
Abstract
Inflammation is an important factor in Alzheimer’s disease (AD). An NMR measurement in plasma, glycoprotein acetyls (GlycA), captures the overall level of protein production and glycosylation implicated in systemic inflammation. With its additional advantage of reducing biological variability, GlycA might be useful in monitoring the relationship between peripheral inflammation and brain changes relevant to AD. However, the associations between GlycA and these brain changes have not been fully evaluated. Here, we performed Spearman’s correlation analyses to evaluate these associations cross-sectionally and determined whether GlycA can inform AD-relevant longitudinal measurements among participants in the Alzheimer’s Disease Neuroimaging Initiative (n = 1506), with additional linear models and stratification analyses to evaluate the influences of sex or diagnosis status and confirm findings from Spearman’s correlation analyses. We found that GlycA was elevated in AD patients compared to cognitively normal participants. GlycA correlated negatively with multiple concurrent regional brain volumes in females diagnosed with late mild cognitive impairment (LMCI) or AD. Baseline GlycA level was associated with executive function decline at 3–9 year follow-up in participants diagnosed with LMCI at baseline, with similar but not identical trends observed in the future decline of memory and entorhinal cortex volume. Results here indicated that GlycA is an inflammatory biomarker relevant to AD pathogenesis and that the stage of LMCI might be relevant to inflammation-related intervention.
Similar content being viewed by others

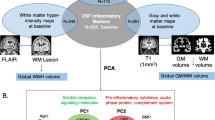

Introduction
Mounting evidence suggests that inflammation and immune dysregulation play a critical role in Alzheimer’s disease (AD) pathogenesis1. Inflammation-associated diseases such as metabolic syndromes, diabetes, and cardiovascular diseases are linked to an increased risk of AD2,3,4,5,6. In addition, disturbances of circulating markers of inflammation, such as cytokines, endocannabinoids, and oxylipins, have been linked to AD and cognitive decline7,8,9,10. In pre-clinical models, the peripheral inflammation stimuli, such as a high-fat diet and chronic lipopolysaccharide administration, can lead to AD-related dysregulations in the central nervous system (CNS), including altered blood–brain barrier functionality, changed cerebrovascular properties, neuroinflammation, and amyloid beta (Aβ) pathologies11,12,13,14,15,16. However, complexity does exist, as factors such as sex differences17,18 and health status19,20 may alter the underlying metabolic connections between peripheral inflammation and brain pathologies. In particular, males and females are susceptible to different inflammatory risk factors throughout their lifetime21. The relationship between peripheral inflammation and brain pathologies between males and females may differ over time or throughout the progression of diseases. Generally speaking, despite the observed linkage of peripheral metabolism to central pathologies13,22,23,24, we still need a better understanding of the crosstalk between peripheral inflammation and AD characteristics in the CNS to guide the improvement of inflammatory-related modifiable actions for reducing AD risks and mitigating its pathological trajectories.
Glycoprotein acetyls (GlycA), a biomarker for systemic inflammation, is informative for the level of vascular aging (e.g., due to the significant correlation between GlycA and an indicator of arterial stiffness, aortic pulse wave velocity25), the presence of cardiovascular risks (e.g., due to the significant correlation between GlycA and future risk of hypertension and metabolic syndrome26, cardiovascular mortality27 and other cardiovascular risk markers28) and the risk of all-cause mortality29. This proton nuclear magnetic resonance (NMR)-based measurement detects the production and further glycosylation of several acute-phase proteins, including alpha-1-acid glycoprotein, alpha-1-antitrypsin, alpha-1-antichymotrypsin, haptoglobin and transferrin30. As such, GlycA level captures the overall burden of both the acute and chronic phases of inflammation28,30 and has been shown to be informative in the context of inflammatory disorders, such as rheumatoid arthritis31,32, lupus33,34, psoriasis35, atherosclerotic cardiovascular diseases27,28, metabolic syndrome26,36, type 2 diabetes37,38, and inflammatory bowel disease39. Compared to the established inflammatory biomarker C-reactive protein (CRP), GlycA may have the advantage of reduced biological variability, as a previous study has shown that the weekly intraindividual and interindividual variations of 23 healthy participants over 5 weeks were substantially lower for GlycA (4.3% and 15.3%, respectively) than for CRP (29.2% and 133.9%, respectively)30. In addition, it was shown that compared to CRP, the detection of GlycA was more sensitive among young healthy individuals, less indicative of acute inflammation but more representative of chronic inflammation26,40. Meanwhile, the inverse relations between cognitive functions and the level of plasma GlycA41,42 or its increases43 over years have been reported in studies of younger adults, but such studies are not entirely informative for neurodegenerative diseases that are usually associated with older ages. Therefore, the relationship between GlycA and AD-related biomarkers in older participants has yet to be established.
To further address these knowledge gaps, we aimed in this study to determine the relationship between peripheral inflammation and AD-related biomarkers. We evaluated: (1) differences in GlycA levels in participants at different diagnosis stages along the AD continuum (“Inflammation level indicated by GlycA was elevated along the AD trajectory”); (2) the correlations between baseline GlycA level and executive function (“Baseline GlycA level was correlated with a future decrease in executive function in participants diagnosed with LMCI at baseline”), memory (“Baseline GlycA level was correlated with a future decrease in memory in participants diagnosed with LMCI at baseline”), brain regional brain volumes (“Baseline GlycA level was correlated with a future decrease in entorhinal cortex volume in participants diagnosed with LMCI at baseline” and “Baseline GlycA correlated with baseline brain structural atrophies specifically in female participants diagnosed with LMCI and AD”) and cerebral spinal fluid (CSF) amyloid/tau/neurodegeneration (A/T/N) biomarkers (“Association of plasma GlycA with CSF A/T/N biomarkers”) measured at baseline and measured longitudinally in the follow-up years while considering the influence of diagnosis status and sex on those correlations. To do so, we utilized the data from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) cohort, including baseline NMR measurement of GlycA, baseline and longitudinal executive function composite score, memory composite scores, magnetic resonance imaging (MRI) of brain morphologies and A/T/N biomarkers to perform correlation analysis.
Methods
ADNI study participants and data accessibility
Baseline demographic, clinical, cognition, MRI imaging, genetic, biomarkers, cytokines, and multi-omics data (including peripheral GlycA measurement) of the study participants used in the current study were downloaded from the ADNI database (www.adni-info.org), managed through the Laboratory of Neuro Imaging Image & Data Archive (http://adni.loni.usc.edu/). The protocol procedures were approved by the Institutional Review Boards or Research Ethics Boards of the participating institutions. Participants provided signed written informed consent and permission for data access and analysis. All procedures were performed according to relevant guidelines and regulations. The ADNI cohort, launched in 2003 as a public–private partnership and led by Principal Investigator Michael W. Weiner, MD, tests the primary goal of combining serial MRI and positron emission tomography (PET) imaging, biological markers, and clinical and neuropsychological evaluation to measure the progression of mild cognitive impairment (MCI) and early AD.
