
Abstract
Physella acuta is a freshwater snail native to North America. Understanding the phylogeography and genetic structure of P. acuta will help elucidate its evolution. In this study, we used mitochondrial (COI and 16S rDNA) and nuclear (ITS1) markers to identify the species and examine its genetic diversity, population structure, and demographic history of P. acuta in Thailand. Phylogenetic and network analyses of P. acuta in Thailand pertained to clade A, which exhibits a global distribution. Analysis of the genetic structure of the population revealed that the majority of pairwise comparisons showed no genetic dissimilarity. An isolation-by-distance test indicates no significant correlation between genetic and geographical distances among P. acuta populations, suggesting that gene flow is not restricted by distance. Demographic history and haplotype network analyses suggest a population expansion of P. acuta, as evidenced by the star-like structure detected in the median-joining network. Based on these results, we concluded that P. acuta in Thailand showed gene flow and recent population expansion. Our findings provide fundamental insights into the genetic variation of P. acuta in Thailand.
Similar content being viewed by others

Physella acuta (Draparnaud, 1805) (syn. Physa acuta) is a freshwater snail from the Physidae family. This snail is well known for its remarkable ability to invade new habitats. They are classified as freshwater pulmonate snails1,2. Physella acuta is an invasive snail native to North America1. Physella acuta exhibits remarkable expansiveness owing to its capacity to disperse through various vectors, broad ecological tolerance, swift adaptation to novel habitats, and exceptional fecundity and reproductive potential3,4. Physella acuta has successfully invaded various natural and artificial freshwater habitats worldwide and is now found on all continents except Antarctica1,2.
From a biogeographical perspective, snail invasions have a significant negative effect on the environment, leading to the homogenization of fauna, extinction of vulnerable endemic species, and changes in biotic composition within invaded ecosystems5,6. The rapid displacement of native snails by P. acuta in several countries is a significant ecological concern7,8. Elevated population densities of P. acuta in new habitats affect native fauna and severely threaten plants of economic importance in greenhouses8. Furthermore, apart from its ecological implications, P. acuta has significant zoonotic medical relevance, as it can potentially act as a transitional carrier for several human trematode ailments, including echinostomiasis and fascioliasis9,10. Physella acuta has also been associated with outbreaks of cervical dermatitis in humans, serving as an intermediate host for Trichobilharzia, the causative agent of cervical dermatitis in many European countries11.
Physella acuta was first introduced in the northeastern province of Thailand in 2001 and subsequently spread to both the northern and southern provinces by 200512. This snail is commonly found in various habitats, including drainage ditches, rivers, paddy fields, canals, and artificial habitats such as concrete pots or ponds12,13. Previous studies have indicated that P. acuta in Thailand serves as a host for Trichodina unionis, the causative agent of aquatic animal diseases. The disease adversely impacts both captive and wild fish and can negatively affect aquaculture and the economy14. Additionally, P. acuta has been documented to be infected with trematode cercariae, including xiphidiocercariae and echinostome cercariae. These indicate the potential of this invasive snail to act as an intermediate host for trematode cercariae in non-native regions13,15.
Molecular phylogeography and population genetic structure are powerful tools that can reveal crucial aspects of invasion processes, such as migration, gene flow, genetic drift, and population expansion1,2. The mitochondrial cytochrome c oxidase subunit I (COI) and 16S rDNA genes, along with other genes such as the internal transcribed spacer I (ITS1) region, nuclear 18S rRNA, and 28S rRNA genes are commonly used for genetic analysis in P. acuta snails1,2,16,17,18. However, mitochondrial markers are often better at detecting subtle genetic subdivisions owing to their reduced effective population size and lack of recombination1,19. Therefore, mitochondrial markers are often used to characterize population genetic structures and identify source populations1,2.
Efforts have been made to explain the population genetic structure of P. acuta on both regional20,21 and global scales1,2. Previous studies have revealed that the genetic diversity and phylogeography of P. acuta can be classified into two clades, A and B. Clade A exhibits a global distribution, whereas clade B is confined to the western United States1. However, sample sizes from some Asian countries have been too small to assess the population genetic structure, indicating that further investigation would be useful1,21. The COI gene has been used in previous studies to identify clades of P. acuta discovered in Thailand1,21, but the underlying population genetic structure and demographic history have never been explored. Therefore, the present study aimed to shed light on the phylogeography and genetics of P. acuta in Thailand in the context of previous research using mitochondrial and nuclear sequences. We aimed to provide broad insights into the genetic diversity, genetic structure, and demographic history of P. acuta in Thailand. These findings establish a foundation for understanding the genetic diversity of P. acuta and contribute to developing guidelines for controlling invasive snails in Thailand.
