
Abstract
A series of benzoquinoline-employing heterocycles was synthesized by treating 3-chlorobenzo[f]quinoline-2-carbaldehyde with N-phenyl-3-methylpyrazolone, 4-aminoacetophenone, 1,2-diaminoethane, and 2-cyanoethanohydrazide. Also, pyridine, chromene, α,β-unsaturated nitrile, thiosemicarbazone, and 1,2-bis-aryl hydrazine derivatives were prepared from the cyanoethanohydrazone obtained. The DFT calculations and experiment outcomes were consistent. In vitro screening of their antiproliferative efficacy was examined against HCT116 and MCF7 cancer cell lines. The pyrazolone 2 and cyanoethanohydrazone 5 derivatives exhibited the most potency, which was demonstrated by their molecular docking towards the CDK-5 enzyme. The binding energies of compounds 2 and 5 were − 6.6320 kcal/mol (with RMSD of 0.9477 Å) and − 6.5696 kcal/mol (with RMSD of 1.4889 Å), respectively, which were near to that of co-crystallized ligand (EFP). This implies a notably strong binding affinity towards the CDK-5 enzyme. Thus, pyrazolone derivative 2 would be considered a promising candidate for further optimization to develop new chemotherapeutic agents. In addition, the ADME (absorption, distribution, metabolism, and excretion) analyses displayed its desirable drug-likeness and oral bioavailability properties.
Similar content being viewed by others

Introduction
Designing and synthesizing scaffolds with biological characteristics is one of the primary goals of organic and medicinal chemistry. Heterocycles, including nitrogen, are targeted compounds in synthetic and medicinal chemistry because they serve as a critical scaffold in a range of physiologically active chemicals1,2,3,4,5,6,7,8,9,10. Benzoquinoline scaffolds are among the more abundant aza-arenes present in the environment11,12,13. Thus, benzoquinoline compounds exhibit a diverse set of biological properties and act as templates for the synthesis of numerous medications14,15,16,17. Benzoquinolines are known mutagens in Salmonella typhimurium18, and are also less toxic than quinolines19.
Furthermore, the biological significances of pyrazoles20,21,22, imidazolines23,24, and chromenes25,26,27 are well established. As a result, and in continuation to our work28,29,30,31,32,33, it has been of remarkable interest to design and synthesize various benzo[f]quinoline-carrying heterocycles to evaluate their antiproliferative properties. Computational chemical, molecular docking, and in silico studies were performed to support the findings.
Rationale and design
One of the most important targets for the design of new anticancer drugs is DNA, through which drugs can change their configuration preventing replication and transcription leading to cancer cell growth inhibition. Intercalation is the favored binding mode of almost-flat polyaromatic ligands of larger surface area and applicable steric properties. The nature and size of intercalating chromophores are important parameters that govern the binding modes. Noteworthy, many anticancer drugs in clinical utility (doxorubicin, anthracyclines, mitoxantrone) interact with DNA through intercalation34,35.
In a statement, most intercalating agents may be either positively charged or include basic groups protonating under physiological conditions36. Recently, much consideration has been promoted to design and synthesize novel and efficient DNA-targeted anticancer agents encompassing quinoline scaffold35,37.
Thus, this work reports the molecular design and synthesis of a series of new benzo[f]quinolines (planar polyaromatic ligands) encompassing either side chain, phenyl, or heterocyclic scaffold at position-3 of quinoline core like some known DNA-intercalating agents. The rationale for the design of these substrates can be shown in Fig. 1. For example, amonafide-bearing aminoalkyl side chain showed significant cytotoxic potency via intercalation and topoisomerase II inhibition38. R16 exhibits potent anticancer properties and anti-multidrug resistant (MDR) capability39. Compound 2, a benzo[f]quinoline with a pyrazolone scaffold, displayed the best intercalative properties among synthesized quinolines.

Some DNA-intercalating agents and designed substrates.
Results and discussion
Synthesis
In this work, 3-chlorobenzo[f]quinoline-2-carbaldehyde (1)40 was utilized for the creation of various heterocycles based on the benzo[f]quinoline framework. Thus, treating a basic ethanolic solution of aldehyde 1 with 5-methyl-2-phenyl-2,4-dihydropyrazol-3-one furnished the condensation product, pyrazolone 2 (Scheme 1). IR of 2 disclosed pyrazolone’s carbonyl absorption band. Its 1H NMR indicated a singlet signal for methyl protons. Besides other peaks, its mass spectrum displayed isotopic peak (M + 2) at m/z 399.68 (7%) and molecular ion peak (M+.) at m/z 397.08 (21%).

Condensation of 3-chlorobenzoquinoline-2-carbaldehyde 1 with some nucleophiles.
On the other side, Schiff base 3, imidazoline 4, and cyanoethanohydrazone 5 derivatives were produced by the reactions of aldehyde 1 reacted with 4-acetylaniline, 1,2-diaminoethane, and 2-cyanoethanohydrazide, respectively (Scheme 1). Theres was no aldehydic carbonyl absorption in IR of 3–5. A singlet signal for methyl protons of acetyl group occurred in 1H NMR of 3. Also, two triplet signals for CH2-CH2 protons of imidazoline moiety appeared in 1H NMR of 4. A possible pathway for the formation of imidazoline 4 was first condensation between the aldehydic carbonyl group of 1 with one NH2 group of 1,2-diaminoethane to remove water molecule pursued by 5-endo-trig cyclization by nucleophilic addition of second NH2 group on CH=N moiety, then dehydrogenation.
Absorption bands for NH, C≡N, C=O, and C=N moieties were observed at ν 3255, 2265, 1700, and 1602 cm−1, respectively, in IR of hydrazone 5. Further, its 1H NMR implied its existence as a mixture of anti- and syn-isomers in a ratio of 80.3–19.7%, respectively. As a result, for anti-isomer, it offered a singlet signal of CH2 protons at 4.37 ppm, a singlet signal for methine proton (CH=N) at δ 8.46 ppm; while for syn-isomer, the former signal was at δ 3.89 ppm, and the later signal was at δ 8.64 ppm. Moreover, its mass chart proved its molecular ion peak at m/z = 322.35 (52%), in addition to the isotopic (M + 2) peak at m/z = 324.50 (20%) (cf. Experimental).
