
Abstract
As the world's first oral nuclear export inhibitor, selinexor is increasingly being used in clinical applications for malignant tumors. However, there is no extensive exploration on selinexor's adverse events (ADEs), necessitating a real-word assessment of its clinical medication safety. FAERS data (July 2019–June 2023) were searched for selinexor ADE reports across all indications. Use the system organ class (SOC) and preferred terms (PT) from the medical dictionary for regulatory activities (MedDRA) to describe, categorize, and statistic ADEs. Disproportionality analysis was employed through calculation of reporting odds ratio (ROR) and proportional reporting ratio (PRR). Based on total of 4392 selinexor related ADE reports as the primary suspect (PS), of which 2595 instances were severe outcomes. The predominant ADEs included gastrointestinal disorders, myelosuppression symptoms, and various nonspecific manifestations. 124 signals associated with selinexor ADE were detected, and 10 of these top 15 signals were not included into the instructions. Our study provides real-world evidence regarding the drug safety of selinexor, which is crucial for clinicians to safeguard patients’ health.
Similar content being viewed by others

Introduction
Selinexor is a selective inhibitor of nuclear export (SINE) primarily targeting XPO1 protein1,2. As the first orally administered nuclear export inhibitor around the world, selinexor exerts its anticancer properties by blocking the export of tumor suppressor proteins (TSPs), such as p53, FOXO, RB, etc., from the cell nucleus to cytoplasm3. This behavior maximizes the tumor-suppressing effects of these proteins, which leads to the activation of downstream signaling pathways, ultimately resulting in apoptosis and death of cancer cells. Based on the results of phase 3 clinical trial (BOSTON)4, selinexor was approved by the FDA in July 2019 for use in relapsed or refractory multiple myeloma (RRMM)5. Subsequently, it was further approved for treatment in patients with relapsed or refractory diffuse large B-cell lymphoma (RR-DLBCL) who have received at least two prior therapies, bringing hope for the treatment of hematologic malignancies6,7. As a novel small molecule compound, selinexor has also generated significant interest in the oncology field, offering a potential option for treating various cancers8 with high XPO1 expression. Currently, hundreds of clinical trials are undergoing to further expand the indications of selinexor, including lung cancer9,10, prostate cancer11,12, endometrial cancer13,14, glioblastoma15, and other solid tumors8,16,17, etc.
Just like other pharmaceutical intervention, assessment of selinexor’s safety profile is imperative to ensure patients well-being. Despite its wide therapeutic promise, selinexor’s use has been associated with a range of adverse events (ADEs). Researches on selinexor ADEs indicated that the most common was myelosuppression18, followed by gastrointestinal disorders19,20, and nonspecific side effects19,21. Most descriptions of its ADEs previously rely on clinical trial observations, lacking certain studies based on real-world analysis. Moreover, strict clinical trial inclusion criteria and limited sample size may not fully predict the drug's effects and safety in a broader and more diverse population. Therefore, ADE reports associated with selinexor in actual might extend beyond the previously documented symptoms. In this context, there is an urgent need for more specific researches to assess the safety profiles related to selinexor.
FDA Adverse Event Reporting System (FAERS) is a database for collecting and monitoring adverse reactions on drugs and vaccines, aiding the FDA in assessing the risk–benefit balance of them22, thereby safeguarding public health. Numerous drugs for clinical use have undergone specific analysis via the FAERS database23,24,25, and make majority of patients benefit a lot. The real-world data provided by FAERS can reflect the wider population's experience with medication, offering insights beyond the controlled environment of clinical trials. Additionally, due to the long-term onset, these ADEs that may not be captured in clinical trials could also be identified through prolonged monitoring processes. Consequently, continuous monitoring through FAERS is a key component of post-marketing surveillance, ensuring ongoing evaluation of selinexor's safety as its use in clinical practice evolves.
In summary, selinexor offers a promising therapeutic choice for specific malignancies. Nevertheless, the specific evaluation of its safety use is required. This study aims to investigate selinexor's post-market ADEs through FAERS database, contributing to the evidence of safety profile in clinical.
Methods
Data extraction
All data in this study were sourced from selinexor-related ADE reports collected in the FAERS database. Raw data files were downloaded directly from the FDA official website (https://www.fda.gov). The data extracted for this study span from July 2019 (approved for marketing) to June 2023. The search terms used were “selinexor” and “XPOVIO”. The FAERS database is composed of seven distinct data files, which include information on demographic and administrative (DEMO), drug details (DRUG), reported adverse events (REAC), outcomes for patients (OUTC), sources of reports (RPSR), drug therapy (THER), and indications for drug use (INDI).