Results
Molecular identification of P. acuta
To identify P. acuta, PCR-based analysis and sequencing of the COI (509 bp), 16S rDNA (426 bp), and ITS1 (506–512 bp) genes were performed, together with a BLASTN search of the sequences. A total of 322 samples (161 samples for each gene) of Physella sp. were analyzed for mitochondrial COI and 16S rDNA genes. The nucleotide sequences generated in this study were submitted to GenBank under the accession numbers OR738467-OR738627 and OR738836-OR738996 for COI and 16S rDNA, respectively. The COI and 16S rDNA sequences in this study were mostly similar to P. acuta in the GenBank database, with the highest similarity of 99.41–100% and 99.52–100% for COI and 16S rDNA genes, respectively. Seventy-one random samples (GenBank accession numbers OR738997–OR739067) were analyzed using the ITS1 region and were identified as P. acuta with 97.34–100% identity after a BLASTN search.
Phylogenetic and haplotype network analyses
Based on a previous study on P. acuta, two distinct clades, known as Clade A and B, were identified. In the current phylogenetic tree and haplotype network analyses based on the COI gene, all our P. acuta sequences, representing 14 haplotypes, were grouped into clade A. Clade A was the largest group and the most widely distributed across various regions worldwide, whereas clade B was limited to the western United States (Figs. 1, 2). Genetic distances within clade A and clade B are 0.75% and 2.12%, respectively, while the genetic distance between clade A and B is 3.51%. Additionally, analysis using the Automatic Barcode Gap Discovery (ABGD) method for the COI marker revealed no evident barcode gap, as illustrated in Fig. 3.

Maximum likelihood phylogenetic tree based on COI sequences (509 bp) of 14 P. acuta haplotypes from Thailand, along with an additional 112 sequences from various geographical regions. Support values (ML bootstrap/NJ bootstrap/Bayesian posterior probabilities) are shown at the branch points. A dash (-) on the branches indicates less than 50% support value or that a certain grouping was not seen by that analysis method. Bold letters indicate the sequences obtained in this study. Radix rubiginosa and Orientogalba viridis were utilized as outgroups.

Haplotype network (A) and geographical distribution of clades (B) for P. acuta from 26 countries, based on COI sequences (509 bp). In the haplotype network, the size of the circles represents the proportion of samples found in each haplotype, with small black dots indicating hypothetical missing haplotypes. Each mutation between haplotypes is indicated by a bar.

Results of the Automatic Barcode Gap Discovery (ABGD) analysis conducted on 161 COI sequences of P. acuta.
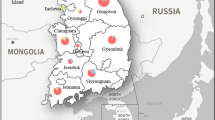
In the haplotype networks, we genetically analyzed 161 individual P. acuta samples from 20 populations in Thailand to examine their relationships with geographical locations within the country. Network analysis of P. acuta based on COI sequences (509 bp) revealed 14 distinct haplotypes (H1–H14) with 14 variable sites (Supplementary Table S1). Among these haplotypes, 13 (92.86%) were unique to specific populations, whereas one (7.14%) was shared by at least two localities. Furthermore, eight populations (40%) comprised multiple haplotypes, and 12 (60%) had a single haplotype. The average haplotype diversity (h) was high at 0.3674, while nucleotide diversity (π) was relatively low, with an average of 0.0010 (Table 1). The analysis of the 16S rDNA (161sequences) identified 13 variable nucleotide sites, which were identified and classified into 13 haplotypes (H1–H13) (Supplementary Table S1). Of these, 10 haplotypes (76.92%) were unique, and three (23.08%) were shared by multiple populations. Among the populations, seven (35%) consisted of multiple haplotypes, whereas 13 (65%) had a single haplotype. Average haplotype and nucleotide diversity values were 0.5498 and 0.0019, respectively (Table 1). Meanwhile, the MJ network of combined mtDNA sequences was classified into 26 haplotypes (H1–H26). Among these, 88.5% (23 haplotypes) were unique, and 11.54% (3 haplotypes) were shared by at least two populations. The estimates of haplotype and nucleotide diversity are shown in Table 1. Haplotype H2 was the most common haplotype for both the COI, 16S rDNA, and mtDNA, shared between populations from different regions (Fig. 4). Additionally, the haplotype network structure of P. acuta in Thailand formed a star-like structure, with the H2 haplotype serving as the prominent central haplotype surrounded by several low-frequency haplotypes (Fig. 4).

Map and haplotype network of P. acuta from 20 provinces in four geographical regions of Thailand based on COI, 16S rDNA, and mtDNA (COI + 16S rDNA) sequences. Each haplotype is represented by a circle, and circle sizes are proportional to haplotype frequency. Colors indicate the geographic origin of the haplotypes. The small black dots indicate hypothetical missing haplotypes. Each mutation between haplotypes is indicated by a bar. The number inside each circle indicates the provinces.
To assess the relationship between P. acuta from Thailand and other countries, we constructed haplotype networks using 273 COI sequences. This dataset includes 161 sequences from the current study and 112 sequences obtained from GenBank. Among the 273 sequences, 75 haplotypes (H1–H75) were identified. The 161 sequences from Thailand used in the current study were represented by 14 haplotypes (H1–H14). In most P. acuta sequences from Thailand, 128 sequences (accounting for 79.50% of all samples in the present study) belonged to the H2 haplotype. Haplotype H2 included sequences from ten countries (in addition to Thailand) across Asia and Europe (Fig. 2).
