
Abstract
The loss of biodiversity in marine populations is one of the consequences of the increased events of extreme environmental conditions in the oceans, which can condition the persistence of populations to future scenarios of climate change. Therefore, it is extremely necessary to explore and monitor the genetic diversity of natural populations. In the Southeast Pacific Ocean (SEPO), specifically on the coast of Chile, the presence of the copepod Acartia tonsa has been indicated solely using morphological evidence, due to the absence of genetic information. In the present work, the genetic diversity, population structure and phylogenetic position within the genus Acartia, of populations identified morphologically as A. tonsa, was evaluated by amplification of the mitochondrial cytochrome c oxidase subunit I and nuclear marker 18 s. Our results showed that the populations identified as A. tonsa correspond to a new monophyletic group endemic to SEPO (GMYC = 1.00; PTP = 0.95). The populations showed moderate to high genetic diversity with an incipient structuring between populations and biogeographic zones. Our results suggest that despite the homogenizing effect of the Humboldt Current, isolation by distance and contrasting environmental conditions at different geographic scales have an important influence on the genetic diversity of zooplankton in the SEPO region.
Similar content being viewed by others


Introduction
A comprehensive understanding of biological diversity, including its individual components, has become a paramount objective within the scientific community studying marine ecosystems. This heightened emphasis from the escalating environmental alterations induced by climate change and exacerbated human activities during the Anthropocene, are leading to a deterioration of biodiversity1,2,3. Given that oceans serve as primary sinks for greenhouse gasses and atmospheric heat, resulting in ongoing ocean acidification and warming4,5, it is reasonable to anticipate that the primary indicators of biodiversity loss will become evident in marine ecosystems6,7,8.
The species distribution and the genetic diversity that populations harbor can serve as indicators of the natural population’s capacity to adapt to environmental variations and extreme conditions in their habitats9,10. Current changes in oceanic conditions have influenced the distribution of numerous marine species, with many either shifting or expanding their ranges searching suitable environments for development11,12. In practical terms, distributional changes can either enhance or restrict the connectivity between populations, potentially reshaping the current and future diversity of natural populations13,14,15. In this context, a recurrent assessment of genetic diversity within natural populations is needed, with a specific focus on endemic and cosmopolitan species.
In marine environments, owing to lower apparent barriers to dispersal, species with high dispersal potential are more likely to display reduced genetic diversity and population structuring, possibly even less than what is observed in terrestrial ecosystems16,17,18. Nevertheless, it has been observed that species with varying degrees of vagility, including copepods19,20, nudibranchs21, shrimps22, and fishes23, exhibit substantial genetic diversity and population structure, often with molecular divergence reaching interspecific levels. Molecular techniques have revealed instances in which species previously believed to have a broad distribution actually consist of multiple species that cannot be distinguished morphologically and harbor a completely unknown cryptic diversity20,24,25.
While it reaffirms the fact that diversity in the marine environment is still underestimated, it also poses a problem because accurate estimations of each component of biodiversity begins with biological classification and species identification. Many of the cosmopolitan species inhabiting the Pacific Ocean were initially described based on populations from the Atlantic Ocean, Indian Ocean, or the Mediterranean Sea, due to the late discovery and description of the biota of the Pacific Ocean and the accidental transport of many species during the maritime trade voyages of the Silk Road (West Pacific coast) and trade with European colonies in the New World (East Pacific coast; see details in26). Consequently, the Pacific Ocean provides an ideal scenario for studying cryptic biodiversity and population divergence.
The calanoid copepod Acartia tonsa Dana, 1849, is classified as a cosmopolitan species, distributed along the coasts of the southeastern Pacific Ocean (SEPO) and commonly inhabiting estuaries and coastlines along the Indian Ocean, Pacific Ocean, Atlantic Ocean, as well as the Mediterranean and Baltic Seas27,28. The Acartia ability to tolerate a broad range of salinity (0.3–36.9 psu) and temperature (− 1 to 32 °C)29,30 enables this species to achieve a wide distribution. Consequently, its ecology and evolution has been extensively studied31,32,33,34. At the central and southern coast of SEPO, the population density of A. tonsa depends mainly on food availability (mainly microplankton), exhibiting a marked seasonality in abundance on the central and southernmost coast of SEPO35,36. A. tonsa is usually the dominant copepod species in embayment’s, estuaries, fjords, sounds, and inner channel systems, acting as an important link between primary producers and higher trophic levels37.
Phylogeographic and phylogenetic studies conducted on A. tonsa along the western coast of the Atlantic Ocean have revealed that populations from both the northern and southern hemispheres exhibit profound molecular divergences, suggesting the presence of a complex of cryptic species in sympatry that have developed prezygotic mechanisms of reproductive isolation38,39,40,41,42,43,44. The broad thermal and salinity tolerances exhibited by several A. tonsa lineages may contribute to differences in their habitat utilization patterns, resulting in niche partitioning39. On a larger geographic scale, the genetic diversity of A. tonsa is strongly influenced by ocean currents and coastal geography44. However, the complexity of recognizing diagnostic characters among congeneric species of the genus Acartia has led to confusion in delineating lineages within A. tonsa populations along the Brazilian coast41,42,45. To date, the presence of A. tonsa in SEPO coastal systems has been identified solely based on morphological characteristics, without molecular confirmation. In this context, it is crucial to emphasize that despite multiple studies on the distribution, physiology, and life history of populations along the coasts of Chile, there are no molecular sequences of A. tonsa available for the SEPO34,46,47,48,49. The SEPO, specifically the coastal domain along the Chilean coast (18–56° S), provides a unique natural setting for studying the population dynamics of planktonic coastal species. The Humboldt Current System (18–42° S), in combination with the coastal upwelling (from the equator to 40° S), the increasing complexity of the coastal geography towards the southernmost localities (fjords and canals of Patagonia) and the contribution of fresh water from rivers and glaciers, which is highest in the southernmost places (south of 40° S), creates favorable scenarios for the emergence of ruptures of population connectivity and local adaptation to specific environmental conditions50,51,52. Many coastal species exhibit phylogenetic breaks along the SEPO associated with biogeographic discontinuities, especially those reported between 30 and 32° S (Acanthina monodon, Scurria scurra, Tegula atra, and Orchestoidea tuberculata in16; Pyura chilensis in53), which coincides with the southern limit of the Peruvian province54. On the other hand, around 42° S, a divergence occurs in the South Pacific Current, leading to the formation of the Humboldt Current to the north and the Cape Horn Current to the south. This natural barrier contributes to the isolation of marine fauna55, while other marine species with higher dispersal capacity show no population structure at a macrogeographic scale (Concholepas concholepas in56; Pleuroncondes monodon, Emerita analoga, and Petrolisthes violaceus in16). Therefore, it is intriguing to investigate whether various discontinuities in oceanographic conditions, geographical distances, and historical events, such as the last maximum glacial period57,58, have played a role in shaping the genetic diversity of this widely distributed copepod species in this ocean region.