Records screened
According to the FDA recommended deduplication reporting method, select the PRIMARY_ID, CASE_ID, and FDA_DT fields of the DEMO table, and sort them in the order of CASE_ID, FDA_DT, and PRIMARY_ID. For reports with the same CASE_ID, keep the one with the largest FDA_DT value. Furtherly, for reports with the same CASE_ID and FDA_DT, keep the one with the largest PRIMARY_ID value.
Data processing
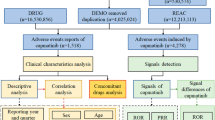
In DEMO data, it documented 7,604,592 of ADE reports associated with selinexor. After excluding duplicates records (n = 1,587,493), the final DEMO was determined (n = 6,017,099). After filtering through the DRUG (n = 34,423), we identified these reports by considering selinexor as the primary suspect drug (PS), while excluding data of secondary suspect (SS) and concomitant (C) drug. Together with REAC (n = 17,894), a total of 4392 selinexor related ADE reports as the PS were ultimately confirmed. The system organ class (SOC) and preferred term (PT) based on the medical dictionary for regulatory activities (MedDRA) facilitated the description, code and classification of the ADEs. Subsequent analysis was conducted as shown in Fig. 1.

Flow chart of the study.
Statistical analysis
The reporting odds ratio (ROR) and proportional reporting ratio (PRR) methods were employed in this study, through a disproportional approach26,27,28. These two methods primarily rely on a four-cell table and calculate the ROR and PRR values using a corresponding formula. A higher numerical value indicates a stronger signal, signifying a stronger association between the target drug and the specific ADE. The calculation formulas and thresholds are presented in Table 1. SAS 9.4 software and GraphPad 8.4 software were employed for database analysis and results statistics.
Results
Annual distribution of selinexor ADE reports
From July 2019 to June 2023, the FDA received a total of 4392 selinexor related ADE reports as the PS. Statistical results indicate an overall upward trend in the number of reports, with specific distributions demonstrated in Fig. 2.

Annual distribution of selinexor ADE reports.
Clinical characteristics distribution of selinexor ADE reports
There were 4392 reports, with detailed distributions of basic clinical characteristics presented in Table 2. The results indicate that a significant portion of patient information was unknown (gender unreported for 69.7%, age unreported for 98%, weight unreported for 87.1%, respectively). Regarding other aspects, the statistics on reporter identification indicate that 62.9% of the reports came from consumers or patients themselves, followed by physicians (22.0%), healthcare professionals (12.0%), pharmacists (1.0%), and non-healthcare professionals (0.8%). Additionally, these received selinexor ADE reports among the countries and regions worldwide, the top five reporting countries were the United States (74.8%), followed by China (4.0%), Canada (3.1%), Israel (2.7%), and Australia (2.6%), respectively (Table 2).
Severe outcomes in selinexor
Among the 4392 selinexor related ADE reports overall, there were 2595 instances of severe outcomes, including hospitalization (55.3%), death (28.9%), life-threatening conditions (2.3%), disability (0.8%), permanent damage (n = 1), and others (12.6%). See Table 3 for details.
Selinexor ADEs involved in SOC
Out of the submitted 4392 reports, a total of 17,483 ADE instances were identified based on different SOC categories. There were predominantly gastrointestinal reactions (n = 3811, 21.80%), followed by general disorders and administration site conditions (n = 3208, 18.35%), investigations (n = 1744, 19.98%) meaning examination relevant abnormalities including various blood tests, biochemical analyses, imaging examinations, etc., as illustrated in Fig. 3.

TOP15 SOCs involved in selinexor ADEs.
Top 15 ADEs Based on PT
Among the 4392 reports, there are 17,894 ADE instances occurrence. We compiled the top 15 ADEs based on PT description, primarily involving gastrointestinal disorders (nausea, anorexia or loss of appetite, diarrhea, vomiting, constipation) for 23.07% (n = 4128), hematologic abnormal investigations (thrombocytopenia, decreased platelet count, anemia) for 6.87% (n = 1229), and other non-specific symptoms (fatigue, lethargy, weight loss, dizziness) for 12.89% (n = 2307), etc. Detailed information is provided in Table 4.