Noteworthy, the benzoquinoline-cyanoethanohydrazone 5 was feasible to be a highly reactive substrate, as the active methylene group can contribute to condensation reactions, in addition to carbonyl and cyano moieties are appropriately situated to build diverse heterocyclic skeletons. Certainly, cyanoethanohydrazone 5 was utilized as synthons for various interesting benzo[f]quinolines via reaction with certain carbon electrophiles and nitrogen nucleophiles (cf. Schemes 2, 3). First, the pyridine candidate 7 was furnished upon conducting 5 with 2-((1,3-diphenylpyrazol-4-yl)methylene)malononitrile (6) under basic medium (Scheme 2). In IR of 7, absorption bands appeared for NH, CN, and C=O. Further in its 1H NMR, singlet signals for imino proton (NH) in the downfield region and C3-H of pyridine moiety occurred.

Interaction of cyanoacetohydrazone 5 with some carbon electrophiles.

Reaction of cyanoethanohydrazone 5 with thiosemicarbazide and hydrazine.
Knoevenagel reaction of 5 with 4-methoxybenzaldehyde in dioxane and piperidine at an ambient temperature provided α,β-unsaturated nitrile 8. In its IR, a lower absorption band for conjugated nitrile was indicated. Also, its 1H NMR lacked the methylene singlet, and offered its being as a mixture of E- and Z-isomers in a ratio of 80:20%, respectively.
Besides under similar conditions, cyclo-condensation of 5 with 2-hydroxybenzaldehyde constructed 2-iminochromene 9. Its IR was devoid of nitrile absorption but offered absorption bands for NH and C=O groups. Further, its 1H NMR provided two NH singlet signals. Furthermore, the molecular ion peak at m/z 426.78 (100%) and isotopic peak (M + 2) at m/z 428.94 (35%) occurred in its mass chart (cf. Experimental).
The interaction of cyanoethanohydrazone 5 with thiosemicarbazide and hydrazine was inspected (Scheme 3). Consequently, treating 5 with thiosemicarbazide in 1,4-dioxane did not produce thiadiazole 10 but formed thiosemicarbazone 1113. Likewise, hydrazinolysis of 5 with hydrazine in ethyl alcohol provided 1,2-bis-hydrazine candidate 1213 instead of pyrazole product 13.
Density functional theory (DFT) simulation
DFT simulation was applied to optimize the molecular configurations of the produced substances, identify both electrophilic and nucleophilic centers, and infer the reactions path2,26. The molecular structures of produced substances are superior to be designed in a stable configuration. Hence, benzo[f]quinoline derivatives' geometry was progressively optimized, and their energy was incessantly diminished until the molecule's energy fluctuations were minimized. The electrophilic-attacking centers are depicted by the HOMO areas of maximal electron density, whereas the nucleophilic-attacking sites are implied by the LUMO regions. The high EHOMO values are prospective to signify a molecule's strong propensity to offer electrons. The optimized, HOMO, and LUMO configurations of substrates 2–9 were portrayed applying ChemBio3D Ultra 14.0 and depicted in Supplementary Fig. 1.
To establish how 3-chlorobenzo[f]quinoline-2-carbaldehyde (1) reacted with certain reagents to produce substrates 2–9, a DFT simulation has been operated to calculate quantum chemical properties including global hardness, softness, chemical potential, global electrophilicity index, nucleophilicity index, ionization potential, electron affinity, and electronegativity (cf. Table 1). Analytical and spectral data proved the assigned structures. Due to the low energy needed to remove an electron from the last occupied orbital, compounds with low energy gap values (ΔE = ELUMO − EHOMO) will display potent inhibition efficiency26,32,41. Quantum chemical parameters calculations were in decent agreement with the antiproliferative efficacy (Table 1)26. The findings pointed out that the energy gap values (ΔE) track the order: 3 < 2 < doxorubicin < 9 < 8 < 7 < 5 < 4. Substrates having low ΔE values are commonly implied to as soft substances, which are more reactive toward radical surface interactions; being efficient in offering electrons clearly to the hole surface.
Chemical softness values decrease in the order of 3, 2, doxorubicin, 9, 8, 7, 5, and 4, respectively, while the hardness values rise in the same order. The scavenging ability toward positive hole, tumor, radical, and oxygen removable was not only dependent on EHOMO values but also, the electron distributions, number of heteroatoms, surface area, and lipophilicity should be measured41,42,43,44. The dipole moment (Debye) and softness (ϭ, eV−1) for most potent substances holding hydrophobic groups were agreed to an outstanding correlation between oxidation inhibition efficiencies. Correspondingly, substances of higher binding energy are of higher potency due to effective interaction between these substances and the receptors’ active sites.
Otherwise, for reactions of carbon-centered electrophiles like 2-((1,3-diphenylpyrazol-4-yl)methylene)malononitrile, 4-methoxybenzaldehyde, and 2-hydroxybenzaldehyde, the LUMO of electrophilic centers (energies of − 4.436 eV, − 4.586 eV, and − 4.774 eV, respectively) reacted with HOMO (energy of − 7.631 eV) of the nucleophilic site of substrate 5 to form new substances 7–9 (cf. Fig. 2). Also, for reactions of thiosemicarbazide and hydrazine, HOMO of nucleophilic centers (energies of − 7.269 eV and − 11.069 eV, respectively) reacted with LUMO of the electrophilic site of substrate 5 (energy of − 3.813 eV) to form substances 11 and 12 (cf. Fig. 2).

The ELUMO of electrophiles and EHOMO of nucleophiles toward EHOMO and ELUMO of cyanoethanohydrazone substrate 5.
In vitro antiproliferative activity
The effects of substances 2–9 on cell viability were screened against colon cancer (HCT116) and breast cancer (MCF7) cell lines with different concentrations (from 1.56 to 100 µM) for each substance (cf. Supplementary Table 1 and Supplementary Figs. 2 and 3) using doxorubicin as a reference and MTT assay45. The findings displayed that the tested substances exhibited varying effects on the viability of cancer cell lines. The most potent substances against the two cell lines were pyrazolone 2 and cyanoethanohydrazone 5.
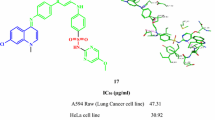
The IC50 (half maximal inhibitory concentration) values were presented in Table 2. The results unveiled variable inhibitory activities by the examined compounds. A very strong effect was depicted by pyrazolone 2 against the two cell lines (IC50 = 7.39 ± 0.5 and 9.24 ± 0.7 µM, respectively). Also, a strong efficacy against the two cell lines was illustrated by substrate 5 (IC50 = 13.46 ± 1.1 and 16.43 ± 1.3 µM, respectively). A moderate effect was shown by compounds 3 and 7. While, weak activity was disclosed by substances 4, 8, and 9.
