
Abstract
Shuttling of specific proteins out of the nucleus is essential for the regulation of the cell cycle and proliferation of both normal and malignant tissues. Dysregulation of this fundamental process may affect many other important cellular processes such as tumor growth, inflammatory response, cell cycle, and apoptosis. It is known that XPO1 (Exportin-1/Chromosome Region Maintenance 1/CRM1) is the main mediator of nuclear export in many cell types. Nuclear proteins exported to the cytoplasm by XPO1 include the drug targets topoisomerase IIα (topo IIα) and BCR-ABL and tumor suppressor proteins such as Rb, APC, p53, p21, and p27. XPO1 can mediate cell proliferation through several pathways: (i) the sub-cellular localization of NES-containing oncogenes and tumor suppressor proteins, (ii) the control of the mitotic apparatus and chromosome segregation, and (iii) the maintenance of nuclear and chromosomal structures. The XPO1 protein is elevated in ovarian carcinoma, glioma, osteosarcoma, pancreatic and cervical cancer. There is a growing body of research indicating that XPO1 may have an important role as a prognostic marker in solid tumors. Because of this, nuclear export inhibition through XPO1 is a potential target for therapeutic intervention in many cancers. The best understood XPO1 inhibitors are the small molecule nuclear export inhibitors (NEIs; Leptomycin B and derivatives, ratjadones, PKF050-638, valtrate, ACA, CBS9106, selinexor/KPT-330, and verdinexor/KPT-335). Selinexor and verdinexor are orally bioavailable, highly potent, small molecules that are classified as Selective Inhibitors of Nuclear Export (SINE). KPT-330 is the only NEI currently in Phase I/II human clinical trials in hematological and solid cancers. Of all the potential targets in nuclear cytoplasmic transport, the nuclear export receptor XPO1 remains the best understood and most advanced therapeutic target for the treatment of cancer.
Similar content being viewed by others

Introduction
Export of mRNA and specific proteins from the nucleus is a key step in intracellular signaling and can lead to cell proliferation or apoptosis [1]. Cancer cells utilize the processes of nuclear-cytoplasmic transport through the nuclear pore complex to stimulate tumor growth and to effectively evade apoptotic mechanisms [1]. It is known that XPO1 (Exportin-1/Chromosome Region Maintenance 1/CRM1) is the main mediator of nuclear export in many cell types. XPO1 interacts with nucleoporins (NUP214 and NUP88) in the nuclear pore complex [2] and transports cargo proteins containing nuclear export signals (NES) out of the cell nucleus [3]. NES are short leucine-rich sequences that can be found in many shuttling proteins, including numerous tumor suppressors and oncogenes [3] (see NESdb database; http://www4.utsouthwestern.edu/chooklab/resources.htm). XPO1 protein mediates cell proliferation through several pathways: (i) the sub-cellular localization of NES-containing oncogenes and tumors suppressor proteins, (ii) the control of the mitotic apparatus and chromosome segregation, and (iii) the maintenance of nuclear and chromosomal structures.
The level of XPO1 protein remains constant throughout the cell cycle [4] and is mainly localized to the nuclear envelope in highly specialized cellular bodies called CRM1 nucleolar bodies (CNoBs) [5],[6]. CNoBs depend on RNA polymerase I activity, indicating a role in ribosome biogenesis [7]. Shuttling of specific proteins out of the nucleus is essential for the regulation of cell cycle and proliferation of both normal and malignant tissues [8]-[11]. Examples of nuclear effectors which are exported into the cytoplasm in cancer include the drug targets topoisomerase IIα [12] and BCR-ABL [13] and tumor suppressor proteins such as Rb [14], APC [15], p53 [16], p21 [17], and p27 [18] (reviewed in Table 1). This makes nuclear export inhibition a potential target for therapeutic intervention in cancer [19],[20].
Prognostic role of XPO1 in solid tumors
The XPO1 protein is elevated in ovarian carcinoma, glioma, osteosarcoma, pancreatic, cervical and gastric cancers and may have an important role as prognostic marker in solid and hematologic tumors [11],[21]-[28]. XPO1 protein expression is increased in osteosarcoma when compared to non-tumor tissue [23]. High serum levels of alkaline phosphatase (ALP) are associated with increased expression of XPO1. From a clinical point of view, elevated expression of XPO1 is associated with increased tumor size and negative histological grade. High XPO1 protein expression is correlated with both poor progression-free (PFS) and overall survival (OS) in human osteosarcoma.
In ovarian [21] and cervical cancer [25], increased XPO1 nuclear and cytoplasmic protein expression was observed in malignant tissues when compared to benign lesions. XPO1 protein was also found differentially expressed in borderline tumors with respect to malignant ovarian cancer [21]. These findings suggest a continuum of expression from benign to malignant lesions encompassing more favorable outcomes for cervical cancer patients [25]. Cytoplasmic XPO1 protein expression was correlated with increased mitotic index, more aggressive tumor growth, advanced tumor stage, and poor OS [25]. XPO1 was shown to export COX-2 from the nucleus [29]. Jang and colleagues suggested that elevated expression of the XPO1 protein may cause COX-2 up-regulation [29]. In cervical cancer cell lines, silencing of the XPO1 protein by RNA interference resulted in increased cell death. This effect was found to be correlated with nuclear retention of p53 [16],[29],[30].
In patients suffering from Stage I and II pancreatic cancer, increased XPO1 protein expression was detected in malignant tissues [24]. Serum CEA and CA19.9 levels, two well-known prognostic markers in pancreatic cancer, correlated with increased XPO1 protein expression in human tissues. In addition, tumor size and presence of distant metastasis also correlated with increased levels of XPO1 protein. Therefore, it may be possible to use high XPO1 expression as a clinical parameter for predicting poor PFS and OS in pancreatic cancer. High XPO1 protein expression was significantly associated with high expression of phospho-serine10-p27, but reduced abundance of p27. Increased XPO1 led to increased cytoplasmic localization and degradation of p27, while phospho-serine10-p27 was resistant to XPO1-mediated nuclear export. Considered together, these results provide direct evidence of XPO1 nuclear export of p27 in pancreatic cancer.
A prognostic role of the XPO1 protein was established in gastric cancer (GC) [28]. A higher XPO1 expression rate (57.8%) was found in tissue derived from malignant lesions when compared to adjacent noncancerous tissues (6.7%). XPO1 protein expression was correlated with increased serum level of CEA, more advanced tumor stages, positive Her2 status, and distant metastasis. Using multivariate analysis, it was determined that high XPO1 expression was an independent indicator for GC survival.