The diagnosis status of the adult participants included cognitively normal (CN), significant memory concern (SMC), early mild cognitive impairment (EMCI), late mild cognitive impairment (LMCI), and AD. The detailed inclusion and exclusion criteria can be accessed at the ADNI documentation website (https://adni.loni.usc.edu/methods/documents/). In brief, CN participants had Mini-Mental State Exam (MMSE) scores of 24–30 (inclusive) and a Clinical Dementia Rating scale (CDR) score of 0 without any memory complaints. Participants with SMC had self-reported subjective memory concerns but MMSE and CDR scores rated as CN (https://classic.clinicaltrials.gov/ct2/show/NCT01231971). Participants with MCI had an MMSE score of 24–30 (inclusive), a CDR score of 0.5, and a memory complaint, yet did not have sufficient levels of cognition and functional performance impairment to be diagnosed with dementia. Among participants with MCI, EMCI and LMCI were differentiated by different levels of abnormal memory functions, which was indicated by the education-adjusted Wechsler Memory Scale-Revised Logical Memory II subscale, with the cut-off value for LMCI being (a) ≤ 8 for 16 or more years of education, (b) ≤ 4 for 8–15 years of education and (c) ≤ 2 for 0–7 years of education. Participants with AD had an MMSE score of 20–26 (inclusive), a CRD of 0.5 or 1.0, a memory complaint, and their cognition and functional performances met the National Institute of Neurological and Communicative Disorders and Stroke/the Alzheimer’s Disease and Related Disorders Association criteria for probable AD. Exclusion criteria included—but were not limited to—major depression and other significant neurological diseases. Other detailed exclusion criteria can be found on the above-mentioned ADNI study documents website. The dataset contained complete info on GlycA, APOE genotypes, sex, body mass index (BMI), age and education (n = 1506), which was detailed in Table S1. Of the participants, 684 (45.4%) were female; 700 (46.5%) were APOE4 carriers, and 153 (10.2%) were APOE4/4 carriers. In addition, 361 (24.0%) were CN participants, 96 (6.4%) had SMC, 280 (18.6%) had EMCI, 481 (31.9%) had LMCI, and 288 (19.1%) had AD. The age at screening (average ± standard deviation) was 73.1 ± 7.1, baseline BMI was 26.9 ± 4.9, and years of education was 15.9 ± 2.9. The detailed protocols for blood and CSF sample collection and storage can be found on the abovementioned ADNI study documents website.
Peripheral GlycA measurement
Peripheral GlycA was measured via the Nightingale Health platform using established protocols44,45,46. In brief, serum samples were stored at − 80 °C and thawed overnight at + 4 °C prior to analysis. The samples were then gently mixed, centrifuged, and mixed with an equal amount of an NMR measurement buffer of 75 mmol/L disodium phosphate in 8:2 H2O/D2O (pH 7.4) with 0.08% sodium 3-(trimethylsilyl)propionate-2,2,3,3-d4 and 0.04% sodium azide44. The prepared samples were then measured using a Bruker AVANCE III HD 500 MHz spectrometer coupled with a SampleJet cooled robotic sample changer and CryoProbe Prodigy TCI, a cryogenically cooled triple resonance probe head45. Quantification results were then obtained from the NMR spectral data via an advanced proprietary software of the Nightingale Health platform46.
In addition, the quantitative result of GlycA was log2-transformed and adjusted with medication data prior to further analyses using an Akaike information criterion (AIC) backward stepwise regression method23,47. The full list of medication classes and the AIC-selected medications were listed in Table S2. Other confounders, such as age, BMI, sex, education or APOE4 were adjusted as specified in the results.
Analysis of CRP in plasma and cerebral spinal fluid
Cytokines, including CRP in plasma and CSF were measured using fluorescence-labeled microspheres via a multiplex proteomics method based on the Luminex xMAP platform, with an established threshold of detection and a dynamic range for each analyte48,49. The detailed sample preparation was provided by the previous publication, which included steps of sample thawing, the introduction of capture microspheres, the addition of multiplexed mixtures of reporter antibodies (biotinylated), multiplex development via the streptavidin–phycoerythrin solution, and final volume adjustment49. Prior to statistical analysis, cytokines were normalized and medication adjusted as above.
MRI brain imaging data acquisition
MRI protocol and data were accessed through the ADNI website (https://adni.loni.usc.edu/). In brief, MRI imaging was acquired using a 1.5 T or 3 T MRI scanner with the sequences of T1 and dual echo T2-weighted imaging (ADNI 1), or a 3 T MRI scanner with fully sampled and accelerated T1-weighted imaging in addition to 2D FLAIR and T2*-weighted imaging (ADNI GO/2). To process MRI scans and extract whole brain and ROI (region of interest)-based neuroimaging measurements determined by automated segmentation and parcellation including volumes and cortical thickness, FreeSurfer V6, a widely employed automated MRI analysis approach, was used50. After the cortical surface was reconstructed, the cortical thickness at each vertex was calculated by taking the Euclidean distance between the grey/white boundary and the grey/cerebrospinal fluid (CSF) boundary at each vertex on the surface.
Cognitive function and memory measurement
A composite score for measuring overall executive functioning (EF) was previously developed and validated51. In brief, the composite score was constructed via an iterative process that included the confirmatory factor analysis to build a model and the reviewing of findings to construct a revised model. The model was built using ADNI baseline data, based on the Category fluency (animals) test, Category fluency (vegetables) test, Trails A and Trails B test, Digit span backwards test, Wechsler Adult Intelligence Scale—Revised Digit Symbol Substitution test, and the five Clock Drawing items test (circle, symbol, numbers, hands, and time).
A composite score for measuring the overall memory (MEM) was previously developed and validated52. In brief, MEM was derived from two versions of the longitudinal Rey Auditory Verbal Learning Test, three versions of the AD Assessment Schedule—Cognition, the MMSE, and Logical Memory data, using a single factor model.
Amyloid/Tau/Neurodegeneration (A/T/N) biomarker measurement in CSF
A/T/N biomarkers, Aβ1-42, t-tau, and p-tau181p in CSF samples were measured via a fully automated Roche Elecsys immunoassay, in which a sample preparation procedure with two incubation steps was used53,54.
Statistical analyses
All statistical analyses were performed in JMP® Pro 16 and 17 (SAS Institute Inc., Cary, NC). We used the standard least-square method for analysis of covariance (ANCOVA) or calculating the residual of variable controlling confounders. ANCOVA was applied to compare the differences in means of GlycA and CRP in both plasma and CSF, followed by Tukey HSD post hoc analysis to evaluate the significant differences among various groups. The effect size of the comparison between any two groups was evaluated using Cohen’s term (dCohen), which was calculated by the differences in means divided by the standard deviation of all measurements in both groups.
To investigate non-linear associations, Spearman’s correlations were performed on residuals of specific measurements while controlling confounders, namely APOE4, age, BMI, education level, sex, diagnosis status and/or medications. For the MRI measurement of brain volumes, we adjusted for the confounders of magnet types of MRI and intracranial volume (log2-transformed). A linear model with sex or diagnosis status interaction with GlycA was additionally applied to test whether the observed associations were different between sexes or among diagnosis statuses.
Additional Spearman’s correlation analyses were performed to evaluate the relationship of baseline GlycA with cognitive functions and brain regional volumes in the future follow-up years with the same confounders adjusted as above, in addition to adjusting to the baseline value of these measurements. The analysis was performed with stratification of diagnosis status at baseline because participants with differing diagnosis status at baseline experienced different trajectories of brain structural and cognitive changes in the follow-up years55, which might result in differential relationships between GlycA and these brain measurements. For multiple comparisons, the false discovery rate (FDR) correction using the Benjamini and Hochberg method (q = 0.2) was applied56.
Ethics approval and consent to participate
The protocol procedures were approved by the Institutional Review Boards or Research Ethics Boards of the participating institutions. Participants provided signed written informed consent and permission for data access and analysis.
Results
Inflammation level indicated by GlycA was elevated along the AD trajectory
To evaluate the level of peripheral inflammation along AD progression, we applied an ANCOVA model that tested GlycA level against diagnosis groups (CN, SMC, EMCI, LMCI and AD) and controlled for the presence of APOE4 allele, age, BMI, education and sex as covariates (Fig. 1a). The sex-diagnosis group interaction was additionally applied to test whether sex influences the pattern of GlycA levels along the disease progression and it was not significant (p = 0.579), and thus the interaction term was removed from further analysis.