These experimental antitumor potentials may be ascribed to the reactive oxygen and nitrogen species represented in amide, lactam, and lactim tautomers46. Perhaps, the N-phenylpyrazolone scaffold (in compound 2) might enhance the lipophilicity, potentially leading to improved binding affinity of the drug to hydrophobic regions of target proteins compared to other compounds46,47. Moreover, this was also in agreement with the theoretical data which showed the small energy gab (ΔE = ELUMO − EHOMO) in pyrazolone 2 (cf. Table 1). That signifies it is more polarized (soft molecule) and reactive than hard ones because it easily offers electrons to an acceptor, so it was more bioactive substrate. The low energy gap values may be due to the groups entering into or extending the conjugation in these molecules in addition to tautomeric structures. The extended conjugation led to higher affinity to generate a face-to-edge aromatic interaction with receptor5. In turn, the inclusion of cyanoethanohydrazone scaffold (in compound 5) might indorse favorable interactions, like H-bond formation through oxygen and nitrogen with specific receptors and target proteins enhancing the drug potency (cf. Fig. 3)48,49. Also, the electron-withdrawing group (like CN) was reported to exhibit a significant effect of improving the ability to inhibit cancer cell proliferation through binding interactions with receptor active sites via van-der Waals and H-bonding50,51.

SAR of the potent compounds.
Apparently, the antitumor action of the nucleoside analog, CNDAC (2′-C-cyano-2′-deoxy-1-β-d-arabinopentofuranosyl-cytosine) was assessed in clinical trials52. Incorporation of CNDAC triphosphate into DNA and extension during replication led to single-strand breakdowns causing cells arrest in G2 phase (Fig. 4). Accordingly, a proposed mechanism of DNA interaction with substances 2 and 5 as compared with CNDAC can be offered in Fig. 5. Also, tautomerism played a worthy role in anticancer agents by raising the functionality and flexibility53.

Pathway of the antitumor action of CNDAC.

A proposed pathway for the antitumor action of compounds 2 and 5 compared to CNDAC.
Molecular docking
A molecular docking approach was performed with the molecular operating environment (MOE 2014.0901) to establish the binding energies of produced compounds toward cyclin-dependent kinase-5 protein (PDB ID: 3IG7) (http://www.rcsb.org/pdb) and govern the interactions between the formed ligands and receptors to compare the produced complexes affinities toward target binding sites of protein54,55. The binding affinity was measured by binding energies (S-score, kcal/mol) and H-bonding. The negative result of the docking score points to the stability of complex. All complexes acquired were docked in the same section of the binding site of the native co-crystallized ligand (EFP) (Tables 3 and 4). Table 3 also summarizes the specific amino-acids contributing to binding interaction between substances 2 and 5 with relevant target proteins. It imparts comprehensive information concerning the binding amino acids and the types of bonds established, like H-acceptor, pi-H, pi-cation, and others, for each substance in connection with the relevant protein target.
Among Table 3, the ligands binding energies were closer to that of co-crystallized ligand (EFP). Substance 2 exhibited a binding energy of − 6.6320 kcal/mol with the smallest RMSD of 0.9477 Å affirming to tightly binding to key nucleobases and amino-acids (through arene-H interactions with GLN 131, VAL 18, and ALA 144) of CDK-5 protein revealing its potential manipulation as DNA intercalator and CDK-5 inhibitor. Substance 5 pointed to a binding energy of − 6.5696 kcal/mol with RMSD 1.4889 Å, which revealed a remarkably strong binding affinity through H-bonding with LYS 20, HIS 84, and LYS 89 of the CDK-5 enzyme.
The docking analysis outcomes of compounds 2 and 5 with the CDK-5 protein can be portrayed in graphical 2D and 3D representations (cf. Table 4). 2D depiction illustrates detailed comprehensions of molecular interactions between prepared ligands and protein. Otherwise, 3D visualization platforms the binding interactions between these ligands and the CDK-5 protein, point up H-bonding interactions highlighted in red. Accordingly, binding affinities and docking interaction results provide an experience for the future development of CDK-5 enzyme inhibitors.
Modeling pharmacokinetics studies
To direct the choice of selecting substrates from a vast collection of prepared substances in the initial stages of drug discovery, pharmacological activities, and advance for an effective drug, ADME profiles comprising physicochemical properties, lipophilicity, and drug-likeness of the produced substrate have been prophesied by SwissADME free web tool56,57. Substrates 1, 2, and 5 were found to follow Lipinski’s rule of five, with a total polar surface area (TPSA) of 29.96, 45.56, and 78.14 Å2, in addition to good lipophilicity, stated by consensus Log Po/w which are in 3.36, 4.92, and 2.97, respectively. They revealed a high GI absorption and showed a good bioavailability score (0.55), as displayed in Supplementary Table 2.
Their skin permeation (Log KP) parameters were − 5.03, − 4.56, and − 5.76 cm/s, respectively, hence enhancing the accessibility of bioactive molecules through the skin. Their cytochrome P450 isoenzymes (CYP1A2/CYP2C19/CYP2C9/CYP2D6/CYP3A4), acting a main part in biotransformation of drugs through O-type oxidation reactions, have been likewise estimated (cf. Supplementary Table 2). The substrates’ bioavailability was also estimated according to their pink area on radar chart (cf. Supplementary Figs. 4–11). Regarding the absorption properties, they illustrated gastrointestinal tract (GIT) absorption upon their existence in the BOILED-EGG white area chart as presented in Supplementary Fig. 12. The substances were completely comprised in the pink area and justified their good predicted oral bioavailability specifically compounds 1, 2, 3, 4, and 5.
Conclusion
Certain benzoquinoline-based heterocycles such as pyrazolone, imidazoline, pyridone, and chromene derivatives, were prepared by treating a 3-chlorobenzo[f]quinoline-2-carbaldehyde with N-phenylpyrazolone and various nitrogen nucleophiles. The behavior of the cyanoethanohydrazone obtained was examined towards certain carbon-centered electrophiles and nitrogen nucleophiles. Molecular docking, DFT, and modeling pharmacokinetics approaches were performed to support the findings. The pyrazolone 2 and cyanoethanohydrazone 5 exhibited the most potency, which was demonstrated by their molecular docking towards the CDK-5 enzyme. The binding energies of substances 2 and 5 were closer to that of co-crystallized ligand (EFP), which implies the notably powerful binding affinity towards CDK-5 enzyme. These substances were completely involved in the pink area and justified their good predicted oral bioavailability. Thus, pyrazolone 2 would be considered a promising candidate for further optimization to develop new chemotherapeutic agents.