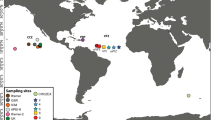
In this study, we investigated the genetic diversity, population structure, and phylogenetic position of morphologically identified populations of A. tonsa in six locations spanning over 3000 km, representing three biogeographic zones: Peruvian Province, Intermediate Area, and Magellanic Province54. These zones are strongly characterized by distinct water masses separated by large current systems, the presence of coastal upwelling, diverse seasonality, and freshwater input from rivers and glaciers (Fig. 1). Our study, for the first time, analyzed genetic sequences of populations identified as A. tonsa along the SEPO, revealing the existence of a previously undiscovered cryptic species within the genus Acartia.

Localities studied along the coast of Chile (SEPO 23–56° S), which are influenced by the Humboldt current system and the Cape Horn current. In this area there are three biogeographic areas, the Peruvian Province where there is a permanent upwelling, the Intermediate Area (transition zone) where the influence of upwelling is seasonal and the Magellanic Province where the influence of fresh water from the fjords predominates.
Results
mtCOI marker
A total of 98 mtCOI sequences were obtained from the studied locations (GenBank Accession Number: OR759115–OR759134), which were manually reviewed and edited. These sequences were aligned, resulting in an alignment of 445 base pairs (bp) in length with 54 polymorphic sites. Overall, the populations exhibited moderate nucleotide diversity (0.017 ± 0.001) and haplotypic diversity (0.756 ± 0.042), with a total of 21 haplotypes. In particular, a high diversity was found in the Intermediate area, localities of Concepcion and Valdivia, with the presence of 10 haplotypes in each one, of which 7 and 5 were private haplotypes (Table 1, Fig. 2). The most common haplotype was present in the three biogeographic zones studied, except for the populations of Concepcion and Magallanes (see details in Fig. 2). Regarding to geographical patterns in the haplotype network, no evident pattern of phylogeographic structuring was observed visually, and the analysis of AMOVA did not identify genetic differences between the biogeographic zones (Фct: 0.289, p value: 0.115), primarily due to a significative genetic variability within (Фst: 0.476, p value: 0.001) and among populations (Фsc: 0.264, p value: 0.003), with 52 and 19% of genetic variation, respectively (Table 2). The pairwise analysis of Фst showed that the populations of the Intermediate area (Concepcion and Valdivia) did not differ from each other, but both exhibited significant differences from the populations present in both biogeographic provinces (Suppl. Table S1). Interestingly, the populations of Antofagasta and Tongoy, present in Peruvian province, do not show significant differences with those of Hualaihue; located 1300 km south at the Magellan province, while Magellan population shows a high differentiation respect to other populations (Suppl. Table S1). Despite this, linearized genetic differentiation according to the geographical distance between populations, showed a positive and significant linear relationship (see details in Suppl. Fig. 1).

Haplotype networks constructed with minimum spanning criteria for COI and 18 s sequences obtained from the studied populations. Haplotypes are represented with a circle; whose size is proportional to its frequency and colors represent its presence in each locality studied. Lines between haplotypes represent mutational steps and black dots represent possible not sampled haplotypes.
Nuclear marker 18 s
For the nuclear marker 18 s, 38 sequences were obtained (GenBank Accession Number: OR771437–OR771462), which were used to create an alignment of 1736 bp, and 161 polymorphic sites. Sequences of the 18 s marker could not be obtained from Magallanes individuals due to the high degradation of their DNA. The populations exhibited a moderate nucleotide diversity (0.011 ± 0.002) and a high haplotype diversity (0.952 ± 0.026), with 28 haplotypes (Table 1). In particular, the Hualaihue population from Magellanic Province, showed a higher number of haplotypes (10), all of which were private haplotypes (see more details in Table 1). The most common haplotype was found only in localities from the Intermediate area, populations of Concepcion and Valdivia, indicating a high presence of unique haplotypes for this nuclear marker in the studied populations (Fig. 2). Regarding the topology of the haplotype network, it was observed that the most common haplotype was centrally located, similar to the star-like pattern, which gives rise to the remaining haplotypes, entirely private. The AMOVA analysis revealed a significant variation within populations, with a 75% of genetic variation (Фst: 0.24, p value: 0.001), and a significative differentiation among biogeographic zones with a 25% of variation (Фct: 0.291, p value: 0.001, see details in Table 2). The pairwise analysis of Фst showed that the main differences were due to the fact that the population of Hualaihue, located in the Magellanic province, differed significantly from the rest of populations; similarity, the populations of Intermediate area, Concepcion and Valdivia did not differ from each other, but showed significant differences with respect to populations present in both biogeographic provinces (see in Suppl. Table S1). Linear regression with linearized genetic differentiation with respect to geographical distance between populations, also showed a positive and significant linear relationship (Suppl. Fig. S1).
Phylogenetic position and species delimitation
Phylogenetic reconstructions by ML and BI were performed with 98 COI sequences previously obtained, plus 59 sequences (157 sequences in total) from species of the genus Acartia available online from GenBank (National Center for Biotechnology Information). Both inferences produced similar tree topologies between themselves and both coincided in recovering the presence of a monophyletic clade, with high bootstrap values (MLb) and posterior probability (Bpp), which grouped all the individuals of the populations studied (Fig. 3). Inside, there are four clades (A–D) that are highly related, where clade A contains individuals of Antofagasta, Tongoy, and Valdivia, the clade B mainly includes individuals of Intermediate area, Concepcion and Valdivia plus some individuals from the other biogeographical provinces. The clade C included only individuals of Magellan, and the clade D, with the largest number of individuals, brings together Hualaihue individuals as well as individuals from Antofagasta, Tongoy, and Valdivia (Fig. 3). The genetic divergence (gd) between these clades, calculated as the sum of the branch lengths of the Bayesian tree, were small and ranged from 0.0147 between the B-C clades to 0.0899 between the A-D clades (Table 3).

Bayesian phylogeny performed with 157 COI sequences of species of genus Acartia sp., including 98 sequences obtained from the populations under study (Antofasta, Tongoy, Concepcion, Valdivia, Hualaihue and Magallanes). Maximum likelihood/Bayesian posterior probability support values were labeled in each node. Putative lineages among populations from Chilean coasts were identified with A-D letters.
The phylogenetic reconstruction revealed with high support (MLb and Bpp) that the studied papulations are a sister clade (gd = 0.232) of a set of species, highly related between them, designated as Acartia arbruta, Acartia californiensis and Acartia sp., all collected from the North Pacific and whose specific identity is subject to discussion (see below). Among the following more closely related Acartia species are A. jilletti (gd = 0.311), A. clausii (gd = 0.366), A. margalefi (gd = 0.449), A. discaudata (gd = 0.413) and A. tonsa (gd = 0.334; more details in Table 3 and Fig. 3). Additionally, a phylogenetic reconstruction was performed by ML and BI with 58 sequences of the 18 s nuclear marker, 38 sequences from the populations studied, and 20 sequences from species of the genus Acartia available online from GenBank (National Center for Biotechnology Information). The phylogenetic reconstructions showed the studied populations as a monophyletic clade and sister to Acartia tonsa, with A. hongi, A. omorii and A. clausii being the next most closely related species (Suppl. Fig. S2). Due to the limited availability of 18S nuclear marker sequences for species of the genus Acartia and the fact that mitochondrial markers are mainly used in the literature, the analysis of interspecific genetic divergence and species delimitation will be performed using only the COI mitochondrial marker.