Top ADE signals based on PT
In the analysis of 4392 selinexor-related ADE reports identified as the PS, using both PRR and ROR screening methods, we identified 124 ADE signals. Notably, 10 of the top 15 signals were neither listed in the prescribing information nor reported in the literature. These signals were illustrated as neurofibrosarcoma (ROR, 144.27; 95% Cl, 61.16–340.32), hypercreatinemia (ROR, 113.92; 95% CI, 57.08–227.37), refractory diffuse large B-cell lymphoma (ROR, 107.67; 95% CI, 77.63–149.33), hypercreatininemia (ROR, 74.00; 95% CI, 37.56–145.79), meibomial gland dysfunction (ROR, 37.57; 95% CI, 15.37–91.81), sarcoma (ROR, 28.20; 95% CI, 14.53–54.73), hypogeusia (ROR, 20.69; 95% CI, 11.06–38.71), abnormal kidney function (ROR, 13.75; 95% CI, 8.74–21.63), bone tumors (ROR, 13.08; 95% CI, 4.88–35.10), iridesma (ROR, 11.31; 95% CI, 3.62–35.32), etc. (Table 5).
Discussion
This study utilized the FAERS database to rigorously evaluate ADEs associated with selinexor's clinical use. Notably, we observed an elevated occurrence of events leading to severe outcomes that could not be ignored. This requires the focus of patient health, regulatory authorities as well as pharmaceutical companies. Based on PT description, major ADEs identified included myelosuppression, gastrointestinal disorders, and non-specific general reactions in our study. Regarding as ADEs involved in SOC, given that the administration manner of oral drugs, the top ADEs were generally refer to be gastrointestinal tract (GI), aligning with the premarketing evaluations. However, some inconsistent and unreported ADE signals emerged compared to previous reports, which deserve more attention.
Hypercreatinemia, typically defined as elevated blood creatine level, is usually associated with issues of musculoskeletal, connective tissue, and metabolism. It has not been explicitly reported in public studies on selinexor, indicating that such an ADE should be closely monitored in following clinical practice. Similar with hypercreatininemia, which is a sign of renal function abnormality, both of them reflect an imbalance of substances related to body metabolism. Yet, the causes for themselves are generally diverse. In FAERS analysis, hypercreatinemia and hypercreatininemia are categorized under varied SOC classes, thus the captured ADE signals differed. Regarding as issue of renal dysfunction, it was notably evidenced by 19 instances of abnormal renal function and 9 instances of hypercreatininemia in our results. It’s well-known that injury of renal may be caused by various factors including disease progression, infections, and the use of other certain medications, such as nonsteroidal anti-inflammatory drugs (NSAIDs)29, statins/fibrates lipid-lowering drugs30, certain antibiotics (aminoglycosides)31, antifungal agents (amphotericin B)32, and antipsychotics (olanzapine)33, etc. Nevertheless, our study focused on statistical analysis of ADEs which were primarily suspected to be related to selinexor, there was no need to consider of concomitant medication. We attempted to explore whether the use of selinexor is associated with renal impairment or not, and found there were only minority of studies elucidated previously34. A clinical trial on RR-DLBCL patients found that selinexor's safety and efficacy to be independent of renal function. The post-hoc analysis of the SADAL study indicates that selinexor remained safe and effective even in patients with impaired renal function35. In assessments of MM, Bader’s study suggested the use of selinexor cannot aggravate the side effects on renal injuries34, regardless of the patient's prior renal function. Furthermore, descriptions of renal function impairment in ADE signals involve many abnormal indicators, including electrolyte level abnormalities. For instance, hyponatremia occurrences were reported frequently in previous studies, with an incidence rate about 30%36. In our study, we did not find specific signals linked selinexor ADEs to electrolyte imbalance. Taken together, the direct association of selinexor with renal function abnormality appears insufficient in clinical applications. We're not sure if it is caused by other concomitant medications, and look forward to further studies in the future.