Materials and methods
General
Melting points (uncorrected) were measured in open capillary tubes on a MEL-TEMP II electrothermal melting point apparatus. The elemental analyses were performed on a Perkin-Elmer 2400 CHN elemental analyzer (Perkin-Elmer, Waltham, MA) at the Faculty of Science, Ain Shams University. The infrared spectra (ν, cm−1) were recorded using the potassium bromide wafer technique on Fourier Transform Infrared Thermo Electron Nicolet iS10 Spectrometer (Thermo Fisher Scientific Inc. Waltham, MA) at Chemistry Department, Faculty of Science, and Faculty of Pharmacy, Ain Shams University. The 1H NMR spectra (δ, ppm) were measured on BRUKER 400 MHz Spectrometer at Faculty of Pharmacy, Cairo University, with tetramethyl silane (TMS) as an internal standard, using DMSO-d6 as a solvent. Mass spectra were carried out on direct probe controller inlet part to single quadrupole mass analyzer in (Thermo Scientific GCMS) MODEL (ISQ LT) using Thermo X-CALIBUR software at the regional center for mycology and biotechnology (RCMB), Al-Azhar University, Cairo, Egypt. Thin-layer chromatography (TLC) was run using TLC aluminum sheets silica gel F254 (Merck, Whitehouse Station, NJ).
4-((3-Chlorobenzo[f]quinolin-2-yl)methylene)-5-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one (2)
A solution of the aldehyde 1 (0.01 mol) and 5-methyl-2-phenyl-2,4-dihydro-3H-pyrazol-3-one (0.01 mol) in absolute ethanol (15 mL) involving piperidine (0.1 mL) was warmed for 10 min. then stirred at room temperature for 4 h. The solid obtained was collected and recrystallized from ethyl alcohol to give beige crystals, mp. 222–224 °C, yield 80%. IR (ν, cm−1): 1686 (C=O). 1H NMR (DMSO, δ, ppm): 2.32 (s, 3H, CH3), 7.16 (t, 1H, Ar–H, J = 7.2 Hz), 7.41 (t, 1H, Ar–H, J = 7.5 Hz), 7.73–8.12 (m, 9H, Ar–H), 8.69 (s, 1H, CH =), 8.97 (s, 1H, C4-H benzoquinoline). EIMS (m/z, %): 399.68 (M + 2, 7), 397.08 (M+., 21), 371.75 (36), 362.94 (21), 343.57 (33), 342.75 (100), 331.98 (41), 312.47 (40), 286.53 (27), 237.69 (57), 230.90 (65), 214.45 (47), 172.73 (49), 159.79 (44), 128.80 (23), 125.91 (63), 111.36 (31), 93.56 (32), 79.24 (56), 67.75 (29). Anal. Calcd. for C24H16ClN3O (397.86): C, 72.45; H, 4.05; N, 10.56; Found: C, 72.32; H, 3.99; N, 10.59%.
1-(4-(((3-Chlorobenzo[f]quinolin-2-yl)methylene)amino)phenyl)ethan-1-one (3)
A solution of the aldehyde 1 (0.01 mol) and 4-acetylaniline (0.01 mol) in absolute ethanol (20 mL) including glacial acetic acid (0.2 mL) was heated under reflux for 4 h. The solid obtained was collected and recrystallized from ethyl alcohol to give pale-yellow crystals, mp. 200–202 °C, yield 83%. IR (ν, cm−1): 1685 (C=O). 1H NMR (DMSO, δ, ppm): 2.61 (s, 3H, CH3), 7.46 (d, 2H, Ar–H, J = 6.9 Hz), 7.79–7.89 (m, 3H, Ar–H), 8.06–8.11 (m, 3H, Ar–H), 8.93 (s, 1H, C4-H benzoquinoline), 9.01 (d, 2H, Ar–H, J = 7.3 Hz), 10.42 (s, 1H, CH = N). EIMS (m/z, %): 360.76 (M + 2, 22), 359.46 (49), 358.17 (M+., 73), 332.70 (38), 329.28 (33), 310.34 (30), 308.46 (100), 290.79 (46), 278.16 (77), 252.14 (79), 232.64 (87), 221.11 (46), 185.77 (75), 177.89 (61), 157.50 (45), 156.74 (93), 133.14 (45), 122.60 (99), 110.71 (23), 88.87 (39), 81.99 (46). Anal. Calcd. for C22H15ClN2O (358.83): C, 73.64; H, 4.21; N, 7.81; Found: C, 73.53; H, 4.15; N, 7.79%.
3-Chloro-2-(4,5-dihydro-1H-imidazol-2-yl)benzo[f]quinoline (4)
A solution of the aldehyde 1 (0.01 mol) and 1,2-diaminoethane (0.01 mol) in absolute ethanol (20 mL) including glacial acetic acid (0.2 mL) was heated under reflux for 3 h. The solid obtained was collected and recrystallized from ethyl alcohol to offer white crystals, mp. 278–280 °C, yield 72%. IR (ν, cm−1): 3150 (NH), 1640 (C=N). 1H NMR (δ, ppm): 2.99 (t, 2H, NHCH2, J = 6.2 Hz), 3.85 (t, 2H, = NCH2, J = 6.2 Hz), 7.79 (br.s, 1H, NH, exchangeable), 7.81–8.07 (m, 5H, Ar–H), 8.83 (d, 1H, Ar–H, J = 7.6 Hz), 9.04 (s, 1H, C4-H benzoquinoline). EIMS (m/z, %): 283.17 (M + 2, 16), 281.56 (M+., 45), 261.90 (40), 254.10 (58), 246.51 (100), 231.36 (23), 218.40 (28), 195.14 (29), 184.61 (40), 148.66 (60), 131.50 (75), 127.43 (73), 108.42 (40), 97.86 (34), 87.52 (51), 81.82 (58), 72.56 (39), 67.24 (41). Anal. Calcd. for C16H12ClN3 (281.74): C, 68.21; H, 4.29; N, 14.91; Found: C, 68.13; H, 4.24; N, 14.93%.