GlycA levels in ADNI participants. GlycA level in (a) groups of various diagnosis statuses, adjusted for sex and (b) sexes, adjusted by diagnosis status. (a) GlycA level was log2-transformed and adjusted for medications, sex, age, APOE4, BMI, and education level. (b) GlycA level was log2-transformed and adjusted for medications, diagnosis status, age, APOE4, BMI, and education level. ANCOVA and Tukey HSD post hoc analysis were performed; levels not labeled with the same letter differed significantly (p < 0.05) (e.g., the groups in the same figure labeled with A and B were significantly different from each other, but the ones labeled with A and AB were not significantly different). Values in the bar graph were shown as the mean residual GlycA level ± standard error. There was no sex-diagnosis status interaction in a full factorial model. AD: Alzheimer’s disease; ANCOVA: Analysis of covariance; BMI: Body mass index; CN: Cognitively normal; EMCI: Early mild cognitive impairment; GlycA: Glycoprotein acetyls; HSD: Honestly significant difference; LMCI: Late mild cognitive impairment; SMC: Significant memory concerns.
GlycA level increased in the participants at more severe disease stages (diagnosis group p < 0.001). In particular, participants with AD had higher GlycA levels compared to CN participants and participants with SMC and EMCI. Participants with LMCI had higher GlycA levels than did participants with SMC and EMCI. In the same model, GlycA was positively associated with BMI (p < 0.001, βestimate = 0.01) and negatively associated with education (p < 0.001, βestimate = − 0.01). GlycA was also higher in females than in males (p < 0.001, dCohen = 0.3, Fig. 1b). GlycA was not influenced by the presence of APOE4 (p = 0.227) or age (p = 0.143).
Additionally, we evaluated the relationship between plasma GlycA and plasma CRP using Spearman’s rank order correlation to compare the NMR-derived inflammation indicator to the commonly used peripheral inflammation marker (Fig. S1A). Moreover, we correlated plasma GlycA and CRP in CSF to evaluate the association between peripheral and central inflammation (Fig. S1B). GlycA was positively associated with CRP in both plasma (p < 0.001, ρ = 0.445) and CSF (p < 0.001, ρ = 0.370). In contrast, CRP in both plasma and CSF did not differ among the different diagnosis groups (p = 0.192 and p = 0.832), and only CRP in plasma was higher in females compared to males (p = 0.015, dCohen = 0.2) (Fig. S2), where fewer participants had their CRP measured (n for each group: in blood: n = 50 (CN), 0 (SMC), 0 (EMCI), 337 (LMCI), and 97 (AD); in CSF: n = 74 (CN), 0 (SMC), 0 (EMCI), 133 (LMCI), and 60 (AD)). The lack of significant differences in plasma CRP among diagnosis groups due to reduced data coverage was supported by the result that when adjusting plasma CRP, GlycA levels were no longer significantly different among the various diagnosis groups in the abovementioned ANCOVA model (p = 0.126).
Baseline GlycA level was correlated with a future decrease in executive function in participants diagnosed with LMCI at baseline
To interrogate whether baseline GlycA can predict future cognitive decline, we evaluated the correlation of baseline GlycA with the longitudinal composite scores for EF51. The relationship between baseline GlycA and EF in follow-up years was evaluated using Spearman’s rank order correlation analyses, adjusting for baseline EF, sex, age, APOE4 genotype, BMI, education and follow-up years, and treating participant ID as a random factor. In addition, due to the differential connections observed between central and peripheral changes in different stages of AD progression57, the analyses were performed with stratification of diagnosis status across participants of CN, SMC, EMCI, LMCI, and AD (Table S3). Linear mixed models were used to confirm these analyses (Tables S4 and S5).
ADNI data with the baseline GlycA measurement enabled us to probe cognitive changes for up to a 13-year follow-up (Fig. 2 and Fig. S3). Participants with differing diagnosis status at baseline experienced different trajectories of brain structural and cognitive changes in the follow-up years (Fig. 2). In particular, participants diagnosed with LMCI at baseline experienced the earliest EF declines (defined as the longitudinal EF significantly deviated from Year 1 follow-up) in follow-up years among participants who were not diagnosed with AD at baseline, starting on Year 3 (Fig. 2 and Fig. S3). Therefore, we specifically investigated the correlations between baseline GlycA level and EF in the different years of follow-up among participants diagnosed with LMCI at baseline, while other diagnosis statuses have also been explored (Table S3). The LMCI-GlycA interaction to predict EF was confirmed using a linear mixed model (p = 0.025, Table S4). In participants with LMCI, baseline GlycA was negatively associated with EF in years 3–9 of follow-up (Fig. 3, with year 3 (p = 0.036, ρ = -0.119), year 4 (p = 0.001, ρ = − 0.235), year 5 (p = 0.003, ρ = − 0.267), year 6 (p < 0.001, ρ = − 0.380), year 7 (p = 0.002, ρ = − 0.314), year 8 (p = 0.002, ρ = − 0.389) and year 9 (p = 0.040, ρ = − 0.345)). Additionally, GlycA-EF association among participants with LMCI was not sex-dependent (pinteraction = 0.833). The negative correlation between longitudinal EF and baseline GlycA in participants diagnosed with LMCI at baseline was confirmed using a linear mixed model (Table S5).

Executive function and entorhinal cortex volume decline in ADNI participants. Participants diagnosed with LMCI at baseline started to have the most continuous executive functional decline on and after 3-year follow-up and continuous entorhinal cortex volume decline on and after 2-year follow-up, which was well distinguished from CN participants and participants with SMC and EMCI. Executive function was executive function score adjusted for sex, APOE4, education, BMI at visit and age at screening. The ANCOVA results comparing different time points within each diagnosis group are shown in the heatmap of Fig. S3; the ANCOVA results comparing EF of different diagnoses at baseline are shown in Fig. S4. Entorhinal cortex volume was entorhinal cortex volume adjusted for intracranial volume (log2), magnet type, sex, APOE4, education, BMI at visit and age at screening. The ANCOVA results comparing different time points within each diagnosis group are shown in the heatmap of Fig. S7; the ANCOVA results comparing the entorhinal cortex volume of different diagnoses at baseline are shown in Fig. 5. Results annotated with * were at a significantly different level compared to Year 1 within the diagnosis group (Figs. S3 and S7). AD: Alzheimer’s disease; ANCOVA: Analysis of covariance; BMI: Body mass index; CN: Cognitively normal; EMCI: Early mild cognitive impairment; LMCI: Late mild cognitive impairment; SMC: Significant memory concerns.

Baseline GlycA level associated with future executive function declines in LMCI patients. Baseline GlycA level was associated with executive function composite score (EF) declines in late mild cognitive impairment (LMCI) patients at 3–9 years of follow-up. Spearman’s rank order correlation was performed on residuals of EF and GlycA, and the analyses were stratified by diagnosis status. EF at different years were adjusted for baseline EF level, screening age, follow-up year, BMI at the time, APOE4, sex, and education, treating participant ID as random factors; baseline GlycA level was log2-transformed and adjusted for medication, screening age, baseline BMI, APOE4, sex, and education. All significant results shown in the figure passed FDR correction using the Benjamini and Hochberg method (q = 0.2)56. The analysis was performed on participants with different diagnosis statuses (Table S3), but only in the participants with LMCI was EF at continuous follow-up years negatively associated with baseline GlycA level and thus shown here. In addition, the LMCI-GlycA interaction was confirmed using a linear mixed model (p = 0.025, Table S4). Therefore, the analysis results for the participants with LMCI are shown here. A full factorial linear mixed model was used to show that there were no sex-GlycA interactions controlling DX (p = 0.864). BMI: Body mass index; DX: Diagnosis at baseline; GlycA: Glycoprotein acetyls; ID: Identification.
Of note, only a weak association (p = 0.049, ρ = − 0.172) was observed between the baseline GlycA and baseline EF in female participants with AD (Fig. S4). No associations were detected in females in other diagnostic groups, or in males. Therefore, the lack of sex effects on the GlycA-EF relationship was consistent between cross-sectional and longitudinal observations.