Cluster analysis
Cluster analysis of P. acuta populations in Thailand revealed two major clusters. Cluster 1 comprised sample populations from Chai Nat and Phetchabun. Cluster 2 was subdivided into two sub-clusters. The first sub-cluster included the snail population from Sing Buri, while the second sub-cluster encompassed populations from Uttaradit, Lamphun, Lop Buri, Phichit, Nakhon Nayok, Uthai Thani, Songkhla, Phitsanulok, Chiang Rai, Chiang Mai, Ayutthaya, Nakhon Sawan, Yala, Chanthaburi, Chachoengsao, Sukhothai, and Chon Buri (Fig. 5).

Cluster analysis of genetic relationships among 20 P. acuta populations in Thailand, based COI sequences.
Genetic structure analysis
Population pairwise FST analysis indicated varying degrees of genetic differentiation among P. acuta populations in the 18 provinces sampled in Thailand. Pairwise FST analysis of P. acuta based on COI sequences revealed that 72.55% (111 of 153) were not genetically significantly different. Similarly, analyses based on 16S rDNA and mtDNA revealed that 94 (61.44%) and 80 (52.29%) of the 153 pairwise comparisons, respectively, were not genetically significantly different. The provinces in the eastern, northern, and southern regions were not genetically significantly different from others in their regions. However, in the central region, some provinces displayed significant genetic differences from others within the same region (Fig. 6, Supplementary Table S2). Moreover, an analysis using a Mantel test to assess the relationship between geographical distances and genetic differentiation revealed non-significant findings (r = 0.0115, p = 0.338). Despite detecting a very weak positive correlation between geographic and genetic distances, the association was not statistically significant (R2 = 0.003, p = 0.299) (Fig. 7).

Graph of pairwise FST distance matrices between populations of P. acuta in Thailand based on COI (A),16S rDNA (B), and mtDNA (COI + 16S rDNA) (C) sequences. Darker blue indicates a higher pairwise FST value and lighter blue indicates a lower value. Asterisks (*) indicate FST values with statistically significant differences (P < 0.05).

Correlation of geographic distance (in kilometers) and genetic distance among 161 individuals of 20 populations of P. acuta based on COI sequences.
Demographic history
Mismatch distribution analysis of P. acuta based on COI,16S rDNA, and mtDNA sequences revealed a unimodal mismatch graph with non-significant values for the sum of squares deviation (SSD = 0.0001, P > 0.05 in COI; SSD = 0.0011, P > 0.05 in 16S rDNA; SSD = 0.0017, P > 0.05 in mtDNA) and Harpending’s raggedness index (HRI = 0.1819, P > 0.05 in COI; HRI = 0.0799, P > 0.05 in 16S rDNA; HRI = 0.0349, P > 0.05 in mtDNA). The results of the neutrality tests were highly significant negative values for both Tajima’s D and Fu’s Fs tests (Fig. 8).

Mismatch distribution of P. acuta from Thailand based on COI (A), 16S rDNA (B), and mtDNA (COI + 16S rDNA) sequences. The black dotted lines with circles illustrate the observed frequency of pairwise differences, and the green lines represent the expected values under the sudden population expansion model. Asterisks indicate values levels of statistical significance (P < 0.05).
Discussion
In the present study, we identified the species and phylogenetic clades of P. acuta in many provinces of Thailand using mitochondrial markers (COI and 16S rDNA) and nuclear genetic markers (ITS1). We evaluated the phylogeography, genetic diversity, population structure, and demographic history of P. acuta in Thailand. Previous research has identified several clades of P. acuta worldwide, including Thailand1,2,21. Nevertheless, to the best of our knowledge, our data are the first to include 16S rDNA and ITS1 sequences reported from Thailand and provide additional insights into the haplotype distribution, population structure, and demographic history of P. acuta in Thailand.
Phylogenetic analysis of P. acuta from Thailand in the present study resulted in the classification of the species into clade A. This conclusion was further reinforced by the haplotype network analysis. Notably, clade A has been previously documented globally1. These data indicate that the P. acuta Thai strains are closely related to those from multiple countries. Physella acuta, which is native to North America22, was first recorded outside of its native range in the Bordeaux region of France in 1805, more than 200 years ago. It is believed to have been introduced via the cotton trade from Mississippi, USA, during the eighteenth century1. However, within less than a century, populations were reported across Eastern Europe, Africa, Asia, South America, the Middle East, the Caribbean, New Zealand, and Australia1. The invasion of the African continent occurred between the 1940s and 1950s, leading to reported populations in South Africa23, Nigeria24, and Namibia25. In Asia, P. acuta was recorded in Malaysia and Thailand in the 1970s26. Subsequently, during the 1980s, the species spread to numerous other countries in Asia, including Hong Kong, Singapore, and Australasia1. In Thailand, P. acuta was found to be more dispersed in the northeastern provinces in the 2001s, and in the northern and southern provinces in the 2005s12. Since then, P. acuta has been reported to have a widespread distribution across all regions of Thailand, as documented by Tantrawatpan et al.21. Previous studies have illustrated that P. acuta from different countries on many continents has a close evolutionary relationship1,2. Ebbs et al.1 studied the phylogeography and genetics of the globally invasive P. acuta and hypothesized that this snail in Western Europe (France and England) likely maintained its first invasive populations. The phenomenon of close evolutionary relationships among snails from different regions worldwide has frequently been observed, especially in invasive or alien species, such as Pomacea canaliculata27, Indoplanorbis exustus28, and Achatina fulica29.
