
Abstract
Lung adenocarcinoma (LUAD) is the most common histological subtype of lung cancer. In the development and progression of LUAD, epigenetic aberration plays a crucial role. However, the function of RNA N6-methyladenosine (m6A) modifications in the LUAD progression is unknown. The m6A regulator modification patterns in 955 LUAD samples were analyzed comprehensively. Patterns were systematically correlated with the tumor microenvironment (TME) cell-infiltration characteristics. Using principal component analysis algorithms, the m6Ascore was generated to quantify m6A modification patterns in individual tumors. Then, their values for predicting prognoses and therapeutic response in LUAD patients were assessed. Three distinct m6A modification patterns in LUAD were identified. Among them, the prognosis of m6Acluster C was the best, while the prognosis of m6Acluster A was the worst. Interestingly, the characterization of TME cell infiltration and biological behavior differed among the three patterns. To evaluate m6A modification patterns within individual tumors, an m6Ascore signature was constructed. The results showed that the high m6Ascore group was associated with a better prognosis; tumor somatic mutations and tumor microenvironment differed significantly between the high- and low- m6Ascore groups. Furthermore, in the cohort with anti-CTLA-4 treatment alone, patients with a high m6Ascore had higher ICI scores, which indicated significant therapeutic advantage and clinical benefits.
Similar content being viewed by others


Globally, cancer-related morbidity and mortality rate are increasing rapidly. Among them, lung cancer is the leading cause of cancer-related mortality1,2,3. Non-small cell lung cancer (NSCLC) is the most common type of lung cancer, accounting for about 85% of all cases4,5,6. Lung adenocarcinoma (LUAD) is one of the major subtype of NSCLC, accounting for roughly 40% of all lung cancer cases7,8. In recent years, surgical resection, targeted therapy, chemotherapy, and other therapeutic approaches have been proven to improve the survival of NSCLC patients, but the prognosis remains poor with many limitations9,10,11. Therefore, further research is required to understand the underlying tumor biological process and treatment options for LUAD.
N6-methyladenosine(m6A)RNA methylation is regarded to be one of the most significant and abundant forms of RNA modification in eukaryotic cells, which plays a key regulatory role in the cell’s existence12,13,14. The enzymes with writers (methylases), erasers (demethylases), and readers are mostly involved in m6A modifications, representing 0.1%-0.4% of the total adenosine residues. Numerous pieces of researchers have revealed that m6A regulatory factors play important roles in several cancer-related biological processes, including apoptosis, cell proliferation, invasion, and metastasis15,16,17. Zhang et al. demonstrated that the reduction of m6A methylation of RNA activates the oncogenic Wnt/PI3K-Akt signaling pathway, which can promote the malignant phenotype of gastric cancer cells18. By regulating the m6A level of USP7 mRNA, the m6A demethylase FTO can promote lung cancer cell proliferation19. According to Xu et al., the m6A methyltransferase METTL3 promotes cell proliferation by inhibiting SOCS2 to maintain the tumorigenicity of colon cancer. However, the function of m6A modulators in LUAD is still unclear20. Thus, in-depth studies and further investigations are required to understand the underlying mechanism for the m6A regulators in LUAD.
Bioinformatics analysis based on database mining is regarded to be one of the most promising approaches for cancer translational research, with the advancement of gene sequencing technology and the establishment of tumor databases21,22,23. In the current investigation, the leading goal was to evaluate the correlation between the m6A modification pattern and the tumor microenvironment. We revealed three different m6A modification patterns and assessed the clinical features, prognostic value, and immune infiltration of the resulting m6A clusters. In addition, we established a scoring system to quantify the m6A modification patterns and determine its value in predicting the prognosis and therapeutic response of LUAD patients.
Materials and methods
LUAD data source and preprocessing
The Cancer Genome Atlas (TCGA) and Gene-Expression Omnibus (GEO) database were used to acquire gene expression data and clinical annotations for LUAD samples. This analysis was comprised a total of 955 LUAD cases (TCGA-LUAD: 513, GSE68465: 442) sourced from TCGA and GEO databases. Patients without survival information were excluded from this study. In TCGA-LUAD cohorts, fragments per kilobase million (FPKM) were transformed into transcripts per million (TPM) values24,25. Before model validations, normalized expression values were logarithmically transformed and scaled for GEO data sets. The "sva" package in R software was used to examine the batch effect26,27. The clinical information of patients is provided in Table S1.
