 (2001).
Lopes, D. et al. Insights of species-specific polar lipidome signatures of seaweeds fostering their valorization in the blue bioeconomy. Algal. Res. 55, 102242 (2021).
Steinberg, P. D., Schneider, R. & Kjelleberg, S. Chemical defenses of seaweeds against microbial colonization. Biodegradation 8, 211–220 (1997).
Florez, J. Z., Camus, C., Hengst, M. B. & Buschmann, A. H. A functional perspective analysis of macroalgae and epiphytic bacterial community interaction. Front. Microbiol. https://doi.org/10.3389/fmicb.2017.02561 (2017).
Burke, C., Thomas, T., Lewis, M., Steinberg, P. & Kjelleberg, S. Composition, uniqueness and variability of the epiphytic bacterial community of the green alga Ulva australis. ISME J. 5, 590–600 (2010).
Chen, J. et al. Composition and functional diversity of epiphytic bacterial and fungal communities on marine macrophytes in an intertidal zone. Front. Microbiol. 13, 1–13 (2022).
Logares, R. et al. Biogeography of bacterial communities exposed to progressive long-term environmental change. ISME J. 7, 937–948 (2013).
Székely, A. J. & Langenheder, S. The importance of species sorting differs between habitat generalists and specialists in bacterial communities. FEMS Microbiol. Ecol. 87, 102–112 (2014).
Mariadassou, M., Pichon, S. & Ebert, D. Microbial ecosystems are dominated by specialist taxa. Ecol. Lett. 18, 974–982 (2015).
Weiss, A., Costa, R. & Wichard, T. Morphogenesis of Ulva mutabilis (Chlorophyta) induced by Maribacter species (Bacteroidetes, Flavobacteriaceae). Bot. Mar. 60, 197–206 (2017).
Paix, B. et al. A multi-omics analysis suggests links between the differentiated surface metabolome and epiphytic microbiota along the thallus of a mediterranean seaweed holobiont. Front. Microbiol. 11, 1–18 (2020).
Marzinelli, E. M. et al. Continental-scale variation in seaweed host-associated bacterial communities is a function of host condition, not geography. Environ. Microbiol. 17, 4078–4088 (2015).
Dworkin, M., Falkow, S., Rosenberg, E., Schleifer, K. H. & Stackebrandt, E. The Prokaryotes 549–590 (Springer, 2006).
Kappelmann, L. et al. Polysaccharide utilization loci of North Sea Flavobacteriia as basis for using SusC/D-protein expression for predicting major phytoplankton glycans. ISME J. 13, 76–91 (2019).
Lu, D. C., Wang, F. Q., Amann, R. I., Teeling, H. & Du, Z. J. Epiphytic common core bacteria in the microbiomes of co-located green (Ulva), brown (Saccharina) and red (Grateloupia, Gelidium) macroalgae. Microbiome 11, 1–22 (2023).
Gavriilidou, A. et al. Comparative genomic analysis of Flavobacteriaceae: Insights into carbohydrate metabolism, gliding motility and secondary metabolite biosynthesis. BMC Genom. 21, 1–21 (2020).
Sakai, T., Kimura, H. & Kato, I. A marine strain of Flavobacteriaceae utilizes brown seaweed fucoidan. Mar. Biotechnol. 4, 399–405 (2002).
Kim, N. K., Oh, S. & Liu, W. T. Enrichment and characterization of microbial consortia degrading soluble microbial products discharged from anaerobic methanogenic bioreactors. Water Res. 90, 395–404 (2016).
Golberg, A. et al. Macroalgal Biorefineries for the Blue Economy (World Scientific Publishing, 2020).
Stiger-Pouvreau, V., Bourgougnon, N. & Deslandes, E. Carbohydrates From Seaweeds. In Seaweed in Health and Disease Prevention 223–274 (2016) https://doi.org/10.1016/B978-0-12-802772-1.00008-7.
Singh, R. P. & Reddy, C. R. K. Seaweed-microbial interactions: Key functions of seaweed-associated bacteria. FEMS Microbiol. Ecol. 88, 213–230 (2014).
Meyer, K. M., Petersen, I. A. B., Tobi, E., Korte, L. & Bohannan, B. J. M. Use of RNA and DNA to identify mechanisms of bacterial community homogenization. Front. Microbiol. 10, 1–13 (2019).
Albuquerque, L. et al. Truepera radiovictrix gen. nov., sp. nov., a new radiation resistant species and the proposal of Trueperaceae fam. nov.. FEMS Microbiol. Lett. 247, 161–169 (2005).
Mo, Y. et al. Biogeography and co-occurrence patterns of bacterial generalists and specialists in three subtropical marine bays. Limnol. Oceanogr. 66, 793–806 (2021).
Thuiller, W., Lavorel, S. & Araújo, M. B. Niche properties and geographical extent as predictors of species sensitivity to climate change. Global Ecol. Biogeogr. 14, 347–357 (2005).
Clavel, J., Julliard, R. & Devictor, V. Worldwide decline of specialist species: Toward a global functional homogenization?. Front. Ecol. Environ. 9, 222–228 (2011).
Kaur, M., Saini, K. C., Mallick, A. & Bast, F. Seaweed-associated epiphytic bacteria: Diversity, ecological and economic implications. Aquat. Bot. 189, 103698 (2023).
Acknowledgements
This project was funded by the Ph.D. excellence scholarship of the University of Haifa given to O.N. We thank Dr. Maya Ofek-Lalzar from the University of Haifa Bioinformatics Service Unit for her professional assistance.
Author information
Authors and Affiliations
Contributions
O.N. wrote the main manuscript text and prepared the figures. O.N., T.L.K., and N.B. collected the samples and analyzed them. O.N, T.L.K, A.I., and M.R.B. analyzed the results and performed the bioinformatic processing. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Nahor, O., Israel, Á., Barger, N. et al. Epiphytic microbiome associated with intertidal seaweeds in the Mediterranean Sea: comparative analysis of bacterial communities across seaweed phyla. Sci Rep 14, 18631 (2024). https://doi.org/10.1038/s41598-024-69362-y
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-69362-y
- Springer Nature Limited