Baseline GlycA level was correlated with a future decrease in memory in participants diagnosed with LMCI at baseline
To investigate whether memory function follows a pattern similar to executive function, similar analyses to the above were performed on baseline GlycA and longitudinal composite scores for MEM across participants of CN, SMC, EMCI, LMCI, and AD (Table S3)52. Participants diagnosed with LMCI at baseline experienced the earliest continuous cognitive declines (i.e., cognition significantly deviated from Year 1 follow-up) in follow-up years among participants who were not diagnosed with AD at baseline, starting in Year 2 (Fig. S5). In these participants, baseline GlycA was negatively associated with MEM in years 5–8 of follow-up (Fig. S6, with year 5 (p = 0.036, ρ = − 0.183), year 6 (p = 0.037, ρ = − 0.186), year 7 (p = 0.019, ρ = − 0.235) and year 8 (p = 0.036, ρ = − 0.262)). However, these associations did not pass FDR correction and were weaker compared to the associations with EF, which was confirmed using a linear mixed model (Table S5). There was also no significant LMCI-GlycA interaction to predict MEM, which was confirmed using a linear mix model (p = 0.441, Table S4). Additionally, GlycA-MEM association among participants with LMCI was not sex-dependent (pinteraction = 0.471). Unlike baseline EF, there was no association between baseline GlycA and baseline MEM in either sex or in any diagnosis group (p > 0.05).
Baseline GlycA level was correlated with a future decrease in entorhinal cortex volume in participants diagnosed with LMCI at baseline
To identify the structural basis of the MEM and EF patterns observed above, we performed a similar analysis on MRI-measured brain regional volumes with additional adjustment for MRI methods and intracranial volume, across participants of CN, SMC, EMCI, LMCI, and AD (Table S6). We therefore investigated whether baseline GlycA can predict the further decline in brain regional volumes using the above-described approach. Participants diagnosed with LMCI at baseline experienced the earliest continuous memory declines (i.e., memory significantly deviated from Year 1 follow-up) in follow-up years among participants who were not diagnosed with AD at baseline, starting in Year 2 (Fig. 2 and Fig. S7). Negative associations between baseline GlycA and entorhinal cortex volume was observed for the follow-up years 2 (p = 0.011, ρ = − 0.140), 4 (p = 0.040, ρ = − 0.165), 6 (p = 0.024, ρ = − 0.224), 7 (p = 0.026, ρ = − 0.257) and 8 (p = 0.025, ρ = − 0.345) (Fig. 4). The negative correlation between longitudinal entorhinal cortex volume and baseline GlycA in participants diagnosed with LMCI at baseline was confirmed using a linear mixed model (Table S5). The LMCI-GlycA interaction to predict entorhinal cortex volume was confirmed using a linear mixed model (p = 0.005, Table S4). Additionally, GlycA-entorhinal cortex volume association among participants with LMCI was not sex-dependent (pinteraction = 0.333). Similar associations were not observed with other brain regional volumes (Table S6).

Baseline GlycA level associated with future entorhinal cortex volume declines in LMCI patients. Baseline GlycA level was associated with entorhinal cortex volume declines in late mild cognitive impairment (LMCI) patients at the 2nd, 4th, and 6–8th years of follow-up. Spearman’s rank order correlation was performed on residuals of entorhinal cortex volume and GlycA, and the analyses were stratified by diagnosis status. Entorhinal cortex volume at different years was adjusted for baseline entorhinal cortex volume, screening age, follow-up year, BMI at the time, APOE4, sex, and education, treating participant ID as random factors (all MRI volumes were log2-transformed and adjusted for intracranial volume and magnet type); baseline GlycA level was log2-transformed and adjusted for medication, screening age, baseline BMI, APOE4, sex, and education. All significant results shown in the figure passed FDR correction using the Benjamini and Hochberg method (q = 0.2)56. The analysis was performed on participants with different diagnosis statuses (Table S6), but only in the participants with LMCI was entorhinal cortex volume in three continuous follow-up years negatively associated with baseline GlycA level. Therefore, the analysis results for the participants with LMCI are shown here. In addition, the LMCI-GlycA interaction was confirmed with a linear mixed model (p = 0.005, Table S4). A similar full factorial linear mixed model was used to show that there were no sex-GlycA interactions controlling DX (p = 0.564). BMI: Body mass index; DX: Diagnosis at baseline; GlycA: Glycoprotein acetyls; ID: Identification; MRI: Magnetic resonance imaging.
Baseline GlycA correlated with baseline brain structural atrophies specifically in female participants diagnosed with LMCI and AD
To identify the cross-sectional relationship between GlycA and regional brain atrophies measured by MRI, we used Spearman’s rank order correlation with sex and diagnostic group stratification (Table S7, Fig. 5). Bilateral total/mean measurements were used to examine the correlation of brain atrophy with GlycA, with measures in the left and right hemispheres showing similar results (Table S8). In addition, rather than using thickness, we mainly used regional volumes to describe brain atrophy due to its stronger sensitivity58, though we did provide both results (Table S8). Comparisons of volumetric measurements among participants of different diagnosis statuses were performed using an ANCOVA model (Table S9).

GlycA cross-sectionally associated with regional brain volumes in females with SMC, LMCI and AD. The full results are listed in Tables S7 and S9 and the present figure illustrates selected representative examples. (a) Significant associations were found between GlycA and volumetric measurements of brain regions, i.e., entorhinal cortex (EntCtx), hippocampus (Hippo), frontal lobe (Frontal), and cerebral cortex grey matter (Ctx). Spearman’s rank order correlation was performed between the residual of GlycA (log2-transformed, adjusted for medications, APOE4, age, BMI, and education) and residuals of whole brain regional volumes (log2-transformed, adjusted for log2-intracranial volume, magnet strength/scan type, APOE4, age, BMI, education); the analysis was performed stratified by sex and diagnosis groups. All significant results shown in the figure passed FDR correction using the Benjamini and Hochberg method (q = 0.2)56. (b) This panel indicates different levels of these brain volumetric measurements, adjusting for the same confounders without stratifications (Table S9). There were no significant sex-diagnosis stages interactions (pinteraction = 0.284 (EntCtx), 0.244 (Hippo), 0.324 (Frontal) and 0.398 (Ctx)) for brain volumetric measurements and therefore this panel is not divided by sex. N: for males: CN: 178, SMC: 40, EMCI: 154, LMCI: 292, and AD: 154; for females: CN: 183, SMC: 55, EMCI: 125, LMCI: 187, and AD: 131. AD: Alzheimer’s disease; ANCOVA: Analysis of covariance; BMI: Body mass index; CN: Cognitively normal; EMCI: Early mild cognitive impairment; GlycA: Glycoprotein acetyls; LMCI: Late mild cognitive impairment; SMC: Significant memory concerns.
In females with AD, GlycA was negatively associated with the frontal lobe volume (p < 0.001, ρ = − 0.343), cerebral cortex grey matter volume (p = 0.002, ρ = − 0.269), global grey matter mean volume (p = 0.002, ρ = − 0.267), sensory motor volume (p = 0.014, ρ = − 0.214), and parietal lobe volume (p = 0.011, ρ = − 0.222) (Table S7). Except for parietal lobe volume, all of these GlycA-brain morphology associations were specific for females with AD, compared to males with AD. The specificity of the association was measured using a linear factorial model with GlycA-sex interaction, stratified by diagnostic group, with results for AD participants including pinteraction < 0.001 for the frontal lobe volume, pinteraction = 0.008 for cerebral cortex grey matter volume, pinteraction = 0.007 for grey matter mean volume, and pinteraction = 0.020 for sensory motor volume (Table S7).
In addition, in females with LMCI, GlycA was negatively associated with hippocampus volume (p = 0.011, ρ = − 0.186) and entorhinal cortex volume (p = 0.026, ρ = − 0.163). In females with SMC, GlycA was negatively associated with cerebral cortex grey matter volume (p = 0.014, ρ = − 0.329), frontal lobe volume (p = 0.012, ρ = − 0.337), and mean grey matter volume (p = 0.011, ρ = − 0.341). No correlation passing FDR correction was observed in the CN or EMCI diagnostic groups, nor in male participants of any diagnosis status. None of the GlycA-sex interactions in non-AD groups and GlycA-diagnosis interactions in both sexes were significant after FDR correction (Table S7).