E/Z-N′-((3-Chlorobenzo[f]quinolin-2-yl)methylene)-2-cyanoacetohydrazide (5)
A solution of aldehyde 1 (0.01 mol) and 2-cyanoethanohydrazide (0.01 mol) in absolute ethanol (20 mL) containing glacial acetic acid (0.2 mL) was refluxed for 3 h. The solid obtained while heating was collected and recrystallized from ethyl alcohol to give yellow crystals, mp. > 300 °C, yield 79%. IR (ν, cm−1): 3255 (NH), 2265 (C≡N), 1700 (C=O), 1602 (C=N). 1H NMR (δ, ppm): (anti- and syn-isomers, 80.3:19.7%); 7.77–8.08 (m, 6H, Ar-H), 12.15 (br.s, 1H, NH, exchangeable); for anti-isomer: 4.37 (s, 2H, CH2), 8.46 (s, 1H, CH=N), 8.99 (s, 1H, C4-H benzoquinoline); for syn-isomer: 3.89 (s, 2H, CH2), 8.64 (s, 1H, CH = N), 8.91 (s, 1H, C4-H benzoquinoline). EIMS (m/z, %): 324.50 (M + 2, 20), 322.35 (M+., 52), 319.35 (64), 278.81 (62), 261.11 (42), 254.75 (100), 218.28 (17), 200.65 (35), 188.65 (38), 164.30 (45), 160.79 (41), 100.42 (25), 81.66 (73). Anal. Calcd. for C17H11ClN4O (322.75): C, 63.26; H, 3.44; N, 17.36; Found: C, 63.15; H, 3.39; N, 17.39%.
E/Z-1-(((3-Chlorobenzo[f]quinolin-2-yl)methylene)amino)-4-(1,3-diphenyl-1H-pyrazol-4-yl)-6-imino-2-oxo-1,2,3,6-tetrahydropyridine-3,5-dicarbonitrile (7)
A mixture of 5 (1 mmol) and 2-((1,3-diphenyl-1H-pyrazol-4-yl)methylene)malononitrile (6) (1 mmol) in 1,4-dioxane (15 mL) including triethylamine (0.1 mL) was refluxed for 5 h. The solid obtained upon cooling was collected, and recrystallized from ethyl alcohol to give brown crystals, mp. 290–292 °C, yield 68%. IR (ν, cm−1): 3146 (NH), 2205 (C≡N), 1667 (C=O). 1H NMR (δ, ppm): 3.40 (s, 1H, CH-CN), 7.49–8.23 (m, 16H, Ar–H), 8.60 (s, 1H, CH=N), 9.00 (s, 1H, C4-H benzoquinoline), 9.25 (s, 1H, C5-H pyrazole), 12.20 (br.s, 1H, NH, exchangeable). EIMS (m/z, %): 619.92 (M + 2, 9), 617.34 (M+., 26), 582.75 (100), 548.69 (62), 547.13 (72), 467.48 (77), 418.68 (44), 396.73 (61), 366.35 (59), 347.56 (54), 313.27 (76), 280.73 (58), 215.82 (62), 186.81 (48), 176.12 (62), 167.28 (83), 113.84 (47), 101.23 (61), 92.29 (29), 65.25 (62). Anal. Calcd. for C36H21ClN8O (617.07): C, 70.07; H, 3.43; N, 18.16; Found: C, 70.00; H, 3.39; N, 18.19%.
E/Z-N'-((3-Chlorobenzo[f]quinolin-2-yl)methylene)-2-cyano-3-(4-methoxyphenyl)acrylohydrazide (8)
A solution of 5 (1 mmol) and 4-methoxybenzaldehyde (1 mmol) in 1,4-dioxane (15 mL) containing piperidine (0.1 mL) was stirred at room temperature for 5 h. The deposited solid was collected and recrystallized from ethyl alcohol to give yellow crystals, mp. 286–288 °C, yield 81%. IR (ν, cm−1): 3275 (NH), 2211 (C≡N), 1697 (C=O). 1H NMR (δ, ppm): (E- and Z-isomers, 80:20%); 7.18 (d, 2H, Ar–H, J = 6.7 Hz), 7.79–8.09 (m, 9H, Ar–H + CH =); for E-isomer: 3.88 (s, 3H, OCH3), 8.29 (s, 1H, CH=N), 8.99 (s, 1H, C4-H benzoquinoline), 12.18 (br.s, 1H, NH, exchangeable); for Z-isomer: 4.37 (s, 3H, OCH3), 8.48 (s, 1H, CH=N), 9.02 (s, 1H, C4-H benzoquinoline), 12.31 (br.s, 1H, NH, exchangeable). EIMS (m/z, %): 442.07 (M + 2, 6), 440.23 (M+., 20), 366.98 (91), 318.98 (59), 233.22 (17), 203.62 (100), 155.03 (47), 110.16 (28), 98.47 (59), 92.70 (81), 73.96 (52), 65.03 (37), 59.43 (38). Anal. Calcd. for C25H17ClN4O2 (440.89): C, 68.11; H, 3.89; N, 12.71; Found: C, 68.02; H, 3.82; N, 12.68%.
E/Z-N'-((3-Chlorobenzo[f]quinolin-2-yl)methylene)-2-imino-2H-chromene-3-carbohydrazide (9)
A solution of cyanoacetamide derivative 5 (1 mmol) and 2-hydroxybenzaldehyde (1 mmol) in 1,4-dioxane (15 mL) containing piperidine (0.1 mL) was refluxed for 4 h. The deposited solid was collected and recrystallized from ethyl alcohol to produce white crystals, mp. 248–250 °C, yield 73%. IR (ν, cm−1): 3301 (NH), 1692 (C=O), 1636 (C=N). 1H NMR (δ, ppm): 7.05 (d, 1H, Ar–H, J = 6.5 Hz), 7.27 (t, 1H, Ar–H, J = 6.7 Hz), 7.37–8.09 (m, 8H, Ar–H), 8.48 (s, 1H, C4-H chromene), 8.73 (s, 1H, CH=N), 9.00 (s, 1H, C4-H benzoquinoline), 9.22 (br.s, 1H, = NH, exchangeable), 13.87 (br.s, 1H, NH, exchangeable). EIMS (m/z, %): 428.94 (M + 2, 35), 426.78 (M+., 100), 401.14 (63), 389.20 (52), 388.56 (47), 358.57 (57), 309.58 (37), 277.86 (57), 256.84 (63), 222.27 (43), 187.22 (50), 166.10 (68), 143.64 (47), 124.20 (39), 66.17 (31). Anal. Calcd. for C24H15ClN4O2 (426.86): C, 67.53; H, 3.54; N, 13.13; Found: C, 67.41; H, 3.47; N, 13.16%.