In terms of species delimitation, the two analytical approaches applied, Generalized Mixed Yule Coalecent (GMYC) and Poisson Tree Process (PTP) with 157 COI sequences used in phylogenetic reconstruction support with a high probability that the differentiation of the monophyletic clade with the populations of the coasts of Chile reaches the species level (GMYC = 1.00, PTP = 0.95) with respect to other species included in phylogeny.
Discussion
The recognition of diagnostic traits is the first step in species identification and it is often the only one, because it is assumed that diversification is accompanied by an evident phenotypic change. The lack of capacity to differentiate morphologically similar species is a problem that has become more recurrent and relevant in taxonomy and manifested in the present study, because cryptic species are more common than expected59,60.
Our research showed that along the Chilean coast (SEPO region between 23° and 53° S), the use of key traits for the identification of the copepod A. tonsa identified individuals who formed a monophyletic group distinct from any species of the genus Acartia, which had not been previously reported and which seems to be endemic to the coasts of the Southeastern Pacific Ocean (SEPO). This is not the first time that the morphological traits of A. tonsa cryptically harbor the presence of other species, either previously described41,42,45 or not previously discovered61, as is well known for the cryptic species complex within A. tonsa in the NW Atlantic Ocean39,40. Recently, a study incorporating genetic data, morphology and phenology, identified the presence of a new cryptic species, designated as Acartia arbruta, which had previously been morphologically confused with A. tonsa in the Northeast Pacific Ocean61. This new species, both in the work in which it was described61 and in our phylogenetic analyses, was positioned in a clade in which the specific identity of its members was not well defined and indicated as Acartia sp. (see details in44). This background is relevant to understand the diversification of the genus Acartia in the Pacific Ocean, because the clade of A. arbruta was composed entirely of OTU (Operative Taxonomic Unities) collected from the Northeast Pacific Ocean, which was the sister clade of the populations included in our study, which so far are the only representatives of the SEPO. The genus Acartia is comprised of 65 recognized species62,63, of which only a dozen species belonging to the subgenera Acanthacartia, Acartia, and Odontacartia are present in the Pacific Ocean. However, recent discoveries of A. arbruta61, A. (Odontacartia) edentata63, Acartia (O.) nadiensis64, and the new monophyletic group reported by us in this study showed that the diversity of the genus Acartia in the Pacific Ocean is still underestimated, mainly due to confusion with species previously described in other oceans. On the other hand, our results and the presence of new endemic species in the Pacific Ocean for the genus Acartia61,63,64 suggest a wide diversification of the genus along the SEPO that needs to be studied.
In our phylogenetic analysis, the populations studied here together with the species A. arbruta and A. jilletti formed a monophyletic clade of endemic species to the Pacific Ocean, whereas species A. nadiensis, described from Fiji coast, was most closely related to A. southwelli, a species endemic to the Indian Ocean, and other Acartia species with a broader distribution (Fig. 3). Therefore, in view of this limited evidence, we could propose that the arrival of the genus Acartia to the Pacific Ocean has occurred on at least two occasions: an older one, from which diversified the species that inhabit the Indo-Pacific and Northwest Pacific Ocean, and another more recent event, from which diversified species inhabiting New Zealand coasts, Northeast and Southeast Pacific Ocean. It is necessary to consider that the experimental design of our research was not developed to test the diversification of the genus Acartia, so it should be appropriately studied.
Phylogenetic reconstructions using the COI marker revealed the presence of four closely related mitochondrial lineages (A–D, Fig. 3) within the studied populations, which were distributed across biogeographic areas, but were not present in all populations studied. In this sense, in all biogeographic areas, three of the four mitochondrial lineages were found, as well as in the populations of Antofagasta, Tongoy, and Valdivia we found that three of the four mitochondrial lineages were in sympatry. In contrast, in the populations of Concepcion and Magallanes, only one mitochondrial lineage was found. It is possible that the presence of a single endemic lineage in Magallanes population was produced by isolation by distance from the other populations studied, but the case of the population of Concepcion was different, because it is located between populations that showed three of the four lineages. In addition, lineage D, which was the most abundant and was present from Hualaihue to northern Chile (Antofagasta), but was not present in Concepcion, which could be due to special environmental conditions in the area. Although our study represents the first assessment of genetic diversity within the Concepcion and Magallanes populations, more sampling and research efforts are needed to confirm the existence of a single mitochondrial lineage. In large and panmictic populations, a small number of haplotypes are expected to dominate in frequency and the vast majority of genetic diversity is expected to be captured with small sample sizes, as observed in the haplotype accumulation curves. However, the use of DNA barcoding has shown that the genetic diversity of marine populations is underestimated, especially in populations that are difficult to identify and sample65,66,67. Although increasing the sampling effort is not the definitive solution, because ecological characteristics of the species studied, such as the reproductive strategy, mitochondrial inheritance mode or spatial and temporal distribution and abundance of haplotypes are capable of influencing diversity estimates and the required sampling effort67,68,69,70,71. We had the possibility of partially exploring the temporal variability of the Concepcion population, with the aim of finding previously undetected mitochondrial haplotypes, but it was interesting that although the population of Concepcion was sampled on two occasions, more than 10 years apart (see Suppl. Table S2), lineages A and D were not found. The environmental variability in Concepcion appears to be influenced by seasonal upwelling, the input of fresh water and organic matter from rivers72,73, a wide continental shelf and the presence of frontal structures acting as plankton retention and accumulation zone74. These environmental factors may exert selective pressures that favors the dominance of the most common lineage within the Intermediate Area (lineage B, Fig. 3). In populations of Acartia tonsa that inhabit the northwest Atlantic Ocean, it has been observed that in common garden conditions, the traits of thermal tolerance and body size present seasonal variation that was probably product of a genetic differentiation mediated by drastic seasonality in environmental conditions75. Previous studies conducted within the same A. tonsa populations had revealed that salinity exerts a significant influence on population genetics, leading to ecological divergence and niche partitioning among mitochondrial lineages39. This demonstrates that various environmental conditions in the localities of origin, including seasonal variability, can exert selection pressure on the genetic polymorphism abundance in planktonic organisms, even at reduced geographical scales.