In Thailand, we identified 14 COI, 13 16S rDNA, 26 mtDNA haplotypes in 161 P. acuta samples from 20 different populations. Our number of P. acuta haplotypes was relatively high compared to that in a previous study. Tantrawatpan et al.21 reported genetic variation in P. acuta in 12 provinces of Thailand based on the COI gene and identified seven different haplotypes. The variation in haplotype numbers between our study and a previous study21 may be due to the smaller sample size in the previous study in comparison to our current study, or it could be due to the high mutation rate of mitochondrial DNA30.
Our haplotype distribution results on P. acuta in Thailand revealed that most P. acuta haplotypes in Thailand were distinct haplotypes discovered in particular regions. In contrast, a mere fraction of the haplotypes were shared, accounting for only 7.14% (COI), 23.08% (16S rDNA), 11.54% (mtDNA) of the total samples analyzed. Haplotype H2 was the most common haplotype for both the COI,16S rDNA, and mtDNA shared between populations from different regions. When assessing the relationship between P. acuta from Thailand and those from other countries, we found that haplotype H2 was shared among P. acuta populations from 10 countries across Asia and Europe. Haplotype H2 is widespread and may be the ancestral haplotype of the P. acuta Thai strain. Our findings are in concordance with those of a previous study by Ebbs et al.1, who reported that the invasive haplotypes of P. acuta were found only in clade A. The presence of shared haplotypes observed in this study indicates gene flow or migration between distant populations31.
Genetic diversity indices of P. acuta from Thailand showed that the overall haplotype diversity was high, but nucleotide diversity was low. Higher values of haplotype diversity relative to nucleotide diversity indicate rapid expansion from the founding populations or continued expansion32. Additionally, this indicates the adaptation of snails to enable the colonization of diverse habitats33. This phenomenon has been previously observed in other invasive snails in Thailand, including P. canaliculata and P. maculata27.
In this study, we found that the population genetic structure of P. acuta in many provinces of Thailand showed that most comparisons were not genetically significantly different. This observation suggests that gene flow within the P. acuta population in Thailand may have led to genetic homogeneity34. However, in the central region, certain provinces displayed significant genetic distinctions from others within the same region. Furthermore, cluster analysis of P. acuta populations identified two major clusters: cluster 1 comprised Chai Nat and Phetchabun populations, while cluster 2 exhibited two sub-clusters. The first sub-cluster encompassed the Sing Buri population, while the second sub-cluster consisted of populations from multiple provinces. Numerous factors influence genetic structure and population distribution. An isolation-by-distance test revealed no significant correlation between genetic and geographical distances among P. acuta populations, indicating that gene flow is not restricted by distance. The genetic structure could be influenced by various factors such as habitat type, climate, the presence of predators, interspecific competition, and various other environmental factors in different habitats35,36,37.
The mismatch distribution analysis of P. acuta in Thailand revealed a unimodal mismatch graph with non-significant values for the sum-of-squares deviation and Harpending’s raggedness index. These results suggest sudden population expansion38. This expansion is consistent with the star-like structure observed in the median-joining network, with the most common haplotypes in the star’s center39. Additionally, population expansion was supported by highly significant negative values in both Tajima’s D and Fu’s Fs tests. Our findings suggested that P. acuta is invasive and is undergoing population expansion in Thailand. However, its limited dispersal ability, anthropogenic activity, hydrological connectivity, and flash floods have facilitated its expansion into new areas20,22. In recent years, human activities, such as water transport, shipment of exotic plants, aquarium trade, and industrial aquaculture, have significantly accelerated the dispersal of this snail22. In particular, in the ornamental trade, as reported by Ng et al.12, P. acuta was discovered on aquatic plants that were being transported for sale in an ornamental pet trade shop located in a market in Bangkok. Ornamental trade may have contributed significantly to their spread in Thailand. Moreover, P. acuta can disperse through river basins through water flow, particularly in flooded rice fields, allowing snails to move freely20. Bird-mediated natural dispersal mechanisms can effectively disperse snails over large distances and through natural barriers in various areas40. Life-cycle characteristics, such as high proliferation rates, a wide range of ecological tolerance, and rapid adaptation to new environments, are regarded as pivotal factors contributing to the successful dispersal of P. acuta into new areas3,41. P. acuta can self-fertilize and reproduce multiple times per year, coupled with rapid juvenile growth, leading to a rapid increase in its population and outcompeting other snails42. The capacity for self-fertilization would have enabled the rapid spread of the original haplotypes from the initial snail populations, as seen in the invasion from the United States to Africa2.