Unsupervised clustering for m6A regulators
The m6A -related literature revealed twenty-three m6A regulators, including 8 writers (METTL3, METTL14, METTL16, WTAP, VIRMA, ZC3H13, RBM15, RBM15B), 13 readers (YTHDC1, YTHDC2, YTHDF1, YTHDF2, YTHDF3, HNRNPC, FMR1, LRPPRC, HNRNPA2B1, IGFBP1, IGFBP2, IGFBP3, RBMX), and 2 erasers (FTO, ALKBH5)28,29,30. Univariate Cox model was used to analyze the correlation between m6A regulatory variables and prognosis. To identify the different m6A modification patterns and classify patients, the "ConsensuClusterPlus" package was used to conduct the above steps and 1000 times repetitions for guaranteeing the stability of clustering31.
Gene set variation analysis (GSVA) and functional annotation
To study the differences in biological processes between m6Aclusters and m6A modification patterns, the "GSVA" R package was used to perform GSVA enrichment analysis32,33,34. The R package "clusterProfiler" was used for functional annotation and the gene set file (c2.cp.kegg.v7.2.symbols.gmt) was obtained from the MSigDB database (https://www.gsea-msigdb.org), with the cutoff value of FDR < 0.05.a.
Immune cell infiltration estimation
To assess the relative abundance of each cell infiltration in distinct m6A subtypes and the amount of immune cell infiltration we employed the ssGSEA (single-sample gene-set enrichment analysis) program35,36.
Gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses
Using the "clusterProfiler" (version 3.0.4; https://www.rdocumentation.org/packages/clusterProfiler/versions/3.0.4), "enrichplot" (version 1.13.1.994; https://www.rdocumentation.org/packages/enrichplot/versions/1.13.1.994) and "ggplot2" (version 3.3.5; https://www.rdocumentation.org/packages/ggplot2/versions/3.3.5) packages of R software, GO and KEGG enrichment analysis on the gene set was performed37,38,49,50,51,52.
Generation of m6Ascores
We constructed a set of scoring system to evaluate the m6A modification pattern of individual patients with LUAD-the m6A gene signature, and we termed it as m6Ascore. The process of establishing the m6A scoring system was as follows: the DEGs identified from different m6Aclusters were firstly normalized among all samples and the overlap genes were extracted. Differential analysis and Venn diagram showed that there are 15 common differential genes among the three m6Aclusters. Then, we performed univariate Cox regression analysis for each gene. These genes with a significant prognosis were extracted for the next step of the analysis. Then we perform principal component analysis (PCA) to calculate the m6Ascore using the following formula:
where i is the expression of the m6A phenotype-associated genes39,40.
Generation of ImmuneScore, StromalScore, and ESTIMATEScore
Through the "estimate" package of R software the ratio of the immune stromal components of each sample in the tumor microenvironment was estimated, and ImmuneScore, StromalScore, and ESTIMATEScore, which was positively correlated with the ratio of immune, stromal, and the sum of both, respectively41,42.
Correlation of m6A-scoring signature with genome mutations, clinical information, and Immunity
The associations between the high- and low-m6Ascore groups and mutation and clinical status were investigated based on the m6A-scoring signature. In addition, ssGSEA was used to quantify the subset of tumor-infiltrating immune cells between the two groups and to assess their immunological differences. The Cancer Immunome Database (TCIA) was used to download the Immune checkpoint inhibitor (ICI) Immunophenoscore (IPS) for immunotherapy. IPS is a good predictor of CTLA-4 and PD-1 blocking responsiveness and thus was used to predicts the response to immunotherapy between the two groups43,44.