It is important to note that brain region atrophy was not uniform along AD disease progression (Fig. 5b, Table S9). While areas such as the entorhinal cortex and hippocampus had significantly decreased volumes compared to CN as early as the EMCI stage (p < 0.05), other volumetric measurements—such as those in cerebral cortex grey matter and the frontal lobe—only started to have significantly decreased volumes compared to CN in LMCI. Consistent with this, the correlations between GlycA and hippocampus volume and between GlycA and entorhinal cortex volume were found in an earlier diagnosis stage in females (i.e., LMCI) compared to GlycA correlation with cerebral cortex grey matter volume and frontal lobe volume, which were found in a later stage (i.e., AD). However, such disease state patterns were not inclusive, meaning that not all brain anatomical region changes along disease progression coincided with their correlation with GlycA. An example of this is the early change of medial temporal lobe volume at the EMCI stage without a significant correlation with GlycA, found in participants with any diagnosis status.
Association of plasma GlycA with CSF A/T/N biomarkers
We investigated the relationship between GlycA in plasma and A/T/N biomarkers in CSF using Spearman’s rank correlation as described above. No association was observed between plasma GlycA and A/T/N biomarkers in CSF, including the biomarkers Aβ 42, pTau/Aβ 42, pTau, total Tau, and total Tau/Aβ 42 (Table S10). GlycA in plasma was also not associated with A/T/N biomarkers in CSF in all participants in the continuous future follow-up years (Table S11).
Discussion
The current study describes associations between the level of peripheral inflammation and AD-related structural and cognitive changes in the CNS. Importantly, these peripheral-central correlations manifest primarily in the later stages of the progression toward AD, which emphasizes the diagnosis-status-specific heterogeneity in the relationship between peripheral inflammation, cognition, and brain atrophy. Our findings point to peripheral inflammation as a risk factor for future cognitive decline and brain atrophy in both males and females, which opens the door to the identification of a population at risk as well as potential therapeutic interventions relevant to further inflammatory-related cognitive decline.
Abnormal cognition and memory decline are important markers of AD pathogenesis59, and the structural and functional degeneration of the entorhinal cortex often indicate the early changes in AD development, which implies the particular vulnerability of this region to AD pathogenesis60,61. Accordingly, our results have shown that the peripheral inflammation at baseline correlates with decreasing executive functions, decreasing memory and decreasing entorhinal cortex volume in the follow-up years. The decreased entorhinal cortex volume may provide an inflammatory-related structural basis to explain the observed declines in brain functions that are associated with inflammation.
In addition, such correlations between baseline GlycA and future declines in executive functions, memory and entorhinal cortex volume are restricted to the participants diagnosed with LMCI at baseline. It happened to coincide with the observation that among non-AD participants, the LMCI diagnosis group had the earliest brain function and structural decline in the follow-up years. Mechanistically, peripheral cytokines can directly enter the CNS and/or interact with the blood–brain barrier through multiple pathways13,62. Meanwhile, under pathological conditions, peripheral immune cells can migrate across the blood–brain barrier63,64,65 and modulate microglia function66,67, a key player in CNS inflammation and AD development. As results, peripheral inflammation stimuli can modulate the expression of inflammation-related genes in the brain19 and exacerbate CNS neurodegeneration53. Furthermore, brain inflammation has been suggested to have differential effects along the AD progression20. For example, in the AD rodent model of APPPS1 mice, the lack of neuroinflammation-related gene TREM2 expression (i.e., TREM2 deficiency) lowered amyloid pathologies at the earlier stage of disease development but worsened it at the later stage. Specifically at the later stage, TREM2 deficiency reduced the proliferation of myeloid cells and the gene transcription levels related to inflammation, such as IL-1β and TNF-α20. These studies support the notion that certain inflammation components can be protective at the beginning of the progression toward AD, but detrimental to CNS homeostasis at the later stages20,68. Such a notion may explain the relationship between baseline GlycA and AD-related biomarkers in the follow-up years for participants with LMCI. It may also explain the cross-sectional correlation between baseline GlycA and baseline AD-related biomarkers in the female participants with LMCI and AD. This is due to the consideration that the depletion of estrogens after menopause may lead to additional vulnerability to oxidative stress in women69,70, and thus that the relationship between oxidative stress and neurodegeneration may differ in older females compared to males71.
Sex differences were observed in the cross-sectional correlation between peripheral inflammation and brain structures. This was different from the relationship between baseline peripheral inflammation and brain structures in the follow-up years, in which no sex-dependent effect was observed. While causation cannot be inferred from our observations, such a difference in peripheral-central connections between cross-sectional and longitudinal analysis may be due to the difference between acute and chronic inflammation, both of which can be captured by GlycA28,30. Sexual dimorphism in neurological disorders has been associated with differential microglia sub-populations, including disease-associated microglia and activated response microglia72,73,74, as well as the microglial involvement in AD-related pathologies such as neuroinflammation75, Aβ accumulation72 and tau pathologies75,76,77. In a rodent study, the identical acute peripheral inflammatory stimuli caused a more substantial pro-inflammatory response in the aged brains of females compared to males18. However, it is not clear whether the aged brains of males and females respond to the chronic peripheral stimuli in a similar manner19,78, considering that males and females overall manifest different fluctuations in peripheral inflammation over a lifetime21. Further longitudinal studies on aged models or populations with chronic inflammation are therefore warranted to clarify the potential sex effect of inflammation on AD development.
Additionally, while we observed the correlation between GlycA and brain structure, and between GlycA and cognitive changes, there was no correlation between GlycA and CSF biomarkers for Aβ and tau pathologies. Similar observations on the lack of correlations between peripheral inflammatory indicators and AD pathologies were reported before, which can be accounted for by factors such as the temporal dynamics of these inflammatory cytokines and AD progression (reviewed in7). Moreover, additional regulation of peripheral inflammation and AD pathologies must occur in the CNS, which is supported by the evidence found in animal research: for example, the level of amyloid deposition in the brain is negatively associated with CSF Aβ level79. However, in an amyloid precursor protein knock-in mouse model, peripheral inflammation increased Aβ deposition in the brain80, but interrupted Aβ clearance from CSF to blood80. Such an interaction could explain the lack of changes in CSF Aβ under the peripheral inflammation stimuli80, consistent with our current observation. In addition, peripheral inflammation affects the transmission of Tau proteins among brain regions81, but how these processes collectively regulate the level of Tau pathology in CSF is not clear. Overall, the mechanisms that regulate the complex crosstalk between peripheral inflammation and different AD pathologies warrant further investigation.
It should be pointed out that our analyses were limited to observational findings instead of causative ones. Replications on independent cohorts are required for confirmation. In addition, the comorbidity of AD pathologies and the diagnosis status of inflammatory diseases such as diabetes and metabolic syndrome were not discussed here, considering that GlycA captured the systemic inflammation level28,30 regardless of the specific diagnosis statuses of those inflammatory diseases. Nevertheless, the comorbidity of diabetes and AD-related pathologies has been reported in a previous study performed using the ADNI data82. Furthermore, some of the stratifications resulted in a relatively small sample size per group (e.g. SMC), but these stratifications remain to be used considering the different risks of dementia in different populations83,84. Further analyses are warranted to investigate the extent of this population heterogeneity and the possibility of combining subgroups to achieve higher statistical power. Meanwhile, other factors that can contribute to population heterogeneity, such as race and ethnicity, cannot be investigated well with the current dataset but can be addressed through future opportunities using cohorts involving diverse populations (e.g. ADNI4, which is currently under recruitment). Additionally, other composite scores to measure different cognition domains, such as language composite (LAN) and visuospatial composite scores (VS) are available in ADNI85, but the systemic inspection of their relationship with GlycA was outside of this paper’s scope.