Physella acuta is known to displace native snails and become the dominant species over very short periods of time7,23. The geographical expansion of P. acuta may have important parasitological implications as it serves as a natural intermediate host for numerous trematodes, including Fasciola hepatica10, Echinoparyphium sp.43, Echinostoma revolutum9, and Trichobilharzia physellae11. Notably, Trichobilharzia, which belongs to the family Schistosomatidae and is also known as the avian schistosome, is a causative agent of avian cervical dermatitis worldwide44. The presence of the parasitosis vector in a geographical area facilitates the establishment of the parasite life cycle and increases the chances of rapid infection by secondary intermediate and definitive hosts45. Furthermore, P. acuta invasion accelerates the spread of parasites because of their high expansiveness, resulting in the occurrence of a parasitic life cycle in non-endemic areas46. Therefore, the invasion of P. acuta may be key to the successful spread of several parasites into new areas.
Conclusions
In the present study, we confirmed that P. acuta from Thailand belongs to Clade A. Genetic diversity and population genetic structure analyses suggested gene flow within the P. acuta population in Thailand. Haplotype network analysis revealed a star-like structure with the most common haplotypes in the star’s center, a characteristic of the recent demographic expansion of populations. Population expansion was also supported by the unimodal graph of the mismatch distribution analysis and the highly negative values of the neutrality indices. Our findings suggested that P. acuta is invasive and is undergoing population expansion in Thailand. Understanding the invasion and levels of gene flow of P. acuta intermediate hosts within Thailand can be valuable not only for studying its genetic divergences but also for surveillance in areas vulnerable to the circulation of trematode parasites.
Materials and methods
Ethic and biosafety statement
The research involving invertebrate animals (snails) in this study received ethical approval from the Center for Animal Research at Naresuan University under Project Ethics Approval No. NU-AQ640803. The experimental protocols for biosafety and biosecurity were approved by the Naresuan University Institutional Biosafety Committee under Project Approval No: NUIBC MI 64-09-34.
Snail collection and identification
Physella acuta snails were randomly collected from 25 distinct locations across 20 provinces in central, eastern, northern, and southern Thailand. Details of these locations are listed in Table 2. Snail samples were obtained from their natural habitats, including paddy fields, lotus ponds, and canals. The collection method involved handpicking and scooping. The snails were then transported at ambient temperature to the Department of Microbiology and Parasitology, Faculty of Medical Science, Naresuan University, Phitsanulok, Thailand. The snails were cleaned with tap water and identified using standard morphological criteria described by Paraense and Pointier47, Wethington et al.48, and Ng et al.12. After identification, the snail bodies were separated from their shells, and approximately 25 mg of foot tissue from each individual was excised and stored at − 20 °C for future DNA analysis.
DNA extraction, amplification, and DNA sequencing
Genomic DNA was extracted from individual P. acuta samples using the NucleoSpin® Tissue Kit (Macherey–Nagel, Duren, Germany), following the manufacturer’s instructions. The quality of the genomic DNA was assessed by electrophoresis on a 0.8% agarose gel in 1 × TBE buffer at 100 V for 35 min, and the DNA was stored at − 20 °C until further analysis. Polymerase chain reaction (PCR) was performed to amplify a portion of the mitochondrial COI and 16S rDNA genes and the ITS1 region using the primers specified in Table 3. The PCR reaction (total volume 30 µl) consisted of 15 µl of OnePCR Ultra (Bio-helix, New Taipei, Taiwan), 1.5 µl of each primer at 5 µM (0.25 µM), 9 µl of distilled water, and 3 µl of the DNA template (20–200 ng). PCR was performed using a Biometra TOne Thermal Cycler (Analytik Jena AG, Jena, Germany), as shown in Table 3. The PCR products were purified using a NucleoSpin® Gel and PCR Clean-Up Kit (Macherey–Nagel, Düren, Germany) according to the manufacturer’s instructions. Purified PCR products were analyzed by 1.2% agarose gel electrophoresis at 100 V, stained with ethidium bromide, destained with distilled water, and visualized and photographed under UV light. Purified PCR products were sequenced by Macrogen Inc. (Seoul, Korea).
Phylogenetic and haplotype analyses
The COI, 16S rDNA, and ITS1 genes were manually reviewed and edited using SeqMan II (DNASTAR, Madison, WI, USA). Nucleotide sequences were aligned and trimmed using the ClustalW algorithm in MEGA (version 7.0 program)51. All sequences obtained in this study were BLASTed against GenBank (http://blast.ncbi.nlm.nih.gov/Blast.cgi) for species identification. A phylogenetic tree was constructed using the maximum likelihood (ML), neighbor-joining (NJ), and Bayesian inference (BI) methods. ML phylogenetics was estimated using the Tamura 3-parameter model52, whereas the NJ tree was estimated using the Kimura 2-parameter model53 with bootstrap support of 1000 resamplings using MEGA version 7.0. BI analysis was conducted using the MrBayes 3.2.0 program54. Bayesian posterior probabilities (BPPs) were estimated via Markov chain Monte Carlo (MCMC) analysis with 2,000,000 generations. Trees were sampled every 1000th generation, resulting in 2000 trees. Additionally, genetic species delimitation was conducted using mtDNA COI sequences through the Automatic Barcode Gap Discovery (ABGD) method55. The online version of ABGD (http://wwwabi.snv.jussieu.fr/public/abgd/abgdweb.html) was employed with default settings and the K2P distance model.