Statistical analysis
A t-test was used for variables with a normal distribution, and a non-parametric test (Wilcoxon rank rank-sum test) was employed for variables with a non-normal distribution when comparing data between two groups. One-way ANOVA and Kruskal–Wallis tests were used as parametric and non-parametric methods, respectively for data from more than two groups. A Chi-square test was used to examine the correlation between m6A modification patterns and clinical features. The P values were corrected for multiple comparisons via the Benjamini and Hochberg (BH). The best cut-off score between the two groups of high and low m6Ascore was derived by the surv-cutpoint function. The mutation landscape in patients was shown using the waterfall function of the "maftools" package. The R packages "survival" and "survminer" were used for survival analysis45. Unless specified, P-value < 0.05 was statistically significant. All data processing was done in R 4.1.0 software.
Results
The landscape of genetic variation of m6A regulators in LUAD
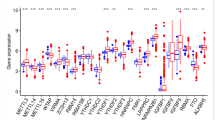
The flow chart diagram of this study is presented in Fig. S1. The somatic mutations and copy counts of 23 m6A regulators were summarized in LUAD. Among 561 samples, we found 115 experienced mutations of m6A regulators, with a frequency of 20.5%. The mutation frequency of ZC3H13 was highest, followed by FMR1 (Fig. 1A). Further analysis showed that there was no significant mutation co-occurrence relationship between ZC3H13 and other m6A regulators (Fig. S2). The study of CNV alteration frequency revealed that 23 regulators had a common CNV modification, with the majority of them focusing on copy number amplification, while RBM15, ZC3H13, METTL16, and YTHDC2 had a high frequency of CNV deletion (Fig. 1B). The location of CNV alterations on the chromosome for m6A regulators is shown in Fig. 1C. In addition, compared with normal tissues, the expression levels of METL3, VIRMA, RBM15, YTHDF1, YTHDF2, LRPPRC, HNRNPA2B1, IGFBP3, RBMX, FTO, and ALKBH5 were significantly up-regulated in LUAD, and the median value of the boxplot was higher, WTAP, METTL16, METTL14, and ZC3H13 were significantly down-regulated (P < 0.05, Fig. 1D).

Genetic variation of m6A regulators in LUAD. (A) Genetic alteration for the queried m6A regulators. (B) Frequency of CNVs in m6A regulators. Blue dots represent CNV amplification; purple dots represent CNV deletion. (C) The location of the CNV alteration of the m6A regulators changes on 23 chromosomes in the TCGA-LUAD cohort. Red square represent more samples with increased copy number of the gene. Blue square represent more samples with missing copy number of the gene. (D) Comparison of gene expression of 23 m6A regulators in LUAD with normal tissue (*P < 0.05; **P < 0.01; ***P < 0.001). LUAD : Lung adenocarcinoma.
m6A RNA methylation modification patterns mediated by 23 regulators in LUAD
One meta-cohort was formed by combining two LUAD datasets (TCGA-LUAD, GSE68465) with existing OS data and clinical information (Table S1). The univariate Cox regression analysis was used to screen for m6A regulators associated with prognosis in LUAD (Fig. 2A). The findings revealed that WTAP, ZC3H13, RBM15, HNRNPC, LRPPRC, HNRNPA2B1, IGFBP1, and IGFBP3 were risk factors for poorer prognosis. The interaction between the m6A regulators is displayed in Fig. 2B. Based on the expression levels of 23 m6A regulatory genes, the "ConsensusClusterPlus" R package was used to classify patients with qualitatively different m6A modification patterns, K = 3 is the optimal number of clusters determined by the consensus clustering algorithm, and three different modification patterns were determined (Fig. 2C), named m6Acluster A, m6Acluster B, and m6Acluster C. Furthermore, it was also found that most of the three m6A modified subtypes are in a state of separation, but there is also some overlap in the middle. Therefore, the PCA method based on m6A-related genes may have limitations for some patients, which requires our attention (Fig. 2D). In the results of prognostic analysis, we found that m6Acluster C has a higher 50% survival rate and a better survival advantage (P = 0.01, Fig. 2E).

m6A RNA methylation modification patterns mediated by 23 regulators in LUAD. (A) Univariate Cox regression models were used to assess the prognoses based on 23 m6A regulators. (B) Interactions between m6A regulators in LUAD. The size of the circles represents the effect of each modulator on prognosis; larger circles represent a greater effect on prognosis (p-values: 1–0.0001). The association between the m6A regulators is shown by the connecting line; negative correlations are in blue and positive correlations are in pink. (C) Heat map of the matrix of co‐occurrent proportions for LUAD samples (K = 3). (D) Principal component analysis (PCA) analysis of m6A methylation modification pattern. (E) The overall survival of m6A methylation modification pattern using Kaplan–Meier curves.