For decades, the presence of A. tonsa has been documented along the coast of Chile, with numerous ecological studies conducted on these populations34,46,47,48,49,76. In these studies, it was expected that A. tonsa populations from the Chilean coast would exhibit a range of physiological and life history traits characteristic of cosmopolitan species, such as A. tonsa found in other oceans and seas worldwide. These traits include a broad tolerance to environmental fluctuations and the production of large cohorts27,77,78. Ecology and physiology studies have shown that populations previously identified as A. tonsa off the coast of Chile, including populations studied here, used mechanisms of tolerance and plasticity to face extreme environmental conditions34,49, and populations of different biogeographical areas showed signs of adaptation to local conditions of their habitat34. When we analyzed the genetic variation of both mitochondrial and nuclear markers between populations and biogeographic areas, we observed high genetic variability within populations, which was consistent with the levels of genetic diversity (Table 1). However, when the genetic variance was partitioned between populations, we observed that only the mitochondrial marker showed a significant effect, whereas the nuclear marker showed a significant differentiation between biogeographic areas. This could be due to the considerable presence of private haplotypes, and that the 18 s marker showed that the Peruvian, Intermediate and Magallanes Provinces did not share haplotypes between them (Fig. 2). On the other hand, although a series of privates’ mitochondrial haplotypes were observed, the most abundant haplotypes were shared and present in more than one biogeographic area, even was possible to find the same haplotype (the most common) in all biogeographic zones (Fig. 2). In general, in the Humboldt current system (SEPO region) it has been observed that species with a prolonged larval phase showed a high genetic diversity associated with a low structuring of their populations, with highly diffused haplotypes capable of tolerating changes in environmental conditions and being present in more than one biogeographic area16,56.
The populations studied were composed of some haplotypes and lineages, mainly mitochondrial, capable of being present in different localities, overcoming biogeographic breaks, and their contrasting environmental conditions. However, when we compared the most contrasting populations, we observed that both markers (nuclear and mitochondrial) showed that the localities of Concepcion and Magellan showed the highest significant differences in Фst with respect to the other localities. In this sense, the nucleotide differences between populations were significantly greater with increasing distance between locations (Suppl. Fig. S1), suggesting that isolation by geographic distance played a role in the differentiation between the populations studied, as expected for the Magallanes population (isolated 1200 km from the nearest locality, Hualaihue). It is important to consider that the low number of samples from Magallanes population can influence phylogenetic relationships with other populations and population structure patterns, however a clear pattern of isolation by distance was previously reported for the gastropod Concholepas concholepas79, which is endemic to the coast of Chile, with a larval phase duration of four months in South-central Chile80 and that can reach 12 months in Patagonia81. In addition, the presence of significant differences between populations and biogeographic areas, similar to what was found in our study (Table 2 and Suppl. Table S1), suggests that environmental heterogeneity hinders connectivity among populations79.
In conclusion, we report the presence of a new species, molecularly recognized for the genus Acartia that is endemic to SEPO, specifically from the coasts of Chile, and that so far is morphologically indistinguishable from Acartia tonsa. The populations of this new species are more closely related phylogenetically to other endemic species of genus Acartia to the Pacific Ocean than to Acartia tonsa. Additionally, our results suggest that although connectivity between the populations studied here is possible mainly due to the action of Humboldt current system, the geographical distances and contrasting environmental conditions34,52 may be the main structures of the genetic diversity of the copepod populations that inhabit the SEPO region.
Materials and methods
Study area and individual collection
In this study, we studied six locations along the Chilean coast covering three biogeographic zones previously proposed in54, spanning over 3000 km (Fig. 1), where the presence of the copepod A. tonsa has been described. Samples were collected at: (A) Antofagasta (23.6° S 70.4° W) and Tongoy (30.2° S 71.5° W), both situated in the Peruvian Province (4–30° S) which is strongly influenced by year round upwelling and the absence of rivers, (B) off Concepcion (36.5° S 72.9° W) and at the Valdivia estuary (39.8° S 73.2° W), both located in the Intermediate area (transition zone) strongly affected by seasonal upwelling (spring and summer) and high river discharges (autumn and winter). (C) Hualaihue (42° S 72.6° W) and Magallanes (53.1° S 70.9° W), both immersed in the Magellan Province (42–56° S), with a strong influence of a continuous discharge of fresh water along a dismembered coast by fjords and channels with glacial relicts of the last glacial maximum (see details in Fig. 1).
Our research group and other authors have repeatedly characterized the variability of the coastal ocean environment in the localities under study52,82,83,84, accounting for considerable differences between localities in temperature, salinity, PCO2, food availability related to the different biogeographical zones. Broadly speaking, higher temperatures prevail in the Peruvian province, which decrease with increasing latitude84. Regarding salinity, this is higher in the Peruvian province and decreases in the other provinces, being directly dependent on the contribution of fresh water from the rivers, which increases in winter82,84. In the Peruvian province and in the intermediate area, episodic events of high ocean acidification (PCO2 ~ 1800 μatm) can be found, mainly associated with upwelling areas and river discharges52,83. Finally, it has been observed that the concentration of food is higher and less variable in estuarine localities of the intermediate area than in upwelling areas of the Peruvian province84. Detailed information on the main environmental conditions of each population studied in Suppl. Table S2.
Zooplankton samples were collected at each location using gently oblique hauls with a WP2 net (50 cm diameter, 200-µm mesh size), equipped with a non-filtering 1 L cod-end, targeting depths ranging from 10 to 7 m. In the particular case of the Magallanes locality, zooplankton samples were collected from a depth of 50 m to the surface with a filtering cod-end (more details in Suppl. Table S3). The collected samples were preserved in 95% ethanol at − 20 °C. Subsequently, plankton samples were taken to the laboratory for analysis where the identification and sorting of A. tonsa females were conducted using taxonomic keys85.
DNA extraction, amplification of molecular markers and genetic analysis
Specimens were selected for genetic analysis, and genomic DNA was extracted from a single individual using the Mollusc DNA Kit (Omega), following the manufacturer's protocols and with the modifications suggested in20. Subsequently, the concentration of the extracted DNA was quantified, and the presence of contaminants was verified using a Nanodrop. Finally, the DNA was stored at − 20 °C.
To perform the genetic analyses, two genes were employed: the mitochondrial gene encoding the cytochrome c oxidase subunit I (COI) and the nuclear gene encoding the small subunit of the ribosome (18S). The primers used to amplify each gene fragment, along with the observed and expected fragment lengths and the hybridization temperatures during PCR amplification cycle are specified in supplementary materials Table S4. PCR amplification was conducted in a total volume of 30 μL, where included 3 μL of 5X Green GoTaq G2 Flexi Buffer, 3 μL of 25 mM MgCl2, 0.5 μL of dNTPs (10 mmol L−1), 0.5 μL of each primer (100 pmol µL−1), 0.045 units of GoTaq G2 Flexi DNA Polymerase (Promega), and 5 μL of DNA from each individual. The PCR products were verified by electrophoresis on 1.2% agarose gel. Subsequently, the amplified products were sent to Macrogen for sequencing. To avoid ambiguous readings of certain nucleotides, the PCR products were sequenced in both directions (forward and reverse).
Sequence alignment and phylogenetic analyses
The sequences from both strands of the same individual were aligned to confirm the nucleotide bases and to generate a consensus sequence using the Geneious R11.1.5 (2018). Additionally, species verification was performed using consensus sequences in BLASTn to avoid misidentification of the specimens. The absence of stop codons was confirmed. The final alignments for both gene fragments were conducted using MUSCLE alignment algorithm86. The inclusion of A. tonsa sequences additional to those generated by us was carried out following a two-step approach: First, we searched for similarity (BLASTn) between our sequences and the sequences available in Genbank and the most similar ones were included. Then (second step), sequences generated in previous research were collected, where the relationship or phylogenetic identity of A. tonsa populations had been evaluated. In this case, we included those that were identified as misidentifications and also taxonomic identity corrections38,39,40,41,42,43,44,45,61,63,64. This approach allowed us to include the largest number of A. tonsa populations from different oceans and seas. In this search, our sequences were the only ones generated from the SEPO region, because the closest ones came from populations on the North Atlantic coasts44 or sequences of North Pacific populations misidentified as A. tonsa61.