To assess the relationship between P. acuta haplotypes from different geographic areas in Thailand and across the world, a median-joining network was constructed using PopART v1.756. For the COI sequences, we generated a phylogeny and two median-joining networks to assess the genetic relationships among P. acuta populations within Thailand and compared them to populations in other countries. The grouping classifications in our phylogenetic tree and haplotype network were the same as those used by Ebbs et al.1.
Genetic diversity, population genetic structure, and demographic history
Genetic diversity parameters, such as the number of segregation sites, number of haplotypes, haplotype diversity (h), and nucleotide diversity (π), were calculated using DnaSP version 557 and Arlequin version 3.5.1.258. Pairwise population FST calculations were performed using Arlequin software. To mitigate the influence of limited sample size, populations consisting of only one sample were excluded from the FST analysis. Neutrality indices (Fu’s Fs and Tajima’s D) were used to assess the hypothesis of selective neutrality using Arlequin. Mismatch distribution analysis was performed using the DnaSP and Arlequin programs to evaluate demographic equilibrium and examine sudden population expansion. The sum of squares deviation (SSD) and Harpending’s raggedness index59 were employed to assess the deviation from sudden or spatial expansion models using the Arlequin program. Cluster analysis was conducted using R software to explore the genetic structure of P. acuta populations in Thailand.
To assess the genetic relationships between the COI haplotypes of P. acuta in Thailand and those from other geographic regions, we added 112 sequences obtained from GenBank from outside Thailand. Genetic relationships between COI haplotypes were established using the median-joining (MJ) network algorithm60 in PopART v1.7.
Association between geographical distance and genetic divergence
To evaluate whether geographical distance between populations explained genetic differentiation, we examined the relationship between genetic divergence and geographic distances (isolation by distance, IBD) with a Mantel test in software R using the package Vegan61. Additionally, we generated scatter plots to examine the relationships between individual pairwise genetic and geographical distances.
Data availability
The sequences analysed during the current study are available in the GenBank with accession numbers.
OR738467–OR738627 (COI), OR738836–OR738996 (16S rDNA), and OR738997–OR739067 (ITS1).
The authors confirm that the data supporting the findings of this study are available within the article.
References
Ebbs, E. T., Loker, E. S. & Brant, S. V. Phylogeography and genetics of the globally invasive snail Physa acuta Draparnaud 1805, and its potential to serve as an intermediate host to larval digenetic trematodes. BMC Evol. Biol. 18, 103 (2018).
Lawton, S. P., Allan, F., Hayes, P. M. & Smit, N. J. DNA barcoding of the medically important freshwater snail Physa acuta reveals multiple invasion events into Africa. Acta Trop. 188, 86–92 (2018).
Núñez, V. Fecundity and survival advantages of an exotic gastropod compared to a native species. Am. Malacol. Bull. 29, 95–103 (2011).
Spyra, A., Cieplok, A., Strzelec, M. & Babczyńska, A. Freshwater alien species Physella acuta (Draparnaud, 1805)—A possible model for bioaccumulation of heavy metals. Ecotoxicol. Environ. Saf. 185, 109703 (2019).
Cowie, R. Invertebrate invasions on Pacific Islands and the replacement of unique native faunas: A synthesis of the land and freshwater snails. Biol. Invasions 3, 119–136 (2001).
Strayer, D. L. Alien species in fresh waters: Ecological effects, interactions with other stressors, and prospects for the future. Freshw. Biol. 55, 152–174 (2010).
Dobson, M. Replacement of native freshwater snails by the exotic Physa acuta (Gastropoda: Physidae) in southern Mozambique; a possible control mechanism for schistosomiasis. Ann. Trop. Med. Parasitol. 98, 543–548 (2004).
Zukowski, S. & Walker, K. F. Freshwater snails in competition: alien Physa acuta (Physidae) and native Glyptophysa gibbose (Planorbidae) in the River Murray, South Australia. Mar. Freshw. Res. 60, 999–1005 (2009).
Kanev, I. Life-cycle, delimitation and redescription of Echinostoma revolutum (Froelich, 1802) (Trematoda: Echinostomatidae). Syst. Parasitol. 28, 125–144 (1994).
Dreyfuss, G., Vignoles, P., Abrous, M. & Rondelaud, D. Unusual snail species involved in the transmission of Fasciola hepatica in watercress beds in central France. Parasite 9, 113–120 (2002).
Helmer, N. et al. First record of Trichobilharzia physellae (Talbot, 1936) in Europe, a possible causative agent of cercarial dermatitis. Pathogens 10, 1473 (2021).