Molecular signals associated with XPO1 inhibition
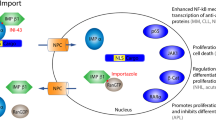
The physical separation of the genome from the cytoplasm by the nuclear envelope (NE) is a hallmark of the eukaryotic cell requiring the transport of macromolecules across the nuclear membrane to mediate their normal functions (Figure 1a). It is known that intracellular localization is deregulated in cancer [10],[11] (Figure 1b). Targeting nucleo-cytoplasmic transport could restore normal localization and function of tumor suppressor and oncoproteins (Figure 1c). The targeting of XPO1 by nuclear export inhibitors (NEI) induces apoptosis in cancer cell lines and slows tumor growth in xenograft mouse models. There are many different mechanisms which achieve this in cancer cells [16]-[18],[31]-[61]. These mechanisms are highlighted in Table 1.

Nucleo-cytoplasmic transport as therapeutic target in cancer. The balance of nuclear transport in (a) normal and (b) cancer cells. (c) Inhibition of nuclear export by NEIs in cancer cells.
Small molecule nuclear export inhibitors (NEIs) and anti-cancer activity
Of all the potential targets in nuclear-cytoplasmic transport, the nuclear export receptor XPO1 remains the most promising therapeutic target. Figure 2 summarizes some of the small molecule nuclear export inhibitors (NEIs) described in this review. Leptomycin B (LMB; Figure 2a) is the first specific NEI discovered [62]. LMB is a small molecule capable of disrupting protein-protein interactions that are typically difficult to target. When therapeutically evaluated in a single Phase I clinical trial in humans, LMB was found to exhibit severe dose-limiting toxicity, resulting in profound anorexia and malaise, which are potentially off-target effects [63]. In attempts to reduce the potential off-target effects of LMB, different derivatives were developed with improved pharmacological properties (Figure 2b) [64]-[72]. Several different natural products as well as semi-synthetic and synthetic compounds were identified, although they have not been tested in humans.

Structures of Nuclear Export Inhibitors (NEIs).
The long LMB lactone polyketide almost fills up the NES groove lengthwise and XPO1 adopts a conformation that is an intermediate space between open and closed conformations [73]. The polyketide of LMB interacts hydrophobically with the same XPO1 groove residues that contact NES sequences. LMB analogs, anguinomycins (Figures 2c and d) display selective cytotoxicity against transformed cells at picomolar concentrations [65]. A truncated anguinomycin analog was still capable of blocking nuclear export [64]. Based on these findings, goniothalamin (Figure 2e) was identified as a nuclear export inhibitor [66]. A medicinal chemistry approach using a modified LMB yielded several semi-synthetic LMB derivatives which maintained the high potency of LMB, but were up to 16-fold less toxic than LMB in vivo[64]. The most potent derivative, KOS-2464 (Figure 2f), showed substantial efficacy in multiple mouse xenograft models, without affecting normal lung fibroblasts [64]. Furthermore, the treatment of several p53 wild-type cell lines with this potent derivative led to the up-regulation and nuclear localization of p53 [64]. These data suggest that toxicity associated with LMB may be linked to off-target effects and provides proof that nuclear export can be inhibited with manageable toxicities in vivo.
Anti-cancer/anti-fungal XPO1 inhibitors have been isolated from myxobacterium Sorangium cellulosum in a soil sample collected in Cala Ratjada (Mallorca, Spain) by Höfle, Reichenbach and others in 1995 [67]-[71]. Ratjadones (Figure 2g and h) have similar chemical structures to LMB and employ an identical molecular mechanism to inhibit XPO1 [67]-[69]. Cells treated with ratjadones manifest a significant increase in the size of their nuclei, further indicating an effective block of nuclear export [68]. Cell-cycle analysis of these cells showed that ratjadone compounds arrest the cells in G1 phase [70]. Synthetic ratjadone analog C (Figure 2h) inhibits nuclear export of topo IIα and sensitizes drug-resistant human multiple myeloma cells to the topo IIα inhibitors doxorubicin and etoposide when used at nanomolar concentrations [72]. Therefore, blocking XPO1 nuclear export may sensitize cancer cells either by preventing export of additional tumor suppressors or by preventing cell cycle progression. To date, ratjadones compounds have not been tested in vivo.
Daelemans et al. identified the synthetic small molecule PKF050-638 (Figure 2i) with a molecular mass of 292.7 kDa that reversibly disrupts XPO1-NES interaction in the micromolar range and demonstrates strict structural requirements for its activity [74]. Structural studies on PKF050-638 indicated XPO1 inhibition and highlighted that the activity of these compounds was not solely correlated to the targeted cysteine in XPO1. This suggests that more structural elements in the NES binding domain are involved [74].
Other natural compounds that bind to Cysteine 528 of XPO1were identified, including valtrate (Figure 2j) and acetoxychavicolacetate (ACA; Figure 2k) isolated from Valeriana fauriei and Alpiniagalangal, respectively [75],[76]. Although valtrate and ACA were developed as anti-viral compounds, they might be useful as anti-cancer agents.
An orally-active synthetic small molecule, CBS9106 (Figure 2l), which reversibly blocks XPO1-mediated nuclear export, is currently being developed as a preclinical anti-cancer agent [77]. Its mechanism of action remains to be fully elucidated. CBS9106 is able to reduce XPO1 protein levels without affecting its mRNA expression. This effect is reversed by adding bortezomib, suggesting that CBS9106-mediated XPO1 inhibition results in proteasome-dependent XPO1 degradation. XPO1 protein degradation mediated by CBS9106 encompasses are large portion of the inhibitory activity of this compound [77],[78]. In cells, CBS9106 caused a reversible arrest of the cell cycle and induced apoptosis in a time- and dose-dependent manner across a broad spectrum of cancer cells. Oral administration of CBS9106 suppressed tumor growth and prolonged survival in myeloma-bearing mice without causing significant weight loss [77]. A reduced level of XPO1 protein was also observed in tumor xenografts isolated from CBS-9106-treated mice. Toxicology studies will need to be completed in order to determine whether this promising pre-clinical candidate could be advanced to human clinical trials.
Selective Inhibitors of Nuclear Export (SINE™)
The use of novel computational methods has recently facilitated the discovery of orally-bioavailable and highly-potent small molecules classified as Selective Inhibitors of Nuclear Export (SINE) [79]. The most advanced molecule in this series, selinexor (KPT-330; Figure 2m) is currently in Phase I/II clinical trials [80]-[89]. X-ray crystal structures of various SINE compounds were elucidated and indicate that they covalently bind to Cysteine 528 of XPO1 in a slowly-reversible and highly-selective manner (KPT-185; Figure 2n) [90],[91]. SINE compounds showed broad activity when tested in in vitro cytotoxicity experiments in hematological and solid tumor cell lines. In these experiments, the IC50 values ranged from 20 to 2000 nM with 95% of the cells tested having IC50 values below 500 nM. There was minimal toxicity to normal cells, indicating that the cytotoxicity of SINE compounds specifically targets malignant cells [90],[91].