Finally, the largest number of sequences of different species of the genus Acartia were included, in order to establish the phylogenetic position of our populations with respect to the species A. tonsa within the genus Acartia.
Phylogenetic analyses were performed using maximum likelihood (ML) and Bayesian inference (BI) methods for both analyzed gene fragments. The GTR + G model was selected using Bayesian information criterion (BIC) implemented in Mega11 program87 for COI and 18S, respectively. Bayesian analyses were performed using MrBayes V3.2.788. The Markov Chain Monte Carlo chains (MCMC) were run in three simultaneous runs with default heating values, along with pre-established evolutionary models, for 1.0 × 106 generations, sampling tree topologies every 1000 generations for both genes. The first 25% of the trees were discarded as burn-in, and a chain standard deviation of > 0.001 was set to indicate analysis stability. ML inferences were performed following the respective evolutionary model for each gene, using 10,000 bootstraps and 1000 replicates with the IQ-TREE V2.2 web server (http://iqtree.cibiv.univie.ac.at/;89). Indices of genetic diversity were calculated for the studied populations, including the number of haplotypes, haplotype diversity, nucleotide diversity, polymorphic sites, and private haplotypes. These calculations were performed using the DnaSp V.6 software90. Furthermore, population structuring by location was evaluated using Analysis of Molecular Variance (AMOVA), and pair-wise differentiation between populations (Fst) was determined using Arlequin V.3.5 software91) after 10,000 number of iterations. To visualize the haplotypes based on their presence in different locations and the relationships among DNA sequences, a haplotype network was constructed for each molecular marker using the PopArt software92. This construction was based on the minimum spanning network criterion93 with 1000 bootstrap replicates.
Species delimitation
Two different analytical methods, the Generalized Mixed Yule Coalescent (GMYC) probability method and the Poisson Tree Process (PTP) model, were used for species delimitation within the Acartia genus. In the GMYC analysis, an ultrametric tree of the COI gene was employed, and the BEAST v2.7.494 was used. The following input parameters were selected to represent branching patterns: the GTR substitution model, three categories for gamma-distributed rate variation, local random clock, and Yule model. Four independent runs were conducted, each with 100 million iterations and sampling every 10,000. The convergence of the runs was evaluated using Tracer v1.695. The Maximum Credibility Clade (MCC) tree was determined using Tree Annotator in BEAST v2.7.494 after discarding the first 25% of the trees. The number of delimited species was inferred for each MCC tree using the "gmyc" library96 in R97. Additionally, species delimitation was performed using the PTP method through the bPTP web server (http://species.h-its.org/ptp/). Bayesian Inference (BI) phylogeny of the COI gene was used as an input for PTP analysis, and 3 × 105 MCMC sampling generations were conducted, with a thinning value of 100 and a burn-in of 25%. The convergence of the MCMC was visually confirmed between the output files.
Data availability
DNA sequence data reported in this publication are available through NCBI under accession numbers: mtCOI (OR759115-OR759134), 18 s (OR771437-OR771462).
References
Panetta, A. M., Stanton, M. L. & Harte, J. Climate warming drives local extinction: Evidence from observation and experimentation. Sci. Rep. 4(2), eaaq1819. https://doi.org/10.1126/sciadv.aaq1819 (2018).
Gruber, N., Boyd, P. W., Frölicher, T. L. & Vogt, M. Biogeochemical extremes and compound events in the ocean. Nature 600(7889), 395–407. https://doi.org/10.1038/s41586-021-03981-7 (2021).
Starko, S. et al. Marine heatwaves threaten cryptic coral diversity and erode associations among coevolving partners. Sci. Rep. 9(32), eadf0954. https://doi.org/10.1126/sciadv.adf0954 (2023).
Doney, S. C. The dangers of ocean acidification. Sci. Am. 294(3), 58–65 (2006).
Johnson, G. C. & Lyman, J. M. Warming trends increasingly dominate the global ocean. Nat. Clim. Change 10(8), 757–761. https://doi.org/10.1038/s41558-020-0822-0 (2020).
Smale, D. A. et al. Marine heatwaves threaten global biodiversity and the provision of ecosystem services. Sci. Rep. 9(4), 306–312. https://doi.org/10.1038/s41558-019-0412-1 (2019).
Magel, J. M., Dimoff, S. A. & Baum, J. K. Direct and indirect effects of climate change-amplified pulse heat stress events on coral reef fish communities. Ecol. Appl. 30(6), e02124. https://doi.org/10.1002/eap.2124 (2020).
Gurgel, C. F. D., Camacho, O., Minne, A. J., Wernberg, T. & Coleman, M. A. Marine heatwave drives cryptic loss of genetic diversity in underwater forests. Curr. Biol. 30(7), 1199–1206. https://doi.org/10.1016/j.cub.2020.01.051 (2020).
Lavergne, S. & Molofsky, J. Increased genetic variation and evolutionary potential drive the success of an invasive grass. Proc. Natl. Acad. Sci 104(10), 3883–3888. https://doi.org/10.1073/pnas.0607324104 (2007).
Pauls, S. U., Nowak, C., Bálint, M. & Pfenninger, M. The impact of global climate change on genetic diversity within populations and species. Sci. Rep. 22(4), 925–946. https://doi.org/10.1111/mec.12152 (2013).
Johnson, C. R. et al. Climate change cascades: Shifts in oceanography, species’ ranges and subtidal marine community dynamics in eastern Tasmania. J. Exp. Mar. Biol. Ecol. 400(1–2), 17–32 (2011).
Poloczanska, E. S. et al. Responses of marine organisms to climate change across oceans. Front. Mar. Sci. https://doi.org/10.3389/fmars.2016.00062 (2016).
Lo Brutto, S., Arculeo, M. & Stewart Grant, W. Climate change and population genetic structure of marine species. Chem. Ecol. 27(2), 107–119. https://doi.org/10.1080/02757540.2010.547486 (2011).
Inoue, K. & Berg, D. J. Predicting the effects of climate change on population connectivity and genetic diversity of an imperiled freshwater mussel, Cumberlandia monodonta (Bivalvia: Margaritiferidae), in riverine systems. Glob. Change Biol. 23(1), 94–107. https://doi.org/10.1111/gcb.13369 (2017).
Mathiesen, S. S. et al. Genetic diversity and connectivity within Mytilus spp. in the subarctic and Arctic. Sci. Rep. 10(1), 39–55 (2017).
Haye, P. A. et al. Phylogeographic structure in benthic marine invertebrates of the southeast Pacific coast of Chile with differing dispersal potential. PLoS ONE 9(2), e88613. https://doi.org/10.1371/journal.pone.0088613 (2014).
Carrete Vega, G. & Wiens, J. J. Why are there so few fish in the sea?. Proc. R. Soc. B Biol Sci. 279(1737), 2323–2329. https://doi.org/10.1098/rspb.2012.0075 (2012).