Ng, T. H. et al. Correcting misidentifications and first confirmation of the globally-invasive Physa acuta Draparnaud, 1805 (Gastropoda: Physidae) in Thailand and Laos. Bioinvasions Rec. 7, 15–19 (2018).
Wiroonpan, P., Chontananarth, T. & Purivirojkul, W. Cercarial trematodes in freshwater snails from Bangkok, Thailand: Prevalence, morphological and molecular studies and human parasite perspective. Parasitology 148, 366–383 (2021).
Wiroonpan, P. & Purivirojkul, W. New record of Trichodina unionis (Ciliophora, Trichodinidae) from freshwater gastropods in Bangkok, Thailand. Parasite 26, 47 (2019).
Ardpairin, J. et al. Preliminary survey of larval trematodes in freshwater snails of Phitsanulok Province in lower morthern Thailand. Iran. J. Parasitol. 17, 268–276 (2022).
Klussmann-Kolb, A. et al. From sea to land and beyond—New insights into the evolution of euthyneuran Gastropoda (Mollusca). BMC Evol. Biol. 8, 57 (2008).
Benelli, G. et al. Mediterranean essential oils as effective weapons against the West Nile vector Culex pipiens and the Echinostoma intermediate host Physella acuta: what happens around? An acute toxicity survey on non-target mayflies. Parasitol. Res. 114, 1011–1021 (2015).
Young, M. K., Smith, R., Pilgrim, K. L. & Schwartz, M. K. Molecular species delimitation refines the taxonomy of native and nonnative physinine snails in North America. Sci. Rep. 11, 21739 (2021).
Bayha, K. M. et al. Worldwide phylogeography of the invasive ctenophore Mnemiopsis leidyi (Ctenophora) based on nuclear and mitochondrial DNA data. Biol. Invasions 17, 827–850 (2015).
van Leeuwen, C. H. et al. How did this snail get here? Several dispersal vectors inferred for an aquatic invasive species. Freshw. Biol. 58, 88–99 (2013).
Tantrawatpan, C. et al. Genetic variation of the lymnaeid Radix rubiginosa (Michelin, 1831) (Mollusca, Gastropoda) and other freshwater snails in Thailand examined using mitochondrial COI sequences. Molluscan Res. 43, 1–9 (2023).
Vinarski, M. V. The history of an invasion: Phases of the explosive spread of the physid snail Physella acuta through Europe, Transcaucasia and Central Asia. Biol. Invasions 19, 1299–1314 (2017).
Appleton, C. C. Alien and invasive fresh water gastropoda in South Africa. Afr. J. Aquat. Sci. 28, 69–81 (2003).
Fashuyi, S. A. Freshwater gastropod molluscs in Ondo state, Nigeria. J. Afr. Zool. 104, 165–170 (1990).
Curtis, B.A. Freshwater macro-invertebrates of Namibia. In The Status and Conservation of Wetlands in Namibia (eds Simmons, R.E. et al.). (Madoqua, 1991).
Sullivan, J., Dondero, T., Palmieri, J. & Palmieri, M. Occurrence of Physastra sumatrana (Mollusca: Pulmonata) in Malaysia. Southeast Asian J. Trop. Med. Public Health 8, 408 (1977).
Dumidae, A. et al. Population genetics analysis of a Pomacea snail (Gastropoda: Ampullariidae) in Thailand and its low infection by Angiostrongylus cantonensis. Zool. Stud. 60, 31 (2021).
Saijuntha, W. et al. Phylogeographic genetic variation of Indoplanorbis exustus (Deshayes, 1834) (Gastropoda: Planorbidae) in South and Southeast Asia. One Health 12, 100211 (2021).
Dumidae, A. et al. Low genetic diversity and the phylogeny of Achatina fulica, an intermediate host of Angiostrongylus cantonensis in Thailand, inferred from 16S mitochondrial sequences. Infect. Genet. Evol. 92, 104876 (2021).
Vandewoestijne, S., Baguette, M., Brakefield, P. M. & Saccheri, I. J. Phylogeography of Aglais urticae (Lepidoptera) based on DNA sequences of the mitochondrial COI gene and control region. Mol. Phylogenet. Evol. 31, 630–646 (2004).
Koopman, W. J. M. et al. Linked vs unlinked markers: multilocus microsatellite haplotype-sharing as a tool to estimate gene flow and introgression. Mol. Ecol. 16, 243–256 (2007).
Roderick, G. K. Geographic structure of insect populations: gene flow, phylogeography, and their uses. Annu. Rev. Entomol. 41, 325–352 (1996).
Liu, X., Zhou, Y., Ouyang, S. & Wu, X. Phylogeographic patterns and demographic history of Pomacea canaliculata and Pomacea maculata from different countries (Ampullariidae, Gastropoda, Mollusca). Nat. Conserv. 36, 71–92 (2019).
Szalanski, A. L. et al. Mitochondrial and ribosomal internal transcribed spacer 1 diversity of Cimex lectularius (Hemiptera: Cimicidae). J. Med. Entomol. 45, 229–236 (2008).