SINE compounds were able to overcome the protective micro-environment effects in studies using multiple myeloma and chronic lymphocytic leukemia cell viability assays conducted in the presence of bone marrow stromal cells (BMSC) [90],[92]. The in vivo efficacy of SINE compounds was established in numerous pre-clinical murine xenograft, orthotopic, primagraft, and leukemograft models [26],[61],[90]-[102]. SINE compounds displayed single-agent activity and provided a statistically significant survival advantage in hematological malignancies, specifically in models of non-Hodgkin lymphoma, chronic lymphocytic leukemia, acute myeloid leukemia, acute lymphocytic leukemia, and multiple myeloma [26],[90]-[96]. Selinexor also demonstrated robust single-agent efficacy in solid tumour xenografts including kidney, pancreas, prostate, breast, lung, melanoma, colon, gastric, ovarian, neuroblastoma, and sarcomas [61],[97]-[105]. Marked synergy was observed when selinexor was used with a variety of chemotherapies and targeted therapies including platinum and taxanes [60], topoisomerase I and II inhibitors [53],[106], dexamethasone [87], cytarabine [107], proteasome inhibitors [53], and various tyrosine kinase inhibitors (TKIs) [108].
A structurally-related SINE compound, verdinexor (KPT-335; Figure 2o), is currently being developed for canine lymphoma [109]-[112]. Verdinexor has shown potent cytotoxic activity in canine NHL and melanoma cells when administered 2-3 times a week to companion dogs with spontaneously-occurring B- and T-cell lymphomas. Lymphomas are some of the most common malignancies in companion dogs and the diseases are characterized by rapid progression; dogs may live up to only a few weeks if left untreated [109]. In Phase I/II canine clinical trial in companion dogs with NHL (naïve or relapsed), verdinexor was orally administered at doses of 1.0 – 1.75 mg/kg and was generally very well-tolerated, with anorexia as the most common toxicity indicator [112]. In the Phase II study, verdinexor displayed single-agent activity with an overall objective response rate of 34% (20/58 dogs). This included 19 partial responses and one complete response (in a dog with T-cell lymphoma).
First human Phase I studies of selinexor in advanced hematological and solid malignancies were initiated as of June 2012 (clinicaltrials.gov). Selinexor was orally administered 2-3 times per week in doses between 3 and 85 mg/m2. Selinexor was rapidly absorbed and showed dose-proportional pharmacokinetics with no accumulation. Preliminary results suggest that selinexor is generally well-tolerated, with nausea, anorexia and fatigue being the primary side-effects. Side-effects were mostly Grade I and II in nature, reversible and manageable with supportive care. In line with the pre-clinical animal model results mentioned above, preliminary signals of efficacy were observed particularly with hematological malignancies including AML, CLL, NHL, and MM [80]-[82]. For solid malignancies, selinexor also displayed single-agent activity in prostate [88], ovarian [84],[85], cervical [84], and colorectal [83],[84] cancers and showed prolonged disease control in patients with head and neck cancer [89], as well as sarcoma [86]. Based on the encouraging results from the Phase I studies, several Phase II studies of selinexor have been initiated in patients with AML, GBM, melanoma, prostate, ovarian, cervical, and endometrial malignancies (a complete list of the on-going clinical trials is available at clinicaltrials.gov). Taken together, these results suggest that selinexor can be safely administered for prolonged periods to heavily pre-treated, relapsed and/or refractory patients with manageable side-effect profiles.
Conclusions
Nuclear import and export is a highly-coordinated process involving numerous proteins and large complexes working in concert at the nuclear envelope. This process delicately balances cell growth and death mechanisms in cells. One of the central proteins in nuclear export is XPO1, or exportin-1. XPO1 mediates cell proliferation through several pathways: (i) the subcellular localization of NES cargoes, (ii) the control of the mitotic apparatus and chromosome segregation, and (iii) the maintenance of nuclear and chromosomal structures. Nuclear export through XPO1 is up-regulated in different cancer types and may be used as prognostic indicator. XPO1 dysregulation of intracellular localization of crucial suppressors and oncogenic proteins (p53, Rb, FOXO, p21, IκB, NFκB, and others) contributes to cancer development and progression. Because of this, XPO1 represents a pre-clinical and clinical target under active investigation.
To date, numerous small molecule NEIs have been developed. Selinexor represents the most advanced pharmacological agent currently being evaluated in Phase I/II human clinical trials in hematological and solid cancers. Although interfering with nuclear-cytoplasmic transport machinery could be detrimental to all active cells, SINE compounds have been shown to preferentially suppress or eliminate tumor cells and spare normal cells both in the pre-clinical and clinical setting. Continued evaluation of selinexor will establish the safety of targeting nuclear export through XPO1 and help pave the way for targeting other keystone cellular processes.
References
Tran EJ, King MC, Corbett AH: Macromolecular transport between the nucleus and the cytoplasm: Advances in mechanism and emerging links to disease. Biochim Biophys Acta. 1843, 2014: 2784-2795.
Fornerod M, Ohno M, Yoshida M, Mattaj IW: CRM1 is an export receptor for leucine-rich nuclear export signals. Cell. 1997, 90: 1051-1060.
Fung HY, Chook YM: Atomic basis of CRM1-cargo recognition, release and inhibition. Semin Cancer Biol. 2014, 27: 52-61.
Kudo N, Khochbin S, Nishi K, Kitano K, Yanagida M, Yoshida M, Horinouchi S: Molecular cloning and cell cycle-dependent expression of mammalian CRM1, a protein involved in nuclear export of proteins. J Biol Chem. 1997, 272: 29742-29751.
Huang TT, Kudo N, Yoshida M, Miyamoto S: A nuclear export signal in the N-terminal regulatory domain of IkappaBalpha controls cytoplasmic localization of inactive NF-kappaB/IkappaBalpha complexes. Proc Natl Acad Sci U S A. 2000, 97: 1014-1019.
Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S: Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc Natl Acad Sci U S A. 1999, 96: 9112-9117.
Ernoult-Lange M, Wilczynska A, Harper M, Aigueperse C, Dautry F, Kress M, Weil D: Nucleocytoplasmic traffic of CPEB1 and accumulation in Crm1 nucleolar bodies. Mol Biol Cell. 2009, 20: 176-187.
Gravina GL, Marampon F, Sherris D, Vittorini F, Di Cesare E, Tombolini V, Lenzi A, Jannini EA, Festuccia C: Torc1/Torc2 inhibitor, Palomid 529, enhances radiation response modulating CRM1-mediated survivin function and delaying DNA repair in prostate cancer models. Prostate. 2014, 74: 852-868.