Reaction of cyanoacetohydrazone 5 with thiosemicarbazide
A solution of 5 (1 mmol) and thiosemicarbazide (1 mmol) in 1,4-dioxane (15 mL) was refluxed for 6 h. The solid obtained after cooling was collected and recrystallized from an ethyl alcohol/dioxane mixture (2:1) to produce thiosemicarbazone derivative 11 as white crystals, mp. 283–285 °C [Lit.13 283–285 °C].
Reaction of cyanoacetohydrazone 5 with hydrazine hydrate
A solution of 5 (1 mmol) and hydrazine hydrate (1 mmol, 80%) in ethyl alcohol (20 mL) was refluxed for 4 h. The solid obtained after cooling was collected and recrystallized from ethanol to produce 1,2-bis((3-chlorobenzo[f]quinolin-2-yl)methylene)hydrazine (12) as yellow crystals, mp. > 300 °C [Lit.13 mp. > 300 °C].
Cytotoxicity activity
Cell lines
Mammary gland breast cancer (MCF7) and colorectal carcinoma colon cancer (HCT116) cell lines were used for screening. The cell lines were obtained from ATCC via a holding company for biological products and vaccines (VACSERA). Doxorubicin was used as a reference anticancer drug for comparison.
Chemical reagents
RPMI-1640 medium, MTT, and DMSO were provided by Sigma Co. (USA), and Fetal Bovine serum was obtained from GIBCO (UK).
MTT assay
The mentioned cell lines were used to explore the inhibitory effects of substances on cell growth using the MTT assay45. This colorimetric assay was based on the conversion of the yellow tetrazolium bromide (MTT) to a purple formazan derivative by mitochondrial succinate dehydrogenase in viable cells. The stock samples of the substances were diluted with RPMI-1640 medium to desired concentrations ranging from 1.56 to 100 μM. The final concentration of DMSO in each sample did not exceed 1% (v/v). Doxorubicin was used as a positive control and DMSO was employed as a negative control. Cell lines were cultured in RPMI-1640 medium with 10% fetal bovine serum. Antibiotics added were 100 units/mL penicillin and 100 µM streptomycin at 37 °C in a 5% CO2 incubator. The cell lines were seeded in a 96-well plastic plate at a density of 1.0 × 104 cells/well at 37 °C for 48 h under 5% CO2. After incubation, the cells were treated with different concentrations of substances and incubated for 48 h. After that, 20 µL of MTT solution at 5 mg/mL was added and incubated for 4 h. DMSO in volume of 100 µL was added into each well to dissolve the purple formazan formed. The colorimetric assay was measured and recorded at absorbance of 570 nm using a plate reader (EXL 800, USA). The relative cell viability in percentage was calculated as (A570 of treated samples/A570 of untreated sample) × 100. The IC50 values were determined according to the equation of Boltzmann sigmoidal concentration–response curves utilizing the non-linear regression fitting model58.
Statistical analysis
The bioassay was repeated in triplicate. The data obtained were presented as means ± standard error of the means (SEM) (n = 3) using SPSS 13.0 program (SPSS Inc., Chicago, IL). Differences between groups were considered statistically significant at p values < 0.05.
Data availability
All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
Upadhyay, K., Dodia, N., Khunt, C., Rupesh, C. R. & Shah, A. K. Synthesis and biological screening of pyrano[3,2-c]quinoline analogues as anti-inflammatory and anticancer agents. ACS Med. Chem. Lett. 9(3), 283 (2018).
Elgubbi, A. S., El-Helw, E. A. E., Alzahrani, A. Y. & Ramadan, S. K. Synthesis, computational chemical study, antiproliferative activity screening, and molecular docking of some thiophene-based oxadiazole, triazole, and thiazolidinone derivatives. RSC Adv. 14(9), 5926–5940 (2024).
Youssef, Y. M., Azab, M. E., Elsayed, G. A., El-Sayed, A. A. & El-Helw, E. A. E. Synthesis and antiproliferative screening of some heterocycles derived from 4-((5-Chloro-3-methyl-1-phenyl-1H-pyrazol-4-yl)methylene)-2-phenyloxazol-5(4H)-one. Polycycl. Aromat. Compds. 43(8), 7152–7163 (2023).
Kato, J. K., Ito, Y., Ijuin, R., Aoyama, H. & Yokomatsu, T. Novel strategy for synthesis of substituted benzimidazo[1,2-a]quinolines. Org. Lett. 15, 3794 (2013).
Ramadan, S. K. et al. Synthesis, SAR studies, and insecticidal activities of certain N-heterocycles derived from 3-((2-chloroquinolin-3-yl)methylene)-5-phenylfuran-2(3H)-one against Culex pipiens L. larvae. RSC Adv. 12(22), 13628 (2022).
Kaddah, M. M. et al. Synthesis and biological activity on IBD virus of diverse heterocyclic systems derived from 2-cyano-N’-((2-oxo-1,2-dihydroquinolin-3-yl)methylene)acetohydrazide. Synth. Commun. 51(22), 3366 (2021).
Abdelrahman, A. M., Fahmi, A. A., Rizk, S. A. & El-Helw, E. A. E. Synthesis, DFT and antitumor activity screening of some new heterocycles derived from 2,2’-(2-(1,3-diphenyl-1H-pyrazol-4-yl)ethene-1,1-diyl)bis(4H-benzo[d][1,3]oxazin-4-one). Polycycl. Aromat. Compds. 43(1), 721–739 (2023).
Kaddah, M. M., Fahmi, A. A., Kamel, M. M., Rizk, S. A. & Ramadan, S. K. Rodenticidal activity of some quinoline-based heterocycles derived from hydrazide-hydrazone derivative. Polycycl. Aromat. Compds. 43(5), 4231 (2023).
Ramadan, S. K., Abd-Rabboh, H. S., Gad, N. M., Abou-Elmagd, W. S. I. & Haneen, D. S. Synthesis and characterization of some chitosan-quinoline nanocomposites as potential insecticidal agents. Polycycl. Aromat. Compds. 43(8), 7013 (2023).
Halim, K. N., Rizk, S. A., El-Hashash, M. A. & Ramadan, S. K. Straightforward synthesis, antiproliferative screening, and density functional theory study of some pyrazolylpyrimidine derivatives. J. Heterocycl. Chem. 58(2), 636 (2021).
Umar, A. et al. Design, synthesis, in vitro antiproliferative effect and in situ molecular docking studies of a series of new benzoquinoline derivatives. J. King Saud Univ. Sci. 34, 102003 (2022).