Prasankok, P., Sutcharit, C., Tongkerd, P. & Panha, S. Biochemical assessment of the taxonomic diversity of the operculate land snail, Cyclophorus fulguratus (Gastropoda: Cyclophoridae), from Thailand. Biochem. Syst. Ecol. 36, 900–906 (2008).
Fernandez, M. A., Thiengo, S. C., Bezerra, F. S. M. & Alencar, L. M. S. Current distribution of the exotic freshwater snail Helisoma duryi (Gastropoda: Planorbidae) in Brazil. Nautilus 124, 44–50 (2010).
Saito, T. et al. A comprehensive phylogeography of the widespread pond snail genus Radix revealed restricted colonization due to niche conservatism. Ecol. Evol. 11, 18446–18459 (2021).
Roger, A. R. & Harpending, H. 1992: Population growth makes waves in the distribution of pairwise genetic differences. Mol. Biol. Evol. 9, 552–569 (1992).
Slatkin, M. & Hudson, R. R. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics 129, 555–562 (1991).
Kopp, K. C., Wolff, K. & Jokela, J. Natural range expansion and human-assisted introduction leave different genetic signatures in a hermaphroditic freshwater snail. Evol. Ecol. 26, 483–498 (2012).
Bernot, R. J., Kennedy, E. E. & Lamberti, G. A. Effects of ionic liquids on the survival, movement, and feeding behavior of the freshwater snail Physa acuta. Environ. Chem. Toxicol. 24, 1759–1765 (2005).
Dillion, R. T. Empirical estimates of reproductive isolation among the Physa species of South Carolina (Gastropoda: Pulmonata: Basommatophora). Nautilus 123, 276–281 (2009).
Pantoja, C. et al. Diversity of echinostomes (Digenea: Echinostomatidae) in their snail hosts at high latitudes. Parasite 28, 59 (2021).
Horák, P. et al. Avian schistosomes and outbreaks of cercarial dermatitis. Clin. Microbiol. Rev. 28, 165–190 (2015).
Lu, X. T. et al. Snail-borne parasitic diseases: An update on global epidemiological distribution, transmission interruption and control methods. Infect. Dis. Poverty 7, 28 (2018).
Lv, S. et al. Invasive snails and an emerging infectious disease: Results from the first national survey on Angiostrongylus cantonensis in China. PLoS Negl. Trop. Dis. 3, e368 (2009).
Paraense, W. & Pointier, J. P. Physa acuta Draparnaud, 1805 (Gastropoda: Physidae): A study of topotypic specimens. Mem. Inst. Oswaldo Cruz. 98, 513–517 (2003).
Wethington, A. R., Wise, J. & Dillon, R. T. Genetic and morphological characterization of the Physidae of South Carolina (Gastropoda: Pulmonata: Basommatophora), with description of a new species. Nautilus 123, 282–292 (2009).
Folmer, O. et al. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 3, 294–299 (1994).
Kessing, B. et al. The simple fool’s guide to PCR (University of Hawaii, 1989).
Kumar, S., Stecher, G. & Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874 (2016).
Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G + C-content biases. Mol. Biol. Evol. 9, 678–687 (1992).
Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542 (2012).
Puillandre, N., Lambert, A., Brouillet, S. & Achaz, G. ABGD, automatic barcode gap discovery for primary species delimitation. Mol. Ecol. 21, 1864–1877 (2012).
Leigh, J. W. & Bryant, D. Popart: Full-feature software for haplotype network construction. Methods Ecol. Evol. 6, 1110–1116 (2015).
Librado, P. & Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452 (2009).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10, 564–567 (2010).
Harpending, H. C. Signature of ancient population growth in a low-resolution mitochondrial DNA mismatch distribution. Hum. Biol. 66, 591–600 (1994).
Bandelt, H. J., Forster, P. & Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48 (1999).
Oksanen, J. et al. Vegan: community ecology package. https://cran.r-project.org/web/packages/vegan/index.html (2018).
Acknowledgements
We would like to thank Naresuan University (NU), and National Science, Research and Innovation Fund (NSRF), Thailand (Grant No. R2566B046) for funding support. This study was also partially supported by the Global and Frontier Research University Fund, Naresuan University (Grant number R2567C003).
Author information
Authors and Affiliations
Contributions
A. D. performed experiments, analyzed data, and wrote the paper. J. A. performed experiments and wrote the paper. S. P. performed experiments and wrote the paper. C. H. performed experiments and wrote the paper. M. N. performed experiments and wrote the paper. A. T. provided materials and wrote the paper. A. V. performed experiments, analyzed data, provided materials, and wrote the paper. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Dumidae, A., Ardpairin, J., Pansri, S. et al. Genetic diversity and population structure of Physella acuta (Gastropoda: Physidae) in Thailand using mitochondrial gene markers: COI and 16S rDNA. Sci Rep 14, 13161 (2024). https://doi.org/10.1038/s41598-024-64184-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-64184-4
- Springer Nature Limited