Gravina GL, Marampon F, Muzi P, Mancini A, Piccolella M, Negri-Cesi P, Motta M, Lenzi A, Di Cesare E, Tombolini V, Jannini EA, Festuccia C: PXD101 potentiates hormonal therapy and prevents the onset of castration-resistant phenotype modulating androgen receptor, HSP90, and CRM1 in preclinical models of prostate cancer. Endocr Relat Cancer. 2013, 20: 321-337.
Turner JG, Sullivan DM: CRM1-mediated nuclear export of proteins and drug resistance in cancer. Curr Med Chem. 2008, 15: 2648-2655.
Takeda A, Yaseen NR: Nucleoporins and nucleocytoplasmic transport in hematologic malignancies. Semin Cancer Biol. 2014, 27: 3-10.
Mirski SE, Sparks KE, Friedrich B, Kohler M, Mo YY, Beck WT, Cole SP: Topoisomerase II binds importin alpha isoforms and exportin/CRM1 but does not shuttle between the nucleus and cytoplasm in proliferating cells. Exp Cell Res. 2007, 313: 627-637.
Abraham SA, Holyoake TL: Redirecting traffic using the XPO1 police. Blood. 2013, 122: 2926-2928.
Ohtani N, Brennan P, Gaubatz S, Sanij E, Hertzog P, Wolvetang E, Ghysdael J, Rowe M, Hara E: Epstein-Barr virus LMP1 blocks p16INK4a-RB pathway by promoting nuclear export of E2F4/5. J Cell Biol. 2003, 162: 173-183.
Henderson BR: Nuclear-cytoplasmic shuttling of APC regulates beta-catenin subcellular localization and turnover. Nat Cell Biol. 2000, 2: 653-660.
Santiago A, Li D, Zhao LY, Godsey A, Liao D: p53 SUMOylation promotes its nuclear export by facilitating its release from the nuclear export receptor CRM1. Mol Biol Cell. 2013, 24: 2739-2752.
Alt JR, Gladden AB, Diehl JA: p21 (Cip1) Promotes cyclin D1 nuclear accumulation via direct inhibition of nuclear export. J Biol Chem. 2002, 277: 8517-8523.
Wang Y, Wang Y, Xiang J, Ji F, Deng Y, Tang C, Yang S, Xi Q, Liu R, Di W: Knockdown of CRM1 inhibits the nuclear export of p27(Kip1) phosphorylated at serine 10 and plays a role in the pathogenesis of epithelial ovarian cancer. Cancer Lett. 2014, 343: 6-13.
Yashiroda Y, Yoshida M: Nucleo-cytoplasmic transport of proteins as a target for therapeutic drugs. Curr Med Chem. 2003, 10: 741-748.
Niu M, Wu S, Mao L, Yang Y: CRM1 is a cellular target of curcumin: new insights for the myriad of biological effects of an ancient spice. Traffic. 2013, 14: 1042-1052.
Noske A, Weichert W, Niesporek S, Roske A, Buckendahl AC, Koch I, Sehouli J, Dietel M, Denkert C: Expression of the nuclear export protein chromosomal region maintenance/exportin 1/Xpo1 is a prognostic factor in human ovarian cancer. Cancer. 2008, 112: 1733-1743.
Shen A, Wang Y, Zhao Y, Zou L, Sun L, Cheng C: Expression of CRM1 in human gliomas and its significance in p27 expression and clinical prognosis. Neurosurgery. 2009, 65: 153-159. discussion 159-160
Yao Y, Dong Y, Lin F, Zhao H, Shen Z, Chen P, Sun YJ, Tang LN, Zheng SE: The expression of CRM1 is associated with prognosis in human osteosarcoma. Oncol Rep. 2009, 21: 229-235.
Huang WY, Yue L, Qiu WS, Wang LW, Zhou XH, Sun YJ: Prognostic value of CRM1 in pancreas cancer. Clin Invest Med. 2009, 32: E315-
van der Watt PJ, Maske CP, Hendricks DT, Parker MI, Denny L, Govender D, Birrer MJ, Leaner VD: The Karyopherin proteins, Crm1 and Karyopherin beta1, are overexpressed in cervical cancer and are critical for cancer cell survival and proliferation. Int J Cancer. 2009, 124: 1829-1840.
Schmidt J, Braggio E, Kortuem KM, Egan JB, Zhu YX, Xin CS, Tiedemann RE, Palmer SE, Garbitt VM, McCauley D, Kauffman M, Shacham S, Chesi M, Bergsagel PL, Stewart AK: Genome-wide studies in multiple myeloma identify XPO1/CRM1 as a critical target validated using the selective nuclear export inhibitor KPT-276. Leukemia. 2013, 27: 2357-2365.
Kojima K, Kornblau SM, Ruvolo V, Dilip A, Duvvuri S, Davis RE, Zhang M, Wang Z, Coombes KR, Zhang N, Qiu YH, Burks JK, Kantarjian H, Shacham S, Kauffman M, Andreeff M: Prognostic impact and targeting of CRM1 in acute myeloid leukemia. Blood. 2013, 121: 4166-4174.
Zhou F, Qiu W, Yao R, Xiang J, Sun X, Liu S, Lv J, Yue L: CRM1 is a novel independent prognostic factor for the poor prognosis of gastric carcinomas. Med Oncol. 2013, 30: 726-
Jang BC, Munoz-Najar U, Paik JH, Claffey K, Yoshida M, Hla T: Leptomycin B, an inhibitor of the nuclear export receptor CRM1, inhibits COX-2 expression. J Biol Chem. 2003, 278: 2773-2776.
Lu M, Zak J, Chen S, Sanchez-Pulido L, Severson DT, Endicott J, Ponting CP, Schofield CJ, Lu X: A code for RanGDP binding in ankyrin repeats defines a nuclear import pathway. Cell. 2014, 157: 1130-1145.
Alt JR, Cleveland JL, Hannink M, Diehl JA: Phosphorylation-dependent regulation of cyclin D1 nuclear export and cyclin D1-dependent cellular transformation. Genes Dev. 2000, 14: 3102-3114.
Moll UM, LaQuaglia M, Benard J, Riou G: Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci U S A. 1995, 92: 4407-4411.
Nikolaev AY, Li M, Puskas N, Qin J, Gu W: Parc: a cytoplasmic anchor for p53. Cell. 2003, 112: 29-40.
Yang H, Zhao R, Yang HY, Lee MH: Constitutively active FOXO4 inhibits Akt activity, regulates p27 Kip1 stability, and suppresses HER2-mediated tumorigenicity. Oncogene. 2005, 24: 1924-1935.
Hu MC, Lee DF, Xia W, Golfman LS, Ou-Yang F, Yang JY, Zou Y, Bao S, Hanada N, Saso H, Kobayashi R, Hung MC: IkappaB kinase promotes tumorigenesis through inhibition of forkhead FOXO3a. Cell. 2004, 117: 225-237.