Asran, M., El-Helw, E. A. E., Azab, M. E., Ramadan, S. K. & Helal, M. H. Synthesis and antioxidant activity of some benzoquinoline-based heterocycles derived from 2-((3-chlorobenzo[f]quinolin-2-yl)methylene)hydrazine-1-carbothioamide. J. Iran. Chem. Soc. 20(12), 3023 (2023).
El-Helw, E. A. E., Asran, M., Azab, M. E., Helal, M. H. & Ramadan, S. K. Synthesis, cytotoxic, and antioxidant activity of some benzoquinoline-based heterocycles. Polycycl. Aromat. Compds. https://doi.org/10.1080/10406638.2023.2270767 (2023) (in Press).
Alizadeh, A. & Rostampoor, A. An efficient synthesis of novel functionalized benzo[h]pyrano[2,3-b]quinolines and pyrano[2,3-b]quinoline derivatives via one-pot multicomponent reactions. J. Iran. Chem. Soc. 19, 1239–1249 (2022).
Kaur, K., Jain, M., Reddy, R. P. & Jain, R. Quinolines and structurally related heterocycles as antimalarials. Eur. J. Med. Chem. 45, 3245–3264 (2010).
Marella, A. et al. Quinoline: A versatile heterocyclic. Saudi Pharm. J. 21, 1–12 (2013).
Radini, I., Elsheikh, T., El-Telbani, E. & Khidre, R. New potential antimalarial agents: Design, synthesis and biological evaluation of some novel quinoline derivatives as antimalarial agents. Molecules 21, 909 (2016).
Adams, E. A., LaVoie, E. J., Hoffmann, D. Mutagenicity and metabolism of azaphenanthrenes: In Polynuclear Aromatic Hydrocarbons, Formation, Metabolism, and Measurement (eds. Cooke, M. C., Dennis, A. J.) 73–87 (Battelle Press, 1983).
LaVoie, E. J. et al. Carcinogenicity of quinoline, 4- and 8-methylquinoline and benzoquinolines in newborn mice and rats. Food Chem. Toxicol. 26(7), 625 (1988).
Balaji, P. N. et al. In-vitro anti-inflammatory and antimicrobial activity of synthesized some novel pyrazole derivatives from coumarin chalcones. Der Pharm. Chem. 3(6), 685 (2012).
Kaddah, M., Fahmi, A. A., Kamel, M. M., Ramadan, S. K. & Rizk, S. A. Synthesis, characterization, computational chemical studies and antiproliferative activity of some heterocyclic systems derived from 3-(3-(1,3-diphenyl-1H-pyrazol-4-yl)acryloyl)-2H-chromen-2-one. Synth. Commun. 51(12), 1798–1813 (2021).
Sayed, G. H., Azab, M. E., Negm, N. A. & Anwer, K. E. Antimicrobial and cytotoxic activities of some novel heterocycles bearing pyrazole moiety. J. Heterocycl. Chem. 55(7), 1615–1625 (2018).
Shalini, K., Sharma, P. K. & Kumar, N. Imidazole and its biological activities: A review. Der Chem. Sin. 1(3), 36–47 (2010).
Youssef, A. M., El-Ziaty, A. K., Abou-Elmagd, W. S. I. & Ramadan, S. K. Novel synthesis of some imidazolyl-, benzoxazinyl-, and quinazolinyl-2,4-dioxothiazolidine derivatives. J. Heterocycl. Chem. 52(1), 278–283 (2015).
Patel, K. et al. Design, synthesis and biological evaluation of some novel 3-cinnamoyl-4-hydroxy-2H-chromen-2-ones as antimalarial agents. J. Med. Chem. Res. 21(8), 1780 (2012).
Ramadan, S. K. & Rizk, S. A. Synthesis, density functional theory, and cytotoxic activity of some heterocyclic systems derived from 3-(3-(1,3-diphenyl-1H-pyrazol-4-yl)acryloyl)-2H-chromen-2-one. J. Iran. Chem. Soc. 19(1), 187 (2022).
Azab, M. E., El-Helw, E. A. E. & Ramadan, S. K. 2-Cyano-N′-[(1,3-diphenyl-1H-pyrazol-4-yl)methylidene]acetohydrazide in the synthesis of nitrogen heterocycles. Russ. J. Org. Chem. 55(12), 1940–1945 (2019).
El-Helw, E. A. E., Abdelrahman, A. M., Fahmi, A. A. & Rizk, S. A. Synthesis, density functional theory, insecticidal activity, and molecular docking of some N-heterocycles derived from 2-((1,3-diphenyl-1H-pyrazol-4-yl)methylene)malonyl diisothiocyanate. Polycycl. Aromat. Compds. 43(9), 8265–8281 (2023).
El-Helw, E. A. E., Alzahrani, A. Y. & Ramadan, S. K. Synthesis and antimicrobial activity of thiophene-based heterocycles derived from thiophene-2-carbohydrazide. Future Med. Chem. 16(5), 439–451 (2024).
Ramadan, S. K., El-Ziaty, A. K. & El-Helw, E. A. E. Synthesis and antioxidant evaluation of some heterocyclic candidates from 3-(1,3-diphenyl-1H-pyrazol-4-yl)-2-(4-oxo-4H-benzo[d][1,3]oxazin-2-yl)propenonitrile. Synth. Commun. 51(8), 1272 (2021).
Hassaballah, A. I., Ramadan, S. K., Rizk, S. A., El-Helw, E. A. E. & Abdelwahab, S. S. Ultrasonic promoted regioselective reactions of the novel spiro 3,1-benzoxazon-isobenzofuranone dye toward some organic base reagents. Polycycl. Aromat. Compds. 43(4), 2973 (2023).
Gad, N. M., Abou-Elmagd, W. S. I., Haneen, D. S. & Ramadan, S. K. Reactivity of 5-phenyl-3-[(2-chloroquinolin-3-yl)methylene]furan-2(3H)-one towards hydrazine and benzylamine: A comparative study. Synth. Commun. 51(9), 1384 (2021).
Abdelrahman, A. M., Fahmi, A. A., El-Helw, E. A. E. & Rizk, S. A. Facile synthesis, biological evaluation, DFT studies and in silico prediction ADME/pharmacokinetics properties of N-(1-(2-chlorobenzo[h]quinolin-3-yl)-1-substituted-vin-2-yl)benzamide derivatives. Polycycl. Aromat. Compds. 43(7), 6597–6614 (2023).
Jafari, F. et al. Design, synthesis and biological evaluation of novel benzo- and tetrahydrobenzo[h]quinoline derivatives as potential DNA-intercalating antitumor agents. Eur. J. Med. Chem. 164, 292–303 (2019).