Cowen, R. K. & Sponaugle, S. Larval dispersal and marine population connectivity. Annu. Rev. Mar. Sci. 1, 443–466. https://doi.org/10.1146/annurev.marine.010908.163757 (2009).
Halbert, K. M., Goetze, E. & Carlon, D. B. High cryptic diversity across the global range of the migratory planktonic copepods Pleuromamma piseki and P. gracilis. PLoS ONE 8(10), e77011. https://doi.org/10.1371/journal.pone.0077011 (2013).
Gonzalez, C. E., Goetze, E., Escribano, R., Ulloa, O. & Victoriano, P. Genetic diversity and novel lineages in the cosmopolitan copepod Pleuromamma abdominalis in the Southeast Pacific. Sci. Rep. 10(1), 1115. https://doi.org/10.1038/s41598-019-56935-5 (2020).
Trickey, J. S., Thiel, M. & Waters, J. M. Transoceanic dispersal and cryptic diversity in a cosmopolitan rafting nudibranch. Sci. Rep. 30(3), 290–301. https://doi.org/10.1071/IS15052 (2016).
Cheng, J. & Sha, Z. L. Cryptic diversity in the Japanese mantis shrimp Oratosquilla oratoria (Crustacea: Squillidae): Allopatric diversification, secondary contact, and hybridization. Sci. Rep. 7(1), 2017. https://doi.org/10.1038/s41598-017-02059-7 (1972).
Li, Y., Zhou, Y., Li, P., Gao, T. & Lin, L. Species identification and cryptic diversity in Pampus species as inferred from morphological and molecular characteristics. Mar. Biodivers. 49, 2521–2534. https://doi.org/10.1007/s12526-019-00976-6 (2019).
Laakmann, S., Blanco-Bercial, L. & Cornils, A. The crossover from microscopy to genes in marine diversity: From species to assemblages in marine pelagic copepods. Philos. Trans. R. Soc. Lond. B. 375(1814), 20190446 (2020).
Pérez-Portela, R., Arranz, V., Rius, M. & Turon, X. Cryptic speciation or global spread? The case of a cosmopolitan marine invertebrate with limited dispersal capabilities. Sci. Rep. 3, 3197. https://doi.org/10.1038/srep03197 (2013).
Darling, J. A. & Carlton, J. T. A framework for understanding marine cosmopolitanism in the Anthropocene. Front. Mar. Sci. 5, 293. https://doi.org/10.3389/fmars.2018.00293 (2018).
Blaxter, J. H., Douglas, B., Tyler, P. A. & Mauchline, J. The Biology of Calanoid Copepods (Academic Press, 1998).
Chaalali, A. et al. Changes in the distribution of copepods in the Gironde estuary: A warming and marinisation consequence?. Estuar. Coast. Shelf Sci. 134, 150–161. https://doi.org/10.1016/j.ecss.2012.12.004 (2013).
Gonzalez, J. G. Critical thermal maxima and upper lethal temperatures for the calanoid copepods Acartia tonsa and A. clausi. Mar. Biol. 27(3), 219–223. https://doi.org/10.1007/BF00391947 (1974).
Svetlichny, L. & Hubareva, E. Salinity tolerance of alien copepods Acartia tonsa and Oithona davisae in the Black Sea. Sci. Rep. 461, 201–208. https://doi.org/10.1016/j.jembe.2014.08.012 (2014).
Jørgensen, T. S. et al. The genome and mRNA transcriptome of the cosmopolitan calanoid copepod Acartia tonsa Dana improve the understanding of copepod genome size evolution. Genome Biol. Evol. 11(5), 1440–1450. https://doi.org/10.1093/gbe/evz067 (2019).
Dam, H. G. et al. Rapid, but limited, zooplankton adaptation to simultaneous warming and acidification. Nat. Clim. Change 11(9), 780–786. https://doi.org/10.1038/s41558-021-01131-5 (2021).
Brennan, R. S. et al. Loss of transcriptional plasticity but sustained adaptive capacity after adaptation to global change conditions in a marine copepod. Nat. Commun. 13(1), 1147. https://doi.org/10.1038/s41467-022-28742-6 (2022).
Aguilera, V. M. & Bednaršek, N. Variations in phenotypic plasticity in a cosmopolitan copepod species across latitudinal hydrographic gradients. Front. Ecol. Evol. 10, 925648. https://doi.org/10.3389/fevo.2022.925648 (2022).
Escribano, R., Hidalgo, P., Fuentes, M. & Donoso, K. Zooplankton time series in the coastal zone off Chile: Variation in upwelling and responses of the copepod community. Prog. Oceanogr. 97, 174–186. https://doi.org/10.1016/j.pocean.2011.11.006 (2012).
Gonzalez, C. E., Escribano, R. & Hidalgo, P. Intra-seasonal variation of upwelling and its effects on copepod community structure off central/southern Chile (2002–2009). Hydrobiologia 758, 61–74. https://doi.org/10.1007/s10750-015-2265-6 (2015).
Kimmel, D. G. & Roman, M. R. Long-term trends in mesozooplankton abundance in Chesapeake Bay, USA: Influence of freshwater input. Mar. Ecol. Prog. Ser. 267, 71–83. https://doi.org/10.3354/meps267071 (2004).
Caudill, C. C. & Bucklin, A. Molecular phylogeography and evolutionary history of the estuarine copepod, Acartia tonsa, on the Northwest Atlantic coast. Hydrobiologia 511, 91–102. https://doi.org/10.1023/B:HYDR.0000014032.05680.9d (2004).
Chen, G. & Hare, M. P. Cryptic ecological diversification of a planktonic estuarine copepod, Acartia tonsa. Mol. Ecol. 17(6), 1451–1468. https://doi.org/10.1111/j.1365-294X.2007.03657.x (2008).
Chen, G. & Hare, M. P. Cryptic diversity and comparative phylogeography of the estuarine copepod Acartia tonsa on the US Atlantic coast. Mol. Ecol. 20(11), 2425–2441. https://doi.org/10.1111/j.1365-294X.2011.05079.x (2011).
Da Costa, K. G., Vallinoto, M. D. & Da Costa, R. M. Molecular identification of a new cryptic species of Acartia tonsa (Copepoda, Acartiidae) from the Northern coast of Brazil, based on mitochondrial COI gene sequences. J. Coast. Res. 36(6), 359–363 (2011).
Costa, K. G. et al. Genetic variability of Acartia tonsa (Crustacea: Copepoda) on the Brazilian coast. J. Plankton Res. 36(6), 1419–1422. https://doi.org/10.1093/plankt/fbu080 (2014).
Plough, L. V., Fitzgerald, C., Plummer, A. & Pierson, J. J. Reproductive isolation and morphological divergence between cryptic lineages of the copepod Acartia tonsa in Chesapeake Bay. Sci. Rep. 597, 99–113. https://doi.org/10.3354/meps12569 (2018).