Regarding issues on tumor signals following the clinical application of selinexor, we could not conclude a definite causal relationship. As shown in the results, our study showed that several ADE signals with tumors were involved, such as neurofibrosarcoma, refractory DLBCL, sarcoma, and bone tumors, etc. Because our analysis did not set a specific application scope, it suggests that patients for potential indications might have experienced progression of their own diseases, due to selinexor is currently under clinical trials for numerous hematologic malignancies and solid tumors. For example, Gounder's et al.37 study assessed the pharmacokinetics, pharmacodynamics, safety, and efficacy of selinexor in patients with advanced soft tissue or bone sarcomas. Another study evaluated the safety of selinexor in patients with relapsed or refractory non-Hodgkin lymphoma (NHL), including subtypes of NHL such as diffuse large B-cell lymphoma, Richter transformation, mantle cell lymphoma, follicular lymphoma, and chronic lymphocytic leukemia38. Additionally, Lewin’s study explored the safety and efficacy of selinexor in combination with doxorubicin in treating advanced soft tissue sarcomas (STS), compassing malignant peripheral nerve sheath tumors (n = 3) and other types of sarcomas (n = 16)39. According to these results, selinexor has shown certain effectiveness in treating various solid tumors as a potential indication. Nevertheless, even if selinexor is highly associated with specific tumor signals, it is difficult to conclude that the drug directly caused the event.
Moreover, selinexor may cause a variety of non-specific side effects as a novel targeting anti-tumor drug. Nagham et al.40 retrospectively analyzed the medical records of 174 patients who received at least one dose of selinexor in combination with chemotherapy or immunotherapy drugs. This cohort monitored the occurrence of the ocular ADEs, with blurred vision (12.64%), followed by dry eye syndrome (12.1%), and progression of cataracts (4.0%). However, there were no discontinuations due to ocular ADEs, leading to the conclusion that selinexor-related ocular ADEs are mild. In our study, meibomian gland dysfunction and iridesma were reported for the first time, suggesting that we can offer distinct insights into selinexor related ADE signals. Another related ADE of taste reduction, the possible physiological mechanisms behind this side effect include the drug's direct toxic effect on taste cells or indirect impacts on the oral environment. Nevertheless, this non-specific ADE of selinexor is rarely reported in clinical, possibly because it is temporary which did not cause enough attention.
The discovery of these potential ADEs underscores the need for a deeper investigation into selinexor's safety profile. Firstly, the identification of these unreported ADEs highlights the importance of continuous drug monitoring post-marketing. Secondly, these findings remind us that with the expanding indications and broaden applications in real-world of selinexor, unexpected ADEs might emerge. Although clinical trials provide crucial data on safety and efficacy, they are usually employed in a strictly controlled environment with limited sample sizes, which may not fully represent a broader and more diverse patient population. By recognizing and understanding these new ADEs, interventions can be implemented to prevent the effects, leading to better patient outcomes and life quality.
However, the FAERS analysis also has its certain limitations. Self-reporting bias, the lack of controlled studies, and the complexity of determining causal relationships are inherent flaws in FAERS analysis41. As a spontaneous reporting system, it means the reports come from numerous sources, and over- or under-reporting of specific information may lead to reporting bias. Therefore, the quality, completeness and accuracy of the reports may be limited. For instance, “N/A” in the chart of our study signifies missing data from the up-loader. This lack of basic information may compromise data integrity, but the generation of ADE signals would not be affected due to the massive real-word data. Besides, ADE reports in FAERS could not establish a causal relationship between drug use and ADEs. In FAERS report, there was no control group to compare ADEs between drug users and non-users. Without a control group, it is difficult to determine whether ADEs caused by specific drug or other factors. Therefore, ADE reports in the FAERS should be interpreted cautiously. Although spontaneous report system provides an inadequate level of evidence hierarchy, it serves as a tool for timely and early assessment of safety issues, especially in new compounds. It's well-known that clinical trials are considered the best source of evidence, but they are often limited by stringent criteria and limited sample sizes. Conversely, the analysis of spontaneous reports reflects these conditions where patients experience primary outcomes within a complex real-world context.
In conclusion, our study offers the first real-world insights into oncological ADEs of selinexor, supplementing its post-marketing safety research and providing reference for its clinical use.
Data availability
Database is available on the FAERS official website. The raw/processed data reproduced these findings can be shared necessarily, which required the approval of the corresponding authors.
References
Azmi, A. S., Uddin, M. H. & Mohammad, R. M. The nuclear export protein Xpo1—from biology to targeted therapy. Nat. Rev. Clin. Oncol. 18, 152–169 (2021).
Gravina, G. L. et al. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J. Hematol. Oncol. 7, 85 (2014).
Syed, Y. Y. Selinexor: First global approval. Drugs 79, 1485–1494 (2019).