Hao, G., Sun, J. & Wei, C. Studies on interactions of carbazole derivatives with DNA, cell image, and cytotoxicity. Bioorg. Med. Chem. 26(1), 285–294 (2018).
Rescifina, A. et al. Recent advances in small organic molecules as DNA intercalating agents: Synthesis, activity, and modeling. Eur. J. Med. Chem. 74, 95–115 (2014).
Calvillo-Paez, V. et al. Synthesis, spectroscopic, physicochemical and structural characterization of tetrandrine-based macrocycles functionalized with acridine and anthracene groups: DNA binding and anti-proliferative activity. Chem. Biol. Interact. 286, 34–44 (2018).
De Isabella, P., Zunino, F. & Capranico, G. Base sequence determinants of amonafide stimulation of topoisomerase II DNA cleavage. Nucleic Acids Res. 23(2), 223–229 (1995).
Wang, Y. Q. et al. Design, synthesis and biological evaluation of substituted 11H-benzo[a]carbazole-5-carboxamides as novel antitumor agents. Eur. J. Med. Chem. 46(12), 5878–5884 (2011).
Fedorowicz, J. et al. Synthesis and fluorescence of dihydro-[1,2,4]triazolo[4,3-a]pyridin-2-iumcarboxylates: An experimental and TD-DFT comparative study. Dyes Pigments 161, 347 (2019).
Jabbari, M. & Jabbari, A. Antioxidant potential and DPPH radical scavenging kinetics of water-insoluble flavonoid naringenin in aqueous solution of micelles. Colloids Surf. A Physicochem. Eng. Asp. 489, 392–399 (2016).
Xi, J. & Guo, R. Effects of pH and micelle on the radical scavenging ability of puerarin. J. Disper. Sci. Technol. 30, 857–864 (2009).
Naseem, B., Shah, S. W. H., Hasan, A. & Shah, S. S. Interaction of flavonoids, the naturally occurring antioxidants with different media: A UV–visible spectroscopic study. Spectrochim. Acta A 75, 1341–1346 (2010).
Heakal, F. E. T. et al. Synthesis, characterization and computational chemical study of novel pyrazole derivatives as anticorrosion and antiscalant agents. J. Mol. Struct. 1147, 714–724 (2017).
Mosmann, T. Rapid colorimetric assay for cellular growth and survival: Application to proliferation and cytotoxic assay. J. Immunol. Methods 65, 55 (1983).
Fathy, U., Gouhar, R. S. & Awad, H. M. Design, synthesis and anticancer activity of some novel 5-(4-methoxyphenyl)-2,4-dihydro-3H-pyrazol-3-one derivatives. Acta Poloniae Pharmaceutica-Drug Res. 74(5), 1427–1436 (2017).
Nassar, I. F., El Farargy, A. F. & Abdelrazek, F. M. Synthesis and anticancer activity of some new fused pyrazoles and their glycoside derivatives. J. Heterocycl. Chem. 55, 1709 (2018).
Mohareb, R. M., Fleita, D. H. & Sakka, O. K. Utilization in the synthesis of coumarin, pyridine, thiazole and thiophene derivatives with antitumor activity. Molecules 16, 16–27 (2011).
Fadda, A. A., Bayoumy, N. M., Soliman, N. N. & Eldiasty, D. M. Cyanoacetamide intermediate in heterocyclic synthesis: Synthesis and biological evaluation of hitherto new dioxoisoindoline heterocyclic derivatives. J. Heterocycl. Chem. 56(2), 597–607 (2019).
Tseng, C. H. et al. Discovery of 2-[2-(5-nitrofuran-2-yl)vinyl]quinoline derivatives as a novel type of antimetastatic agents. Bioorg. Med. Chem. 23(1), 141 (2015).
Miura, K. et al. Chemical and chemotherapeutical studies on the furan derivatives. Xxx. Syntheses and antibacterial activities of 2-(5-nitro-2-furyl)vinyl heterocyclics. Yakugaku Zasshi 83, 771 (1963).
Liu, X. et al. Molecular basis for G2 arrest induced by 2′-C-cyano-2′-deoxy-1-β-d-arabino-pentofuranosylcytosine and consequences of checkpoint abrogation. Cancer Res. 65, 6874 (2005).
Ramadan, S. K., Halim, K. N., Rizk, S. A. & El-Hashash, M. A. Cytotoxic activity and density functional theory studies of some 1,3-diphenylpyrazolyltetrahydropyrimidine derivatives. J. Iran. Chem. Soc. 17, 1575–1589 (2020).
Khatab, H. A., Hammad, S. F., El-Fakharany, E. M., Hashem, A. I. & El-Helw, E. A. E. Synthesis and cytotoxicity evaluation of novel 1,8-acridinedione derivatives bearing phthalimide moiety as potential antitumor agents. Sci. Rep. 13(1), 15093 (2023).
Youssef, Y. M. et al. Synthesis and antioxidant, antimicrobial, and antiviral activity of some pyrazole-based heterocycles using a 2(3H)-furanone derivative. J. Iran. Chem. Soc. 20(9), 2203–2216 (2023).
Daina, A., Michielin, O. & Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 7, 42717 (2017).
El-Sewedy, A., El-Bordany, E. A., Mahmoud, N. F., Ali, K. A. & Ramadan, S. K. One-pot synthesis, computational chemical study, molecular docking, biological study, and in silico prediction ADME/pharmacokinetics properties of 5-substituted 1H-tetrazole derivatives. Sci. Rep. 13(1), 17869 (2023).
Eissa, I. H., El-Naggar, A. M. & El-Hashash, M. A. Design, synthesis, molecular modeling and biological evaluation of novel 1H-pyrazolo[3,4-b]pyridine derivatives as potential anticancer agents. Bioorg. Chem. 67, 43–56 (2016).
Funding
Open access funding provided by The Science, Technology & Innovation Funding Authority (STDF) in cooperation with The Egyptian Knowledge Bank (EKB).
Author information
Authors and Affiliations
Contributions
M.A. did the experiments. E.A.E.E., S.K.R., M.E.A., and M.H.H. supervised the work. A.Y.A.A. offered resources. E.A.E.E. and S.K.R. demonstrated the synthesized compounds and wrote the main manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
El-Helw, E.A.E., Asran, M., Azab, M.E. et al. Synthesis and in silico studies of certain benzo[f]quinoline-based heterocycles as antitumor agents. Sci Rep 14, 15522 (2024). https://doi.org/10.1038/s41598-024-64785-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-64785-z
- Springer Nature Limited