Conclusions
Our study describes connections between peripheral inflammation and cognitive and structural AD-related changes in the brain. Moreover, it highlights the implication of systemic inflammation at earlier time points on the later stage toward AD and highlights the potentially higher sensitivity or susceptibility of females, compared to males, to systemic inflammation-related brain changes. These findings contribute to our understanding of disease heterogeneity and lay the groundwork for more personalized approaches toward disease treatment.
Data availability
The datasets supporting the conclusions of this article are available in the ADNI database (www.adni-info.org), managed through the Laboratory of Neuro Imaging Image & Data Archive (http://adni.loni.usc.edu/). The data is available for authorized investigators.
Abbreviations
- Aβ:
-
Amyloid beta peptide
- AD:
-
Alzheimer’s disease
- ADNI:
-
Alzheimer’s Disease Neuroimaging Initiative
- ANCOVA:
-
Analysis of covariance
- BMI:
-
Body mass index
- CDR:
-
Clinical Dementia Rating scale
- CN:
-
Cognitively normal
- CNS:
-
Central nervous system
- CRP:
-
C-reactive protein
- CSF:
-
Cerebral spinal fluid
- Ctx:
-
Cerebral cortex grey matter
- DX:
-
Diagnosis at baseline
- EF:
-
Executive functioning composite score
- EMCI:
-
Early mild cognitive impairment
- EntCtx:
-
Entorhinal cortex
- Frontal:
-
Frontal lobe
- GlycA:
-
Glycoprotein acetyls
- HSD:
-
Honestly significant difference
- Hippo:
-
Hippocampus
- LMCI:
-
Late mild cognitive impairment
- MEM:
-
Memory composite score
- MMSE:
-
Mini-Mental State Exam
- MRI:
-
Magnetic resonance imaging
- PET:
-
Positron emission tomography imaging
- SMC:
-
Significant memory concern
References
Kinney, J.W., Bemiller, S.M., Murtishaw, A.S., Leisgang, A.M., Salazar, A.M., Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. 2018 (2352–8737 (Electronic)).
Arvanitakis, Z., Wilson, R. S., Bienias, J. L., Evans, D. A. & Bennett, D. A. Diabetes mellitus and risk of Alzheimer disease and decline in cognitive function. Arch Neurol. 61(5), 661–666 (2004).
Kalaria, R. N., Akinyemi, R. & Ihara, M. Does vascular pathology contribute to Alzheimer changes?. J. Neurol. Sci. 322(1), 141–147 (2012).
Hofman, A. et al. Atherosclerosis, apolipoprotein E, and prevalence of dementia and Alzheimer’s disease in the Rotterdam Study. Lancet. 349(9046), 151–154 (1997).
Skoog, I. et al. 15-year longitudinal study of blood pressure and dementia. Lancet. 347(9009), 1141–1145 (1996).
Razay, G., Vreugdenhil, A. & Wilcock, G. The metabolic syndrome and Alzheimer Disease. Arch. Neurol. 64(1), 93–96 (2007).
Brosseron, F., Krauthausen, M., Kummer, M. & Heneka, M. T. Body fluid cytokine levels in mild cognitive impairment and Alzheimer’s disease: A comparative overview. Mol. Neurobiol. 50(2), 534–544 (2014).
Swardfager W, Lanctôt K Fau-Rothenburg L, Rothenburg L Fau-Wong A, Wong A Fau-Cappell J, Cappell J Fau-Herrmann N, Herrmann N. A meta-analysis of cytokines in Alzheimer’s disease. (1873–2402 (Electronic)).
Borkowski, K. et al. Association of plasma and CSF cytochrome P450, soluble epoxide hydrolase, and ethanolamide metabolism with Alzheimer’s disease. Alzheimer’s Res. Therapy. 13(1), 149 (2021).
Borkowski, K. et al. Serum metabolomic biomarkers of perceptual speed in cognitively normal and mildly impaired subjects with fasting state stratification. Sci. Rep. 11(1), 18964 (2021).
Tucsek, Z. et al. Obesity in aging exacerbates blood-brain barrier disruption, neuroinflammation, and oxidative stress in the mouse hippocampus: Effects on expression of genes involved in beta-amyloid generation and Alzheimer’s disease. J. Gerontol. Series A. 69(10), 1212–1226 (2013).
Thériault, P., ElAli, A. & Rivest, S. High fat diet exacerbates Alzheimer’s disease-related pathology in APPswe/PS1 mice. Oncotarget. 7(42), 67808–67827 (2016).
Xie, J., Van Hoecke, L. & Vandenbroucke, R. E. The impact of systemic inflammation on Alzheimer’s disease pathology. Front. Immunol. 12, 5731 (2022).
Kahn, M. S. et al. Prolonged elevation in hippocampal Aβ and cognitive deficits following repeated endotoxin exposure in the mouse. Behav. Brain Res. 229(1), 176–184 (2012).
Lee, J. W. et al. Neuro-inflammation induced by lipopolysaccharide causes cognitive impairment through enhancement of beta-amyloid generation. J. Neuroinflam. 5, 1–14 (2008).
Tejera, D. et al. Systemic inflammation impairs microglial Aβ clearance through NLRP 3 inflammasome. EMBO J. 38(17), e101064 (2019).
Thion, M. S. et al. Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. 172(3), 500–16.e16 (2018).
Murtaj, V. et al. Age and sex influence the neuro-inflammatory response to a peripheral acute LPS challenge. Front. Aging Neurosci. 11, 299 (2019).
Wendeln, A.-C. et al. Innate immune memory in the brain shapes neurological disease hallmarks. Nature. 556(7701), 332–338 (2018).
Jay, T. R. et al. Disease progression-dependent effects of TREM2 deficiency in a mouse model of Alzheimer’s disease. J. Neurosci. 37(3), 637–647 (2017).
Klein, S. L. & Flanagan, K. L. Sex differences in immune responses. Nat. Rev. Immunol. 16(10), 626–638 (2016).
Clarke, J. R. et al. Alzheimer-associated Aβ oligomers impact the central nervous system to induce peripheral metabolic deregulation. EMBO Mol. Med. 7(2), 190–210 (2015).
Batra, R. et al. The landscape of metabolic brain alterations in Alzheimer’s disease. Alzheimer’s Dementia. 19(3), 980–998 (2023).
Arnold MA-O, Nho K, Kueider-Paisley A, Massaro T, Huynh K, Brauner B, et al. Sex and APOE ε4 genotype modify the Alzheimer’s disease serum metabolome. (2041–1723 (Electronic)).
Llauradó, G. et al. Measurement of serum N-glycans in the assessment of early vascular aging (arterial stiffness) in adults with type 1 diabetes. Diabetes Care. 45(10), 2430–2438 (2022).
Chiesa, S. T. et al. Glycoprotein acetyls: A novel inflammatory biomarker of early cardiovascular risk in the young. J. Am. Heart Assoc. 11(4), e024380 (2022).
Kettunen, J. et al. Biomarker glycoprotein acetyls is associated with the risk of a wide spectrum of incident diseases and stratifies mortality risk in angiography patients. Circulat. Genom. Precision Med. 11(11), e002234 (2018).
Mehta, N. N., Dey, A. K., Maddineni, R., Kraus, W. E. & Huffman, K. M. GlycA measured by NMR spectroscopy is associated with disease activity and cardiovascular disease risk in chronic inflammatory diseases. Am. J. Prev. Cardiol. 4, 100120 (2020).
Deelen, J. et al. A metabolic profile of all-cause mortality risk identified in an observational study of 44,168 individuals. Nat. Commun. 10(1), 3346 (2019).
Otvos, J. D. et al. GlycA: A composite nuclear magnetic resonance biomarker of systemic inflammation. Clin. Chem. 61(5), 714–723 (2015).