Grosicki, S. et al. Once-per-week selinexor, bortezomib, and dexamethasone versus twice-per-week bortezomib and dexamethasone in patients with multiple myeloma (Boston): A randomised, open-label, phase 3 trial. Lancet 396, 1563–1573 (2020).
Vogl, D. T. et al. selective inhibition of nuclear export with oral selinexor for treatment of relapsed or refractory multiple myeloma. J. Clin. Oncol. 36, 859–866 (2018).
Kalakonda, N. et al. Selinexor in patients with relapsed or refractory diffuse large B-cell lymphoma (Sadal): A single-arm, multinational, multicentre, open-label, phase 2 trial. Lancet Haematol. 7, e511–e522 (2020).
Cheson, B. D., Nowakowski, G. & Salles, G. Diffuse large B-cell lymphoma: New targets and novel therapies. Blood Cancer J. 11, 68 (2021).
Abdul, R. A. et al. First-in-class, first-in-human phase I study of selinexor, a selective inhibitor of nuclear export, in patients with advanced solid tumors. J. Clin. Oncol. 34, 4142–4150 (2016).
Quintanal-Villalonga, A. et al. Inhibition of XPO1 sensitizes small cell lung cancer to first- and second-line chemotherapy. Cancer Res. 82, 472–483 (2022).
Gupta, A., Saltarski, J. M., White, M. A., Scaglioni, P. P. & Gerber, D. E. Therapeutic targeting of nuclear export inhibition in lung cancer. J. Thorac. Oncol. 12, 1446–1450 (2017).
Quintanal-Villalonga, A. et al. Exportin 1 inhibition prevents neuroendocrine transformation through SOX2 down-regulation in lung and prostate cancers. Sci. Transl. Med. 15, f7006 (2023).
Gravina, G. L. et al. Xpo1/Crm1-selective inhibitors of nuclear export (SINE) reduce tumor spreading and improve overall survival in preclinical models of prostate cancer (Pca). J. Hematol. Oncol. 7, 46 (2014).
Selinexor Delays Wild-Type Tp53 Endometrial Cancer Progression. Cancer Discov. 13, F1 (2023).
Vergote, I. et al. Oral selinexor as maintenance therapy after first-line chemotherapy for advanced or recurrent endometrial cancer. J. Clin. Oncol. 41, 5400–5410 (2023).
Lassman, A. B. et al. A phase II study of the efficacy and safety of oral selinexor in recurrent glioblastoma. Clin. Cancer Res. 28, 452–460 (2022).
Thein, K. Z. et al. Selinexor in combination with standard chemotherapy in patients with advanced or metastatic solid tumors. Exp. Hematol. Oncol. 10, 59 (2021).
Nelson, B. E. et al. Phase 1B study of combined selinexor and eribulin for the treatment of advanced solid tumors and triple-negative breast cancer. Cancer 129, 2201–2213 (2023).
Machlus, K. R. et al. Selinexor-induced thrombocytopenia results from inhibition of thrombopoietin signaling in early megakaryopoiesis. Blood 130, 1132–1143 (2017).
Gounder, M. M. et al. Selinexor in advanced, metastatic dedifferentiated liposarcoma: A multinational, randomized, double-blind, Placebo-Controlled Trial. J. Clin. Oncol. 40, 2479–2490 (2022).
Stephens, D. M. et al. Selinexor combined with ibrutinib demonstrates tolerability and safety in advanced B-cell malignancies: A phase I study. Clin. Cancer Res. 28, 3242–3247 (2022).
Gavriatopoulou, M. et al. Integrated safety profile of selinexor in multiple myeloma: Experience from 437 patients enrolled in clinical trials. Leukemia 34, 2430–2440 (2020).
Yao, X. et al. Mining and visualizing high-order directional drug interaction effects using the faers database. Bmc Med. Inform. Decis. Mak. 20, 50 (2020).
Jing, Y. et al. Association of antibiotic treatment with immune-related adverse events in patients with cancer receiving immunotherapy. J. Immunother. Cancer 10, e003779 (2022).
Krantz, M. J. et al. Ventricular arrhythmias associated with over-the-counter and recreational opioids. J. Am. Coll. Cardiol. 81, 2258–2268 (2023).
Anand, K., Ensor, J., Trachtenberg, B. & Bernicker, E. H. Osimertinib-induced cardiotoxicity: A retrospective review of the Fda Adverse Events Reporting System (FAERS). Jacc-Cardiooncology 1, 172–178 (2019).