Hilmi C, Larribere L, Deckert M, Rocchi S, Giuliano S, Bille K, Ortonne JP, Ballotti R, Bertolotto C: Involvement of FKHRL1 in melanoma cell survival and death. Pigment Cell Melanoma Res. 2008, 21: 139-146.
Zanella F, Renner O, Garcia B, Callejas S, Dopazo A, Peregrina S, Carnero A, Link W: Human TRIB2 is a repressor of FOXO that contributes to the malignant phenotype of melanoma cells. Oncogene. 2010, 29: 2973-2982.
Segura MF, Hanniford D, Menendez S, Reavie L, Zou X, Alvarez-Diaz S, Zakrzewski J, Blochin E, Rose A, Bogunovic D, Polsky D, Wei J, Lee P, Belitskaya-Levy I, Bhardwaj N, Osman I, Hernando E: Aberrant miR-182 expression promotes melanoma metastasis by repressing FOXO3 and microphthalmia-associated transcription factor. Proc Natl Acad Sci U S A. 2009, 106: 1814-1819.
Cornforth AN, Davis JS, Khanifar E, Nastiuk KL, Krolewski JJ: FOXO3a mediates the androgen-dependent regulation of FLIP and contributes to TRAIL-induced apoptosis of LNCaP cells. Oncogene. 2008, 27: 4422-4433.
Demchenko YN, Kuehl WM: A critical role for the NFkB pathway in multiple myeloma. Oncotarget. 2010, 1: 59-68.
Natarajan M, Nayak BK, Galindo C, Mathur SP, Roldan FN, Meltz ML: Nuclear translocation and DNA-binding activity of NFKB (NF-kappaB) after exposure of human monocytes to pulsed ultra-wideband electromagnetic fields (1 kV/cm) fails to transactivate kappaB-dependent gene expression. Radiat Res. 2006, 165: 645-654.
Li W, Wang H, Kuang CY, Zhu JK, Yu Y, Qin ZX, Liu J, Huang L: An essential role for the Id1/PI3K/Akt/NFkB/survivin signalling pathway in promoting the proliferation of endothelial progenitor cells in vitro. Mol Cell Biochem. 2012, 363: 135-145.
Thompson ME: BRCA1 16 years later: nuclear import and export processes. FEBS J. 2010, 277: 3072-3078.
Fong PC, Boss DS, Yap TA, Tutt A, Wu P, Mergui-Roelvink M, Mortimer P, Swaisland H, Lau A, O’Connor MJ, Ashworth A, Carmichael J, Kaye SB, Schellens JH, de Bono JS: Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. N Engl J Med. 2009, 361: 123-134.
Zhang Y, Carr T, Dimtchev A, Zaer N, Dritschilo A, Jung M: Attenuated DNA damage repair by trichostatin A through BRCA1 suppression. Radiat Res. 2007, 168: 115-124.
Rodriguez JA, Span SW, Ferreira CG, Kruyt FA, Giaccone G: CRM1-mediated nuclear export determines the cytoplasmic localization of the antiapoptotic protein Survivin. Exp Cell Res. 2002, 275: 44-53.
Dohi T, Beltrami E, Wall NR, Plescia J, Altieri DC: Mitochondrial survivin inhibits apoptosis and promotes tumorigenesis. J Clin Invest. 2004, 114: 1117-1127.
Colnaghi R, Connell CM, Barrett RM, Wheatley SP: Separating the anti-apoptotic and mitotic roles of survivin. J Biol Chem. 2006, 281: 33450-33456.
Shintani M, Sangawa A, Yamao N, Kamoshida S: Immunohistochemical expression of nuclear and cytoplasmic survivin in gastrointestinal carcinoma. Int J Clin Exp Pathol. 2013, 6: 2919-2927.
Liu JL, Gao W, Kang QM, Zhang XJ, Yang SG: Prognostic value of survivin in patients with gastric cancer: a systematic review with meta-analysis. PLoS One. 2013, 8: e71930-
Serrano-Lopez J, Serrano J, Figueroa V, Torres-Gomez A, Tabares S, Casano J, Fernandez-Escalada N, Sanchez-Garcia J: Cytoplasmic localization of wild-type survivin is associated with constitutive activation of the PI3K/Akt signaling pathway and represents a favorable prognostic factor in patients with acute myeloid leukemia. Haematologica. 2013, 98: 1877-1885.
Gao J, Azmi AS, Aboukameel A, Kauffman M, Shacham S, Abou-Samra AB, Mohammad RM: Nuclear retention of Fbw7 by specific inhibitors of nuclear export leads to Notch1 degradation in pancreatic cancer. Oncotarget. 2014, 5: 3444-3454.
Turner JG, Dawson J, Emmons MF, Cubitt CL, Kauffman M, Shacham S, Hazlehurst LA, Sullivan DM: CRM1 inhibition sensitizes drug resistant human myeloma cells to topoisomerase II and proteasome inhibitors both in vitro and ex vivo. J Cancer. 2013, 4: 614-625.
Falini B, Nicoletti I, Martelli MF, Mecucci C: Acute myeloid leukemia carrying cytoplasmic/mutated nucleophosmin (NPMc+ AML): biologic and clinical features. Blood. 2007, 109: 874-885.
Nakagawa M, Kameoka Y, Suzuki R: Nucleophosmin in acute myelogenous leukemia. N Engl J Med. 2005, 352: 1819-1820. author reply 1819-1820
Park KS, Han BG, Lee KH, Kim DS, Kim JM, Jeon H, Kim HS, Suh SW, Lee EH, Kim SY, Lee BI: Depletion of nucleophosmin via transglutaminase 2 cross-linking increases drug resistance in cancer cells. Cancer Lett. 2009, 274: 201-207.
Imai T, Adachi S, Nishijo K, Ohgushi M, Okada M, Yasumi T, Watanabe K, Nishikomori R, Nakayama T, Yonehara S, Toguchida J, Nakahata T: FR901228 induces tumor regression associated with induction of Fas ligand and activation of Fas signaling in human osteosarcoma cells. Oncogene. 2003, 22: 9231-9242.
Hofmann A, Blau HM: Death of solid tumor cells induced by Fas ligand expressing primary myoblasts. Somat Cell Mol Genet. 1997, 23: 249-257.