Figueroa, N. J., Figueroa, D. F. & Hicks, D. Phylogeography of Acartia tonsa Dana, 1849 (Calanoida: Copepoda) and phylogenetic reconstruction of the genus Acartia Dana, 1846. Mar. Biodivers. 50, 1–20. https://doi.org/10.1007/s12526-020-01043-1 (2020).
Gomes, C. M. et al. Hidden diversity in cryptic Brazilian lineages of Acartia (Copepoda, Calanoida, Acartiidae). J. Sea Res. 141, 21–25. https://doi.org/10.1016/j.seares.2018.08.005 (2018).
Escribano, R. & Pérez, C. S. Variability in fatty acids of two marine copepods upon changing food supply in the coastal upwelling zone off Chile: Importance of the picoplankton and nanoplankton fractions. J. Mar. Biol. Assoc. U.K. 90(2), 301–313. https://doi.org/10.1017/S002531540999083X (2010).
Aguilera, V. M., Vargas, C. A., Lardies, M. A. & Poupin, M. J. Adaptive variability to low-pH river discharges in Acartia tonsa and stress responses to high pCO2 conditions. Mar. Ecol. 37(1), 215–226. https://doi.org/10.1111/maec.12282 (2016).
Ruz, P. M., Hidalgo, P., Yáñez, S., Escribano, R. & Keister, J. E. Egg production and hatching success of Calanus chilensis and Acartia tonsa in the northern Chile upwelling zone (23 S), Humboldt current system. Sci. Rep. 148, 200–212. https://doi.org/10.1016/j.jmarsys.2015.03.007 (2015).
Ruz-Moreno, P. M. et al. Potential effects of shoaling of oxygen minimum zone on the population of Acartia (Acanthacartia) tonsa in the Humboldt current upwelling system (23° S). Sci. Rep. 193, 102372. https://doi.org/10.1016/j.seares.2023.102372 (2023).
Thiel, M., Castilla, J. C., Fernández, M. & Navarrete, S. The Humboldt current system of northern and central Chile. Oceanogr. Mar. Biol. 45, 195–344 (2007).
Escribano, R. & Morales, C. E. Spatial and temporal scales of variability in the coastal upwelling and coastal transition zones off central-southern Chile (35–40 S). Prog. Oceanogr. 92, 1–7. https://doi.org/10.1016/j.pocean.2011.07.019 (2012).
Vargas, C. A. et al. Species-specific responses to ocean acidification should account for local adaptation and adaptive plasticity. Nat. Ecol. Evol. 1(4), 0084. https://doi.org/10.1038/s41559-017-0084 (2017).
Segovia, N. I., Gallardo-Escárate, C., Poulin, E. & Haye, P. A. Lineage divergence, local adaptation across a biogeographic break, and artificial transport, shape the genetic structure in the ascidian Pyura chilensis. Sci. Rep. 7(1), 44559. https://doi.org/10.1038/srep44559 (2017).
Camus, P. A. Biogeografía marina de Chile continental. Rev. Chil. Hist. Nat. 74(3), 587–617. https://doi.org/10.4067/S0716-078X2001000300008 (2001).
Valdovinos, C., Navarrete, S. A. & Marquet, P. A. Mollusk species diversity in the Southeastern Pacific: Why are there more species towards the pole?. Sci. Rep. 26(2), 139–144. https://doi.org/10.1034/j.1600-0587.2003.03349.x (2003).
Cárdenas, L., Castilla, J. C. & Viard, F. A phylogeographical analysis across three biogeographical provinces of the south-eastern Pacific: The case of the marine gastropod Concholepas concholepas. J. Biogeogr. 36, 969–981. https://doi.org/10.1111/j.1365-2699.2008.02056.x (2009).
Provan, J. & Bennett, K. D. Phylogeographic insights into cryptic glacial refugia. Trends Ecol. Evol. 23(10), 564–571. https://doi.org/10.1016/j.tree.2008.06.010 (2008).
Marko, P. B. et al. The expansion-contraction model of Pleistocene biogeography: Rocky shores suffer a sea change?. Sci. Rep. 19(1), 146–169. https://doi.org/10.1111/j.1365-294X.2009.04417.x (2010).
Struck, T. H. et al. Finding evolutionary processes hidden in cryptic species. Sci. Rep. 33(3), 153–163. https://doi.org/10.1016/j.tree.2017.11.007 (2018).
Korshunova, T. et al. Multilevel fine-scale diversity challenges the ‘cryptic species’ concept. Sci. Rep. 9(1), 6732. https://doi.org/10.1038/s41598-019-42297-5 (2019).
Smith, J. A. et al. Acartia arbruta (previously A. tonsa) in British Columbia: A bioindicator of climate-driven ecosystem variability in the northeast Pacific Ocean. Sci. Rep. 43(4), 546–564. https://doi.org/10.1093/plankt/fbab040 (2021).
Razouls, C., De Bovée, F., Kouwenberg, J. & Desreumaux, N. Diversity and geographic distribution of marine planktonic copepods (Sorbonne University, CNRS, accessed 27 July 2023); http://copepodes.obsbanyuls.fr/en (2019).
Srinui, K., Ohtsuka, S., Metillo, E. B. & Nishibori, M. A new species of Acartia (Copepoda, Calanoida) from the Philippines, based on morphological and molecular analyses. ZooKeys 814, 71–94. https://doi.org/10.3897/zookeys.814.24601 (2019).
Lee, S., Soh, H. Y. & Lee, W. A new species in the genus Acartia Dana, 1846 (Crustacea, Copepoda, Calanoida, Acartiidae) from the South Pacific coastal waters of Nadi Bay, Fiji. ZooKeys 893, 69. https://doi.org/10.3897/zookeys.893.38369 (2019).
Zhang, A. B., He, L. J., Crozier, R. H., Muster, C. & Zhu, C. D. Estimating sample sizes for DNA barcoding. Mol. Phylogenet. Evol. 54(3), 1035–1039. https://doi.org/10.1016/j.ympev.2009.09.014 (2010).
Phillips, J. D., Gwiazdowski, R. A., Ashlock, D. & Hanner, R. An exploration of sufficient sampling effort to describe intraspecific DNA barcode haplotype diversity: Examples from the ray-finned fishes (Chordata: Actinopterygii). DNA Barcodes 3(1), 66–73. https://doi.org/10.1515/dna-2015-0008 (2015).
Phillips, J. D., Gillis, D. J. & Hanner, R. H. Incomplete estimates of genetic diversity within species: Implications for DNA barcoding. Ecol. Evol. 9(5), 2996–3010. https://doi.org/10.1002/ece3.4757 (2019).
Fratini, S., Ragionieri, L. & Cannicci, S. Demographic history and reproductive output correlates with intraspecific genetic variation in seven species of Indo-Pacific mangrove crabs. PLoS ONE 11(7), e0158582. https://doi.org/10.1371/journal.pone.0158582 (2016).
Tikochinski, Y., Carreras, C., Tikochinski, G. & Vilaça, S. T. Population-specific signatures of intra-individual mitochondrial DNA heteroplasmy and their potential evolutionary advantages. Sci. Rep. 10(1), 211. https://doi.org/10.1038/s41598-019-56918-6 (2020).