Ormseth, M. J. et al. Utility of a novel inflammatory marker, GlycA, for assessment of rheumatoid arthritis disease activity and coronary atherosclerosis. Arthritis Res. Therapy. 17(1), 117 (2015).
Bartlett, D. B. et al. A novel inflammatory biomarker, GlycA, associates with disease activity in rheumatoid arthritis and cardio-metabolic risk in BMI-matched controls. Arthritis Res. Therapy. 18(1), 86 (2016).
Dierckx T, Chiche L, Daniel L, Lauwerys B, Weyenbergh JV, Jourde-Chiche N. Serum GlycA level is elevated in active systemic lupus erythematosus and correlates to disease activity and lupus nephritis severity. J. Clin. Med. 9(4) (2020).
Chung, C. P. et al. GlycA, a novel marker of inflammation, is elevated in systemic lupus erythematosus. Lupus. 25(3), 296–300 (2015).
Joshi, A. A. et al. GlycA Is a novel biomarker of inflammation and subclinical cardiovascular disease in psoriasis. Circ. Res. 119(11), 1242–1253 (2016).
Gruppen, E. G., Connelly, M. A., Otvos, J. D., Bakker, S. J. & Dullaart, R. P. A novel protein glycan biomarker and LCAT activity in metabolic syndrome. Eur. J. Clin. Invest. 45(8), 850–859 (2015).
Gruppen, E. G., Connelly, M. A. & Dullaart, R. P. Higher circulating GlycA, a pro-inflammatory glycoprotein biomarker, relates to lipoprotein-associated phospholipase A2 mass in nondiabetic subjects but not in diabetic or metabolic syndrome subjects. J. Clin. Lipidol. 10(3), 512–518 (2016).
Akinkuolie, A. O., Pradhan, A. D., Buring, J. E., Ridker, P. M. & Mora, S. Novel protein glycan side-chain biomarker and risk of incident type 2 diabetes mellitus. Arteriosclerosis Thrombosis Vasc. Biol. 35(6), 1544–1550 (2015).
Dierckx, T., Verstockt, B., Vermeire, S. & van Weyenbergh, J. GlycA, a nuclear magnetic resonance spectroscopy measure for protein glycosylation, is a viable biomarker for disease activity in IBD. J. Crohn’s Colitis. 13(3), 389–394 (2019).
Dowd JB, Zajacova A Fau - Aiello AE, Aiello AE. Predictors of inflammation in U.S. children aged 3–16 years. (1873–2607 (Electronic)).
Calice-Silva V, Suemoto CK, Brunoni AR, Bensenor IM, Lotufo PA. Association between GlycA and cognitive function: Cross-sectional results from the ELSA—Brasil study. Alzheimer Disease Associated Disorders. 35(2) (2021).
Tynkkynen J, Chouraki V, van der Lee SJ, Hernesniemi J, Yang Q, Li S, et al. Association of branched-chain amino acids and other circulating metabolites with risk of incident dementia and Alzheimer’s disease: A prospective study in eight cohorts. (1552–5279 (Electronic)).
Cohen-Manheim, I. et al. Increase in the inflammatory marker GlycA over 13 years in young adults is associated with poorer cognitive function in midlife. PLoS One. 10(9), e0138036 (2015).
Soininen, P., Kangas, A. J., Würtz, P., Suna, T. & Ala-Korpela, M. Quantitative serum nuclear magnetic resonance metabolomics in cardiovascular epidemiology and genetics. Circ. Cardiovasc. Genet. 8(1), 192–206 (2015).
Soininen, P. et al. High-throughput serum NMR metabonomics for cost-effective holistic studies on systemic metabolism. Analyst. 134(9), 1781–1785 (2009).
Würtz, P. et al. Quantitative serum nuclear magnetic resonance metabolomics in large-scale epidemiology: A primer on -omic technologies. Am. J. Epidemiol. 186(9), 1084–1096 (2017).
Toledo, J. B. et al. Metabolic network failures in Alzheimer’s disease: A biochemical road map. Alzheimers Dement. 13(9), 965–984 (2017).
Hu, W. T. et al. Plasma multianalyte profiling in mild cognitive impairment and Alzheimer disease. Neurology. 79(9), 897–905 (2012).
Hu, W. T. et al. Novel CSF biomarkers for Alzheimer’s disease and mild cognitive impairment. Acta Neuropathol. 119(6), 669–678 (2010).
Nho K, Kueider-Paisley A, MahmoudianDehkordi S, Arnold M, Risacher SL, Louie G, et al. Altered bile acid profile in mild cognitive impairment and Alzheimer’s disease: Relationship to neuroimaging and CSF biomarkers. (1552–5279 (Electronic)).
Gibbons, L. E. et al. A composite score for executive functioning, validated in Alzheimer’s Disease Neuroimaging Initiative (ADNI) participants with baseline mild cognitive impairment. Brain Imaging Behav. 6(4), 517–527 (2012).
Crane, P. K. et al. Development and assessment of a composite score for memory in the Alzheimer’s Disease Neuroimaging Initiative (ADNI). Brain Imaging Behav. 6(4), 502–516 (2012).
Bittner, T. et al. Technical performance of a novel, fully automated electrochemiluminescence immunoassay for the quantitation of β-amyloid (1–42) in human cerebrospinal fluid. Alzheimer’s Dementia. 12(5), 517–526 (2016).
Shaw LM, Fields L, Korecka M, Waligórska T, Trojanowski JQ, Allegranza D, et al. P2–143: Method Comparison of AB(1–42) Measured in Human Cerebrospinal Fluid Samples by Liquid Chromatography-Tandem Mass Spectrometry, the Inno-Bia ALZBIO3 Assay, and the Elecsys® B-Amyloid(1–42) Assay. Alzheimer’s & Dementia. 2016;12(7S_Part_13):P668-P.
Mofrad SA, Lundervold AJ, Vik A, Lundervold AS. Cognitive and MRI trajectories for prediction of Alzheimer’s disease. (2045–2322 (Electronic)).
Benjamini, Y. & Yekutieli, D. Quantitative trait loci analysis using the false discovery rate. Genetics. 171(2), 783–790 (2005).
Borkowski, K. et al. Integration of plasma and CSF metabolomics with CSF proteomic reveals novel associations between lipid mediators and central nervous system vascular and energy metabolism. Sci. Rep. 13(1), 13752 (2023).
Iannopollo, E. & Garcia, K. Enhanced detection of cortical atrophy in Alzheimer’s disease using structural MRI with anatomically constrained longitudinal registration. Human Brain Mapping. 42(11), 3576–3592 (2021).
Apostolova LG. Alzheimer Disease. (1538–6899 (Electronic)).
Olajide OA-O, Suvanto ME, Chapman CA-O. Molecular mechanisms of neurodegeneration in the entorhinal cortex that underlie its selective vulnerability during the pathogenesis of Alzheimer’s disease. LID-https://doi.org/10.1242/bio.056796 LID-bio056796. (2046–6390 (Electronic)).
Igarashi KM. Entorhinal cortex dysfunction in Alzheimer’s disease. (1878–108X (Electronic)).
Sun, Y., Koyama, Y. & Shimada, S. Inflammation from peripheral organs to the brain: How does systemic inflammation cause neuroinflammation?. Front. Aging Neurosci. 14, 903455 (2022).
Fiala, M. et al. Cyclooxygenase-2-positive macrophages infiltrate the Alzheimer’s disease brain and damage the blood–brain barrier. Eur. J. Clin. Investig. 32(5), 360–371 (2002).
Togo, T. et al. Occurrence of T cells in the brain of Alzheimer’s disease and other neurological diseases. J. Neuroimmunol. 124(1), 83–92 (2002).
Town, T., Tan, J., Flavell, R. A. & Mullan, M. T-cells in alzheimer’s disease. NeuroMol. Med. 7(3), 255–264 (2005).
Dionisio-Santos, D. A., Olschowka, J. A. & O’Banion, M. K. Exploiting microglial and peripheral immune cell crosstalk to treat Alzheimer’s disease. J. Neuroinflam. 16(1), 1–13 (2019).