Srivastava M, Leighton X: Diverse effects of ANXA7 and p53 on LNCaP prostate cancer cells are associated with regulation of SGK1 transcription and phosphorylation of the SGK1 target FOXO3A. Biomed Res Int. 2014, 2014: 193635-
Gravina GL, Ricevuto E, Mancini A, Shechter S, Sandanayaka V, Kauffman MG, Shacham S, Festuccia C: Abstract 1841: selective inhibitors of nuclear export (SINE) activate multiple tumor suppressor pathways and kill prostate cancer cells across multiple genotypes in vitro and in vivo. Cancer Res. 2012, 72: 1841-
Gravina GL, Tortoreto M, Mancini A, Addis A, Di Cesare E, Lenzi A, Landesman Y, McCauley D, Kauffman M, Shacham S, Zaffaroni N, Festuccia C: XPO1/CRM1-selective inhibitors of nuclear export (SINE) reduce tumor spreading and improve overall survival in preclinical models of prostate cancer (PCa). J Hematol Oncol. 2014, 7: 46-
Hamamoto T, Seto H, Beppu T: Leptomycins A and B, new antifungal antibiotics. II. Structure elucidation. J Antibiot (Tokyo). 1983, 36: 646-650.
Newlands ES, Rustin GJ, Brampton MH: Phase I trial of elactocin. Br J Cancer. 1996, 74: 648-649.
Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PB, Murli S: Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009, 69: 510-517.
Bonazzi S, Eidam O, Guttinger S, Wach JY, Zemp I, Kutay U, Gademann K: Anguinomycins and derivatives: total syntheses, modeling, and biological evaluation of the inhibition of nucleocytoplasmic transport. J Am Chem Soc. 2010, 132: 1432-1442.
Wach JY, Guttinger S, Kutay U, Gademann K: The cytotoxic styryl lactone goniothalamin is an inhibitor of nucleocytoplasmic transport. Bioorg Med Chem Lett. 2010, 20: 2843-2846.
Kalesse M, Christmann M, Bhatt U, Quitschalle M, Claus E, Saeed A, Burzlaff A, Kasper C, Haustedt LO, Hofer E, Scheper T, Beil W: The chemistry and biology of ratjadone. Chembiochem. 2001, 2: 709-714.
Koster M, Lykke-Andersen S, Elnakady YA, Gerth K, Washausen P, Hofle G, Sasse F, Kjems J, Hauser H: Ratjadones inhibit nuclear export by blocking CRM1/exportin 1. Exp Cell Res. 2003, 286: 321-331.
Cossey KN, Funk RL: Diastereoselective synthesis of 2,3,6-trisubstituted tetrahydropyran-4-ones via Prins cyclizations of enecarbamates: a formal synthesis of (+)-ratjadone A. J Am Chem Soc. 2004, 126: 12216-12217.
Meissner T, Krause E, Vinkemeier U: Ratjadone and leptomycin B block CRM1-dependent nuclear export by identical mechanisms. FEBS Lett. 2004, 576: 27-30.
Burzlaff A, Kalesse M, Kasper C, Scheper T: Multi parameter in vitro testing of ratjadone using flow cytometry. Appl Microbiol Biotechnol. 2003, 62: 174-179.
Turner JG, Marchion DC, Dawson JL, Emmons MF, Hazlehurst LA, Washausen P, Sullivan DM: Human multiple myeloma cells are sensitized to topoisomerase II inhibitors by CRM1 inhibition. Cancer Res. 2009, 69: 6899-6905.
Sun Q, Carrasco YP, Hu Y, Guo X, Mirzaei H, Macmillan J, Chook YM: Nuclear export inhibition through covalent conjugation and hydrolysis of Leptomycin B by CRM1. Proc Natl Acad Sci U S A. 2013, 110: 1303-1308.
Daelemans D, Afonina E, Nilsson J, Werner G, Kjems J, De Clercq E, Pavlakis GN, Vandamme AM: A synthetic HIV-1 Rev inhibitor interfering with the CRM1-mediated nuclear export. Proc Natl Acad Sci U S A. 2002, 99: 14440-14445.
Li X, Chen T, Lin S, Zhao J, Chen P, Ba Q, Guo H, Liu Y, Li J, Chu R, Shan L, Zhang W, Wang H: Valeriana jatamansi constituent IVHD-valtrate as a novel therapeutic agent to human ovarian cancer: in vitro and in vivo activities and mechanisms. Curr Cancer Drug Targets. 2013, 13: 472-483.
Watanabe K, Takatsuki H, Sonoda M, Tamura S, Murakami N, Kobayashi N: Anti-influenza viral effects of novel nuclear export inhibitors from Valerianae Radix and Alpinia galanga. Drug Discov Ther. 2011, 5: 26-31.
Sakakibara K, Saito N, Sato T, Suzuki A, Hasegawa Y, Friedman JM, Kufe DW, Vonhoff DD, Iwami T, Kawabe T: CBS9106 is a novel reversible oral CRM1 inhibitor with CRM1 degrading activity. Blood. 2011, 118: 3922-3931.
Saito N, Sakakibara K, Sato T, Friedman JM, Kufe DW, VonHoff DD, Kawabe T: CBS9106-induced CRM1 degradation is mediated by Cullin Ring Ligase activity and the Neddylation pathway.Mol Cancer Ther 2014.,
Kalid O, Toledo Warshaviak D, Shechter S, Sherman W, Shacham S: Consensus Induced Fit Docking (cIFD): methodology, validation, and application to the discovery of novel Crm1 inhibitors. J Comput Aided Mol Des. 2012, 26: 1217-1228.