Haye, P. A., Turon, X. & Segovia, N. I. Time or space? Relative importance of geographic distribution and interannual variation in three lineages of the ascidian Pyura chilensis in the Southeast Pacific Coast. Front. Mar. Sci. 8, 657411. https://doi.org/10.3389/fmars.2021.657411 (2021).
Sagorny, C. & von Döhren, J. Occasional sexual reproduction significantly affects the population structure of the widespread, predominantly asexually reproducing marine worm Lineus sanguineus (Nemertea: Pilidiophora). Mar. Biol. 169(7), 92. https://doi.org/10.1007/s00227-022-04077-0 (2022).
Saldías, G. S. & Lara, C. Satellite-derived sea surface temperature fronts in a river-influenced coastal upwelling area off central–Southern Chile. Reg. Stud. Mar. Sci 37, 101322. https://doi.org/10.1016/j.rsma.2020.101322 (2020).
Hernández, A. & Tapia, F. J. Connecting spatial structure in subtidal benthic communities with temporal variability in bottom temperature and dissolved oxygen along an upwelling coast. Estuar. Coast. Shelf Sci. 250, 107166. https://doi.org/10.1016/j.ecss.2021.107166 (2021).
Oerder, V., Bento, J. P., Morales, C. E., Hormazábal, S. & Pizarro, O. Coastal upwelling front detection off Central Chile (365–37° S) and spatio-temporal variability of frontal characteristics. Remote Sens. 10, 690. https://doi.org/10.3390/rs10050690 (2018).
Sasaki, M. C. & Dam, H. G. Genetic differentiation underlies seasonal variation in thermal tolerance, body size, and plasticity in a short-lived copepod. Ecol. Evol. 10(21), 12200–12210. https://doi.org/10.1002/ece3.6851 (2020).
Ruz, P. M., Hidalgo, P., Riquelme-Bugueño, R., Franco-Cisterna, B. & Cornejo, M. Vertical distribution of copepod eggs in the oxygen minimum zone off Mejillones Bay (23 S) in the Humboldt current system. Mar. Ecol. Prog. Ser. 571, 83–96. https://doi.org/10.3354/meps12117 (2017).
Hidalgo, P. et al. Patterns of copepod diversity in the Chilean coastal upwelling system. Deep Sea Res. Part II Top. Stud. Oceanogr. 57(24–26), 2089–2097. https://doi.org/10.1016/j.dsr2.2010.09.012 (2010).
Johnson, W. S. & Allen, D. M. Zooplankton of the Atlantic and Gulf Coasts: A Guide to their Identification and Ecology (JHU Press, 2012).
Cárdenas, L., Castilla, J. C. & Viard, F. Hierarchical analysis of the population genetic structure in Concholepas concholepas, a marine mollusk with a long-lived dispersive larva. Mar. Ecol. 37(2), 359–369. https://doi.org/10.1111/maec.12286 (2016).
Moreno, C. A., Asencio, G. & Ibañez, S. Patrones de asentamiento de Concholepas concholepas (Brugière) (Mollusca: Muricidae) en la zona intermareal rocosa de Valdivia. Chile. Rev. Chil. Hist. Nat. 66, 93–101 (1993).
Molinet, C. et al. Patterns of larval distribution and settlement of Concholepas concholepas (Bruguiere, 1789) (Gastropoda, Muricidae) in fjords and channels of southern Chile. Rev. Chil. Hist. Nat. 78(3), 409–423 (2005).
Garcés-Vargas, J. et al. Tidally forced saltwater intrusions might impact the quality of drinking water, the Valdivia river (40° S), Chile estuary case. Water 12(9), 2387. https://doi.org/10.3390/w12092387 (2020).
Osma, N. et al. Response of phytoplankton assemblages from naturally acidic coastal ecosystems to elevated pCO2. Front. Mar. Sci 7, 323. https://doi.org/10.3389/fmars.2020.00323 (2020).
Aguilera, V. M. et al. Local scale extreme low pH conditions and genetic differences shape phenotypic variation in a broad dispersal copepod species. Front. Mar. Sci 10, 1221132. https://doi.org/10.3389/fmars.2023.1221132 (2023).
Bradford-Grieve, J. M. Fiches d'identification du Zooplancton No. 12. ICES Identification Leaflets for Plankton, 181, 1–19 (1999).
Edgar, R. C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32(5), 1792–1797. https://doi.org/10.1093/nar/gkh340 (2004).
Tamura, K., Stecher, G. & Kumar, S. MEGA11: Molecular evolutionary genetics analysis version 11. Mol. Biol. Evol. 38(7), 3022–3027. https://doi.org/10.1093/molbev/msab120 (2021).
Ronquist, F. et al. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61(3), 539–542. https://doi.org/10.1093/sysbio/sys029 (2012).
Trifinopoulos, J., Nguyen, L. T., von Haeseler, A. & Minh, B. Q. W-IQ-TREE: A fast-online phylogenetic tool for maximum likelihood analysis. Nucleic Acids Res. 44(W1), W232–W235. https://doi.org/10.1093/nar/gkw256 (2016).
Rozas, J. et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34(12), 3299–3302. https://doi.org/10.1093/molbev/msx248 (2017).
Excoffier, L. & Lischer, H. E. Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 10(3), 564–567. https://doi.org/10.1111/j.1755-0998.2010.02847.x (2010).
Leigh, J. W. & Bryant, D. POPART: Full-feature software for haplotype network construction. Methods Ecol. Evol. 6(9), 1110–1116. https://doi.org/10.1111/2041-210X.12410 (2015).
Bandelt, H. J., Forster, P. & Röhl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16(1), 37–48. https://doi.org/10.1093/oxfordjournals.molbev.a026036 (1999).
Bouckaert, R. et al. BEAST 2.5: An advanced software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 15(4), e1006650. https://doi.org/10.1371/journal.pcbi.1006650 (2019).
Rambaut, A., Drummond, A. J. & Suchard, M. Tracer v1. 6 http://beast.bio.ed.ac.uk (2007).
Ezard, T., Fujisawa, T. & Barraclough, T. G. Splits: Species’ limits by threshold statistics. R Package Version 1(11), r29 (2009).
R Core Team R: A language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria. https://www.R-project.org/ (2021).
Acknowledgements
This investigation was funded by Millennium Institute of Oceanography (IMO, Grant ICN120019) and Fondecyt Grant 1210171 to C.A.V. We thank Valeria and Auri for their support.
Author information
Authors and Affiliations
Contributions
A.M. provided funding, performed fieldwork, generated and analyzed DNA sequences, drafted a first version of manuscript and contributed to posterior versions. C.E.G. generated and analyzed DNA sequences and contributed to manuscript. V.M.A. provided funding, performed fieldwork, generated DNA sequences and contributed to manuscript. R.G. provided funding, performed fieldwork and contributed to manuscript. R.E. provided funding. C.A.V. provided funding and contributed to manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Mesas, A., Aguilera, V.M., González, C.E. et al. Molecular evidence for a new endemic species of Acartia (Copepoda, Calanoida) from the Southeast Pacific coast. Sci Rep 14, 12366 (2024). https://doi.org/10.1038/s41598-024-62080-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-62080-5
- Springer Nature Limited