Fisher, Y., Nemirovsky, A., Baron, R. & Monsonego, A. T cells specifically targeted to amyloid plaques enhance plaque clearance in a mouse model of Alzheimer’s disease. PloS One. 5(5), e10830 (2010).
Blasko, I. & Grubeck-Loebenstein, B. Role of the immune system in the pathogenesis, prevention and treatment of Alzheimer’s disease. Drugs Aging. 20, 101–113 (2003).
Mendoza C, Zamarripa CAJ. Menopause induces oxidative stress. Morales-González JA Oxidative stress and chronic degenerative diseases: A role for antioxidants London: Intech Open. 2013:289–316.
Kumar S, Lata K Fau - Mukhopadhyay S, Mukhopadhyay S Fau - Mukherjee TK, Mukherjee TK. Role of estrogen receptors in pro-oxidative and anti-oxidative actions of estrogens: a perspective. (0006–3002 (Print)).
Tenkorang MA, Snyder B, Cunningham RL. Sex-related differences in oxidative stress and neurodegeneration. (1878–5867 (Electronic)).
Guillot-Sestier, M.-V. et al. Microglial metabolism is a pivotal factor in sexual dimorphism in Alzheimer’s disease. Commun. Biol. 4(1), 711 (2021).
Yanguas-Casás, N. Physiological sex differences in microglia and their relevance in neurological disorders. Neuroimmunol. Neuroinflam. 7(1), 13–22 (2020).
Mangold, C. A. et al. Sexually divergent induction of microglial-associated neuroinflammation with hippocampal aging. J. Neuroinflam. 14(1), 141 (2017).
Kodama, L. et al. Microglial microRNAs mediate sex-specific responses to tau pathology. Nat. Neurosci. 23(2), 167–171 (2020).
Ising, C. et al. NLRP3 inflammasome activation drives tau pathology. Nature. 575(7784), 669–673 (2019).
Rangaraju, S. et al. Identification and therapeutic modulation of a pro-inflammatory subset of disease-associated-microglia in Alzheimer’s disease. Mol. Neurodegenerat. 13(1), 24 (2018).
Divangahi, M. et al. Trained immunity, tolerance, priming and differentiation: Distinct immunological processes. Nat. Immunol. 22(1), 2–6 (2021).
Fagan, A. M. et al. Inverse relation between in vivo amyloid imaging load and cerebrospinal fluid Aβ42 in humans. Ann. Neurol. 59(3), 512–519 (2006).
Xie, J. et al. Low-grade peripheral inflammation affects brain pathology in the AppNL-G-Fmouse model of Alzheimer’s disease. Acta Neuropathol. Commun. 9(1), 163 (2021).
Liu Y, Zhang S, Li X, Liu E, Wang X, Zhou Q, et al. Peripheral inflammation promotes brain tau transmission via disrupting blood-brain barrier. Biosci. Rep. 40(2) (2020).
Li, W., Risacher, S. L., Huang, E. & Saykin, A. J. Type 2 diabetes mellitus is associated with brain atrophy and hypometabolism in the ADNI cohort. Neurology. 87(6), 595–600 (2016).
Donovan, N. J. et al. Subjective cognitive concerns and neuropsychiatric predictors of progression to the early clinical stages of Alzheimer disease. Am. J. Geriatr. Psychiatry. 22(12), 1642–1651 (2014).
Jessen, F. et al. Prediction of dementia by subjective memory impairment: Effects of severity and temporal association with cognitive impairment. Arch. General Psychiatry. 67(4), 414–422 (2010).
Choi, S.-E. et al. Development and validation of language and visuospatial composite scores in ADNI. Alzheimer’s Dementia Transl. Res. Clin. Interventions. 6(1), e12072 (2020).
Acknowledgements
The editorial services of Mr. Jon Kilner, MS, MA (Pittsburgh) and the assistance of Ms. Colette Blach, Ms. Lisa Howerton (Duke) and Dr. Alexandra Kueider-Paisley (Duke) are acknowledged here. The list of consortium members of the Alzheimer’s Disease Metabolomics Consortium can be found in the Supplementary Information file. Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in the analysis or the writing of this report. A complete listing of ADNI investigators can be found at: http://adni.loni.usc.edu/wp-content/uploads/how_to_apply/ADNI_Acknowledgement_List.pdf.
Funding
Dr. Kamil Borkowski is funded by the National Institutes of Health Grant P30AG072972. Dr. Kwangsik Nho is funded by Grants U01 AG072177, R01 LM012535, and U19AG074879. The Alzheimer’s Disease Metabolomics Consortium (ADMC) is funded wholly or in part by the following National Institute on Aging grants and supplements, which are components of the Accelerating Medicines Partnership for AD (AMP-AD) and/or Molecular Mechanisms of the Vascular Etiology of AD (M2OVE-AD): NIA R01AG046171, RF1AG051550, RF1AG057452, R01AG059093, RF1AG058942, U01AG061359, U19AG063744, 3U19AG063744-04S1, 1R01AG081322, and FNIH: #DAOU16AMPA awarded to Dr. Rima Kaddurah-Daouk at Duke University in partnership with a large number of academic institutions. Dr. John W. Newman was supported by USDA Project 2032-51530-025-00D. The USDA is an equal opportunity provider and employer. Dr. Peter J. Meikle is supported by an Investigator Grant (2009965) from the National Health and Medical Research Council of Australia. This work was supported by the Victorian Government’s Operational Infrastructure Support Program. Data collection and sharing for this project was supported by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) with National Institutes of Health Grant U01 AG024904 and DOD ADNI with Department of Defense award number W81XWH-12-2-0012. ADNI is funded by the following entities: the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: AbbVie, Alzheimer’s Association; Alzheimer’s Drug Discovery Foundation; Araclon Biotech; BioClinica, Inc.; Biogen; Bristol-Myers Squibb Company; CereSpir, Inc.; Cogstate; Eisai Inc.; Elan Pharmaceuticals, Inc.; Eli Lilly and Company; EuroImmun; F. Hoffmann-La Roche Ltd. and its affiliated company Genentech, Inc.; Fujirebio; GE Healthcare; IXICO Ltd.; Janssen Alzheimer Immunotherapy Research & Development, LLC.; Johnson & Johnson Pharmaceutical Research & Development LLC.; Lumosity; Lundbeck; Merck & Co., Inc.; Meso Scale Diagnostics, LLC.; NeuroRx Research; Neurotrack Technologies; Novartis Pharmaceuticals Corporation; Pfizer Inc.; Piramal Imaging; Servier; Takeda Pharmaceutical Company; and Transition Therapeutics. ADNI clinical sites in Canada were supported by the Canadian Institutes of Health Research. Private sector contributions are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Therapeutic Research Institute at the University of Southern California. ADNI data are disseminated by the Laboratory for Neuro Imaging at the University of Southern California.
Author information
Authors and Affiliations
Consortia
Contributions
N.L. performed the analyses, interpreted the results and drafted and revised the manuscript. K.N., J.W.N., M.A., K.H., P.J.M., K.B. and R.K-D. substantively revised the manuscript. K.B. and R.K-D. also conceptualized the study and interpreted the results. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
Dr. Rima Kaddurah-Daouk and Dr. Matthias Arnold participated in the invention of several patents on applying metabolomics to diagnose and treat CNS diseases. Dr. Kaddurah-Daouk holds equity in Metabolon Inc., Chymia LLC and PsyProtix, which were not involved in the current study. Dr. Arnold holds equity in Chymia LLC and IP in PsyProtix. All other authors declare that they have no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liang, N., Nho, K., Newman, J.W. et al. Peripheral inflammation is associated with brain atrophy and cognitive decline linked to mild cognitive impairment and Alzheimer’s disease. Sci Rep 14, 17423 (2024). https://doi.org/10.1038/s41598-024-67177-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-67177-5
- Springer Nature Limited