Savona M, Garzon R, Brown PN, Yee K, Lancet JE, Gutierrez M, Gabrail N, Mau-Sorensen M, Baz R, Byrd JC, Kuruvilla J, Siegel DS, Shacham S, Rashal T, Yau CY CCRP, McCauley D, Saint-Martin J-R, McCartney J, Landesman Y, Klebanov B, Pond G, Oza AM, Kauffman M, Mirza MR, Stone RM: Phase I trial of selinexor (KPT-330), a first-in-class oral selective inhibitor of nuclear export (SINE) in patients (pts) with advanced acute myelogenous leukemia (AML). Blood. 2013, 122: 1440-
Kuruvilla J, Gutierrez M, Shah BD, Gabrail NY, de Nully Brown P, Stone RM, Garzon R, Savona M, Siegel DS, Baz R, Mau-Sorensen M, Davids MS, Byrd JC, Shacham S, Rashal T, Yau CY, McCauley D, Saint-Martin J-R, McCartney J, Landesman Y, Klebanov B, Pond G, Oza AM, Kauffman M, Mirza MR: Preliminary evidence of anti tumor activity of selinexor (KPT-330) in a phase I trial ofa first-in-class oral selective inhibitor of nuclear export (SINE) in patients (pts) with relapsed / refractory non hodgkin’s lymphoma (NHL) and chronic lymphocytic leukemia (CLL). Blood. 2013, 122: 90-
Chen CI, Gutierrez M, de Nully Brown P, Gabrail N, Baz R, Reece DE, Savona M, Trudel S, Siegel DS, Mau-Sorensen M, Kuruvilla J, Byrd JC, Shacham S, Rashal T, Yau CY CCRP, McCauley D, Saint-Martin J-R, McCartney J, Landesman Y, Klebanov B, Pond G, Oza AM, Kauffman M, Mirza MR: Anti tumor activity of selinexor (KPT-330), a first-in-class oral selective inhibitor of nuclear export (SINE) XPO1/CRM1 antagonist in patients (pts) with relapsed/refractory multiple myeloma (MM) or waldenstrom’s macroglobulinemia (WM). Blood. 2013, 122: 1942-
Mau-Soerensen M, Razak ARA, Mahipal A, Mahaseth H, Gerecitano JF, Shacham S, Yau CYF, Lassen U, Shields AF, McCauley D, Cooksey J, Tan DSP, Rashal T, Shacham E, Landesman Y, Pond G, Oza AM, Kauffman M, Siu L, Mirza MR: Safety and antitumor activity of selinexor (KPT-330), a first-in-class, oral XPO1 selective inhibitor of nuclear export: a phase I study expanded with colon cancer cohort.J Clin Oncol 2014, 32. abstract 482.,
Razak ARA, Soerensen MM, Mahipal A, Shacham S, Yau CYF, Lassen UN, McCauley D, Cooksey J, Tan DSP, Saint-Martin J-R, Landesman Y, Pond G, Oza AM, Kauffman M, Siu LL, Mirza MR: First-in-class, first-in-human phase I trial of KPT-330, a selective inhibitor of nuclear export (SINE) in patients (pts) with advanced solid tumors.J Clin Oncol 2013, 31. abstract 2505.,
Martignetti J, Razak ARA, Chen Y, Gabrail NY, Gerecitano JF, Camacho C, Pereira E, Dottino PR, Shacham S, McCauley D, Rashal T, Saint-Martin J-R, Shacham E, Vincett D, Kauffman M, Mirza MR, Sorensen M: Preclinical and early clinical activity of the oral selective inhibitor of nuclear export (SINE) exportin 1 (XPO1) antagonist KPT-330 (Selinexor) in patients (pts) with platinum-resistant/refractory ovarian cancer (OvCa).J Clin Oncol 2014, 32. abstr 5522.,
Gounder MM, Loong HHF, Nyquist-Schultz K, Baker S, Ustoyev Y, Tanner LR, Shacham S, McCauley D, Rashal T, Saint-Martin J-R, Shacham E, Friedlander S, Tamir S, Marshall T, Yosef L, Kauffman M, Rebello S, Mirza MR, Schwartz GK, Razak ARA: A phase 1b food effect study of the first-in-class, oral, selective inhibitor of nuclear export (SINE) selinexor (KPT-330) in patients (pts) with advanced sarcomas.J Clin Oncol 2014, 32.,
Chen C, Gutierrez M, Brown P, Gabrail M, Baz R, Reece D, Flinn I, Trudel S, Siegel D, Mau-Sorensen M, Kuruvilla J, Carlson R, McCauley D, Shacham E, Saint-Martin JR, McCartney J, Marshall T, Landesman Y, Friedlander S, Pond G, Rebello S, Rashal T, Shacham S, Kauffman M, Mirza M: Anti-tumor activity of selinexor (KPT-330), an oral selective inhibitor of nuclear export (SINE), ± dexamethasone in multiple myeloma preclinical models and translation in patient with multiple myeloma. Haematologica. 2014, 99: 359-
Mahaseth H, Maity SN, Gabrail NY, Mahipal A, Gericitano JF, Shacham S, Rashal T, Klebanov B, Carlson R, Shacham E, Vincent D, Kauffman M, Shields AF, Mirza MR, Araujo JC: Selinexor (KPT-330), an oral selective inhibitor of nucelar export (SINE) shows anti prostate cancer (PRCA) activity preclinically and disease control in patients with chemotherapy refractory, castrate refractory prostate cancer. Ann Oncol. 2014, 25: iv257-
Mahipal A, Gabrail NY, Sukari A, Mahaseth H, Mau-Sorensen M, Shacham S, Saint-Martin J, Friedlander S, Landesman Y, Ellis J, Shacham E, McCartney J, Marshall T, Vincent D, Rashal T, Kauffman M, Mirza MR, Razak ARA: Clinical activity of the oral selective inhibitor of nucelar export (SINE) selinexor (KPT-330) in patients with head and neck squamous cell carcinoma (HN-SCC). Ann Oncol. 2014, 25: iv343-
Lapalombella R, Sun Q, Williams K, Tangeman L, Jha S, Zhong Y, Goettl V, Mahoney E, Berglund C, Gupta S, Farmer A, Mani R, Johnson AJ, Lucas D, Mo X, Daelemans D, Sandanayaka V, Shechter S, McCauley D, Shacham S, Kauffman M, Chook YM, Byrd JC: Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012, 120: 4621-4634.
Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, Mansour MR, Barcelo C, McCauley D, Kauffman M, Shacham S, Christie AL, Kung AL, Rodig SJ, Chook YM, Look AT: Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia. 2013, 27: 66-74.
Tai YT, Landesman Y, Acharya C, Calle Y, Zhong MY, Cea M, Tannenbaum D, Cagnetta A, Reagan M, Munshi AA, Senapedis W, Saint-Martin JR, Kashyap T, Shacham S, Kauffman M, Gu Y, Wu L, Ghobrial I, Zhan F, Kung AL, Schey SA, Richardson P, Munshi NC, Anderson KC: CRM1 inhibition induces tumor cell cytotoxicity and impairs osteoclastogenesis in multiple myeloma: molecular mechanisms and therapeutic implications.Leukemia 2013.,
Etchin J, Sanda T, Mansour MR, Kentsis A, Montero J, Le BT, Christie AL, McCauley D, Rodig SJ, Kauffman M, Shacham S, Stone R, Letai A, Kung AL, Look AT: KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol. 2013, 161: 117-127.
Walker CJ, Oaks JJ, Santhanam R, Neviani P, Harb JG, Ferenchak G, Ellis JJ, Landesman Y, Eisfeld AK, Gabrail NY, Smith CL, Caligiuri MA, Hokland P, Roy DC, Reid A, Milojkovic D, Goldman JM, Apperley J, Garzon R, Marcucci G, Shacham S, Kauffman MG, Perrotti D: Preclinical and clinical efficacy of XPO1/CRM1 inhibition by the karyopherin inhibitor KPT-330 in Ph+ leukemias. Blood. 2013, 122: 3034-3044.
Ranganathan P, Yu X, Na C, Santhanam R, Shacham S, Kauffman M, Walker A, Klisovic R, Blum W, Caligiuri M, Croce CM, Marcucci G, Garzon R: Preclinical activity of a novel CRM1 inhibitor in acute myeloid leukemia. Blood. 2012, 120: 1765-1773.