TME cell infiltration characteristics in distinct m6A modification patterns
To further analyze the difference in immune cell infiltration between different m6A modification patterns, the ssGSEA algorithm was used. It was found that the three m6A modification modes have significant differences in the degree of enrichment of immune cell infiltration. m6Acluster A has more abundant immune cell infiltration, with the highest median boxplot, while m6Acluster B has the worst level (Fig. 3A). Among the three m6A modification modes, m6Acluster A had a higher abundance of immune infiltrating cells, including Activated B cell, Activated CD4 T cell, Activated CD8 T cell, Activated dendritic cell, CD56bright natural killer cell, Immature dendritic cell, MDSC, Macrophage, Neutrophil, Type 1 T helper cell, and Type 17 T helper cell (Fig. 3A). Patients with this m6A modification pattern (m6Acluster A) had the poorest prognosis compared to the other two subtypes of m6A modification patterns (Fig. 2E). Meanwhile, GSVA enrichment analysis was performed to explore the biological behaviors between the m6A modification patterns (Fig. 3B-D). We found that immune-related pathways such as the T cell receptor signaling pathway, toll-like receptor signaling pathway, and natural killer cell-mediated cytotoxicity are significantly enriched in m6Acluster A (Fig. 3B). Glycosaminoglycan biosynthesis-related pathways and ECM receptor interaction were all substantially abundant in m6Acluster B (Fig. 3C). m6Acluster C was significantly enriched in pathways related to metabolism (Fig. 3D).

TME cell infiltration characteristics in distinct m6A modification patterns. (A) The abundance of TME infiltrating cell types in three m6A modification patterns. Statistical p-values are indicated by asterisks (*P < 0.05; **P < 0.01; ***P < 0.001). (B–D) GSVA enrichment analysis showing the activation states of biological pathways in different m6A modification patterns. Heat map for the biological processes; red represents activated pathways and blue represents inhibited pathways. (E) Heat map showing overlapping genes for three m6A methylation modification pattern subtypes. (F) Results of GO (up) and KEGG (down) enrichment.
In addition, 15 overlapping m6A phenotype-related DEGs (IGFBP2, BIRC3, ASCL1, RET, IL18, KCNH2, PIGT, ANXA1, MYOF, RPRM, TMEM59L, SOX2, EGLN3, MYO5C, and FCN3) in the three m6A modification patterns subtypes were identified in this study and performed GO and KEGG enrichment analysis (Fig. 3E). Positive regulation of T − helper cell differentiation, T − helper 2 cell differentiation, regulation of T − helper 2 cell differentiation, positive regulation of T cell proliferation, positive regulation of cell − cell adhesion, positive regulation of cell adhesion, transcription regulator complex, cadherin binding involved in cell − cell adhesion, and Hippo signaling pathway were all enriched in these DEGs (Fig. 3F).
Generation of m6A gene signatures and m6Ascore
Based on the DEGs between the three m6Aclusters, an unsupervised cluster analysis (Fig. 4A) was performed and three m6A modified genome phenotypes, named geneCluster A, geneCluster B, and geneCluster C. Substantial variations in the expression of m6A regulators across these three m6A-modified genomic phenotypes were found (Fig. 4B). geneCluster C had considerably greater levels of METTL3, RBM15B, YTHDF1, YTHDF2, YTHDF3, and IGFBP2 than the other two groups. In addition, a scoring system was devised to determine the pattern of m6A modification in each LUAD patient. To investigate the relationship between m6Ascore and patient prognosis, the "survminer" program was used to obtain the optimum cut-off value and classify patients into high- and low- m6Ascore groups. The m6Ascore group clinical information is shown in table S2. The 50% survival of the high m6Ascore group was significantly higher than that of the low m6Ascore group, and the high m6Ascore was associated with a better prognosis (p < 0.001, Fig. 4C). Most immune cells have a negative association with m6Ascore, according to the findings (Fig. 4D). According to the results of the boxplot, we can find that the median line value of the m6Acluster A group is the lowest, and the patients in the m6Acluster A group have a lower m6ascore and a poor prognosis (Fig. 4E).