Kadoyama, K. et al. Hypersensitivity reactions to anticancer agents: Data mining of the public version of the FDA Adverse Event Reporting System. AERs J. Exp. Clin. Cancer Res. 30, 93 (2011).
Yan, Y. D. et al. Toxicity spectrum of immunotherapy in advanced lung cancer: A safety analysis from clinical trials and a pharmacovigilance system. Eclinicalmedicine. 50, 101535 (2022).
Sakaeda, T., Tamon, A., Kadoyama, K. & Okuno, Y. Data mining of the public version of the FDA Adverse Event Reporting System. Int. J. Med. Sci. 10, 796–803 (2013).
Dunn, M. The role of arachidonic acid metabolites in renal homeostasis. Non-steroidal anti-inflammatory drugs renal function and biochemical, histological and clinical effects and drug interactions. Drugs 33, 56–66 (1987).
Lin, Y. C. et al. Effect of lipid-lowering drugs on renal and cardiovascular outcomes in patients with chronic kidney disease and dyslipidemia: A retrospective cohort study. Clin. Pharmacol. Ther. 114, 1366–1374 (2023).
Andronikou, S., Giapros, V. I., Cholevas, V. I. & Papadopoulou, Z. L. Effect of aminoglycoside therapy on renal function in full-term infants. Pediatr. Nephrol. 10, 766–768 (1996).
Laniado-Laborin, R. & Cabrales-Vargas, M. N. Amphotericin B: Side effects and toxicity. Rev. Iberoam. Micol. 26, 223–227 (2009).
Gardner, D. M., Murphy, A. L., O’Donnell, H., Centorrino, F. & Baldessarini, R. J. International consensus study of antipsychotic dosing. Am. J. Psychiatry 167, 686–693 (2010).
Bader, J. C., Abdul, R. A., Shacham, S. & Xu, H. Pharmacokinetics of selinexor: The first-in-class selective inhibitor of nuclear export. Clin. Pharmacokinet. 60, 957–969 (2021).
Zijlstra, J. M. et al. The association between patient characteristics and the efficacy and safety of selinexor in diffuse large B-cell lymphoma in the SADAL study. Cancers 14, 791 (2022).
Qiu, L. et al. Selinexor plus low-dose dexamethasone in Chinese patients with relapsed/refractory multiple myeloma previously treated with an immunomodulatory agent and a proteasome inhibitor (MARCH): A phase II, Single-Arm Study. Bmc Med. 20, 108 (2022).
Gounder, M. M. et al. Phase Ib study of selinexor, a first-in-class inhibitor of nuclear export, in patients with advanced refractory bone or soft tissue sarcoma. J. Clin. Oncol. 34, 3166–3174 (2016).
Kuruvilla, J. et al. Selective Inhibition of Nuclear Export with Selinexor in Patients with Non-Hodgkin Lymphoma. Blood. 129, 3175–3183 (2017).
Lewin, J. et al. A phase 1B trial of selinexor, a first-in-class selective inhibitor of nuclear export (SINE), in combination with doxorubicin in patients with advanced soft tissue sarcomas (STS). Eur. J. Cancer 144, 360–367 (2021).
Al-Zubidi, N. et al. Overview of ocular side effects of selinexor. Oncologist 26, 619–623 (2021).
Hazell, L. & Shakir, S. A. Under-reporting of adverse drug reactions: A systematic review. Drug Saf. 29, 385–396 (2006).
Funding
This work was partially supported by National Key R&D Program of China (2022YFC2502700); Medical basic-clinical fusion innovation project of the First Affiliated Hospital of Xi'an Jiaotong University (YXJLRH2022008); Institutional Foundation of The First Affiliated Hospital of Xi’an Jiaotong University (2020QN-17).
Author information
Authors and Affiliations
Contributions
Y.L. conceived the study, executed and analyzed the data; R.Y., Y.D., H.F., B.Y. and M.Z. supported with methodology; F.N. and P.H. offered financial support and supervised experiments; Y.L. drafted the manuscript; B.M. and F.N. critically revised the manuscript; F.N. supervised the study. All authors critically reviewed the manuscript and approved the final version for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Liu, Y., Yang, R., Feng, H. et al. Adverse events reporting of XPO1 inhibitor - selinexor: a real-word analysis from FAERS database. Sci Rep 14, 12231 (2024). https://doi.org/10.1038/s41598-024-62852-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-62852-z
- Springer Nature Limited