Azmi AS, Al-Katib A, Aboukameel A, McCauley D, Kauffman M, Shacham S, Mohammad RM: Selective inhibitors of nuclear export for the treatment of non-Hodgkin's lymphomas. Haematologica. 2013, 98: 1098-1106.
Inoue H, Kauffman M, Shacham S, Landesman Y, Yang J, Evans CP, Weiss RH: CRM1 blockade by selective inhibitors of nuclear export attenuates kidney cancer growth. J Urol. 2013, 189: 2317-2326.
Azmi AS, Aboukameel A, Bao B, Sarkar FH, Philip PA, Kauffman M, Shacham S, Mohammad RM: Selective inhibitors of nuclear export block pancreatic cancer cell proliferation and reduce tumor growth in mice. Gastroenterology. 2013, 144: 447-456.
Cheng Y, Holloway MP, Nguyen K, McCauley D, Landesman Y, Kauffman MG, Shacham S, Altura RA: XPO1 (CRM1) inhibition represses STAT3 activation to drive a survivin-dependent oncogenic switch in triple negative breast cancer. Mol Cancer Ther. 2014, 13 (3): 675-686.
Wang S, Han X, Wang J, Yao J, Shi Y: Antitumor effects of a novel chromosome region maintenance 1 (CRM1) inhibitor on non-small cell lung cancer cells in vitro and in mouse tumor xenografts. PLoS One. 2014, 9: e89848-
Salas Fragomeni RA, Chung HW, Landesman Y, Senapedis W, Saint-Martin JR, Tsao H, Flaherty KT, Shacham S, Kauffman M, Cusack JC: CRM1 and BRAF inhibition synergize and induce tumor regression in braf-mutant melanoma. Mol Cancer Ther. 2013, 12: 1171-1179.
Yang J, Bill MA, Young GS, La Perle K, Landesman Y, Shacham S, Kauffman M, Senapedis W, Kashyap T, Saint-Martin JR, Kendra K, Lesinski GB: Novel small molecule XPO1/CRM1 inhibitors induce nuclear accumulation of TP53, phosphorylated MAPK and apoptosis in human melanoma cells. PLoS One. 2014, 9: e102983-
Miyake TM, Pradeep S, Zand B, Dalton HJ, Wen Y, Armaitz Pena GN, Kauffman M, McCauley D, Shacham S, Wiktorowicz JE, Sood AK: Abstract 5541: therapeutic targeting of CRM1 in ovarian cancer. Cancer Res. 2013, 73: 5541-
Attiyeh EF, McKeon-Fish A, Trillo R, Landesman Y, Senapedis W, McCauley D, Kashyap T, Shacham S, Kauffman M, Maris JM: Abstract B36: inhibition of exportin 1 (XPO1) potently suppresses growth of human neuroblastoma cell lines. Cancer Res. 2014, 74: B36-
Nair JS, Tap W, Vasudeva SD, Sicklick JK, Kauffman M, Shacham S, Schwartz GK: Abstract 5210: KPT-330, a selective small molecule inhibitor of nuclear export, is active in bone and soft tissue sarcoma. Cancer Res. 2013, 73: 5210-
Yu X, Ranganathan P, Bhatnagar B, Hofstetter J, Shacham S, Kauffman M, Blum W, Garzon R: XPO1 inhibition using selinexor restores topoisomerase IIA (TOPO IIΑ) localization to the nucleus and sensitize primary refractory and relapsed acute myeloid leukemia (AML) blasts to chemotherapy. Haematologica. 2014, 99: 292-293.
Rettig MP, Holt M, Prior J, Shacham S, Kauffman M, McCauley D, Dipersio JF: Inhibition of nuclear transport modulator Xpo1/CRM1 for the treatment of acute myeloid leukemia (AML). Blood. 2013, 122: 237-
Carlson R, Senapedis W, Landesman Y, Shacham S: The combination of selinexor (KPT-330), a selective inhibitor of nuclear export (SINE), and FLT3 inhibitor quizartinib shows anti-tumor activity in acute myeloid leukemia (AML) in vitro and in vivo. Haematologica. 2014, 99: 290-291.
Marconato L, Gelain ME, Comazzi S: The dog as a possible animal model for human non-Hodgkin lymphoma: a review. Hematol Oncol. 2013, 31: 1-9.
Marconato L: The staging and treatment of multicentric high-grade lymphoma in dogs: a review of recent developments and future prospects. Vet J. 2011, 188: 34-38.
London CA, Bernabe LF, Barnard S, Kisseberth WC, Borgatti A, Henson M, Wilson H, Jensen K, Ito D, Modiano JF, Bear MD, Pennell ML, Saint-Martin JR, McCauley D, Kauffman M, Shacham S: Preclinical evaluation of the novel, orally bioavailable selective inhibitor of nuclear export (SINE) KPT-335 in spontaneous canine cancer: results of a phase i study. PLoS One. 2014, 9: e87585-
London CA, Bernabe LF, Barnard S, Kisseberth W, Borgatti A, Henson M, Wilson-Robles H, Jensen K, Ito D, Modiano J, Bear M, Pennell M, Saint-Martin J-R, McCauley D, Kauffman M, Shacham S: Abstract 3809: evaluation of the novel, orally bioavailable selective inhibitor of nuclear export (SINE) KPT-335 (verdinexor) in spontaneous canine cancer: results of phase I and phase II clinical trials. Cancer Res. 2014, 74: 3809-
Author information
Authors and Affiliations
Corresponding author
Additional information
Competing interests
Dilara McCauley, William Senapedis, Erkan Baloglu and Sharon Shacham are employees of, and hold equity in, Karyopharm Therapeutics, Inc., Newton, MA, USA. The other authors disclosed no potential conflicts of interest.
Authors’ contributions
GLG, DMC, WS and EB participated in the design of this review, performed the selection of literature and interpretation of data, and drafted the manuscript. SS and CF participated in the critical revision of this review. All authors read and approved the final manuscript.
Authors’ original submitted files for images
Below are the links to the authors’ original submitted files for images.
Rights and permissions
This article is published under an open access license. Please check the 'Copyright Information' section either on this page or in the PDF for details of this license and what re-use is permitted. If your intended use exceeds what is permitted by the license or if you are unable to locate the licence and re-use information, please contact the Rights and Permissions team.
About this article

{kind=link}
{kind=link}
Cite this article
Gravina, G.L., Senapedis, W., McCauley, D. et al. Nucleo-cytoplasmic transport as a therapeutic target of cancer. J Hematol Oncol 7, 85 (2014). https://doi.org/10.1186/s13045-014-0085-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s13045-014-0085-1