Generation of m6A signatures and m6Ascores. (A) Consensus clustering of genecluster for k = 3. (B) Gene expression levels of 23 m6A regulators in three m6A modification genomic phenotypes (*P < 0.05; **P < 0.01; ***P < 0.001). (C) The overall survival of m6A scoring signature. (D) Correlation between m6Ascore and immune cell infiltration. (E) m6Ascore in different m6Acluster subgroups.
Clinical and tumor somatic mutation characteristics of m6Ascore cluster in TCGA-LUAD cohort
To further analyze the clinical characteristics based on the m6Ascore, the clinical information of LUAD patients from the TCGA database was obtained (Table S1, S2). The results showed that m6Ascore were higher in the N0-1 stage (p = 0.045, Fig. 5E) and were not significantly correlated with the other clinical stages (Fig. 5A,B,C,D,F). In addition, the association between different types of patients and their prognoses was examined (Fig. 5G–L), and the results showed that a high m6Ascore was related to a better prognosis in most patient categories (age > 55, M0, T1-2, Stage1-2, MALE). Another important finding is that m6Ascore can be used as an independent prognostic indicator for LUAD patients (Fig. S3), and a higher m6Ascore is associated with a better prognosis, which is also consistent with our previous findings (Fig. 4C).

Clinical characteristics of m6Ascore cluster in TCGA-LUAD cohort. (A–F) Correlation analysis between clinical characteristics and m6Ascore. (G–L) Correlation analysis between m6Ascore and prognosis in different patient types.
Then, in the TCGA-LUAD cohort, the "maftools" program was utilized to evaluate the differences in somatic mutation distribution between low- (Fig. 6B) and high-m6Ascores (Fig. 6A). The results showed that the low m6Ascore group had more extensive tumor burden mutation than the high m6Ascore group (p = 0.029, Fig. 6C), and the somatic mutation rate of TP53 in the low m6Ascore group was higher.

Characteristics of tumor somatic mutation in the m6A score cluster in the TCGA dataset. (A) Waterfall plot of tumor somatic mutation for high m6A score. (B) Waterfall plot of tumor somatic mutation for low low m6Ascore. (C) The tumor burden mutation level was higher in the low m6Ascore group.
The role of m6Ascore in tumor microenvironment and immunotherapy
To investigate the function of the m6Ascore in the tumor microenvironment, the R package "estimate" was used to calculate the proportion of immune matrix components in each sample in the tumor microenvironment. An important finding was that compared with the low m6Ascore group, the boxplot of the high m6Ascore group has a significantly higher midline, indicating that the high m6Ascore group has higher scores of ImmuneScore, StromalScore, and ESTIMATEScore (Fig. 7A). The results of clinical correlation analysis showed that with the occurrence and development of tumors (Stage I–IV, T1-T4, M0–M1), the ImmuneScore, StromalScore, and ESTIMATE Score decreased significantly (Fig. S4). In addition, the immunotherapy response of ICI treatment represented by the CTLA-4/PD-1 inhibitors in the high- and low-m6Ascore groups was investigated (Fig. 7B). The results showed that patients in the high m6Ascore group had higher ICI scores in the anti-CTLA-4 treatment alone cohort. In both the anti-PD-1 therapy alone and the combination of the anti-CTLA-4/PD-1 treatment cohorts, patients with a low m6Ascore had higher ICI scores.

The role of m6Ascore in tumor microenvironment and immunotherapy. (A) The score levels of ImmuneScore, StromalScore, and ESTIMATEScore in the high- and low- m6Ascore groups. (B) The relative distribution of immunophenoscore (IPS) was compared between low- and high- m6Ascore groups.
Discussion
m6A methylation is the most common form of mRNA modification which plays an important role in the development and progression of cancer by interacting with several m6A modulators. Previous studies showed that m6A modulator modification is significantly correlated with inflammation, tumor microenvironment, and immune response46,47,48. Thus, investigating the involvement of various m6A modification mechanisms in LUAD will further enhance the understanding of its occurrence and development. Methylation modification patterns, tumor microenvironment infiltration, and the characterization of m6A modulators in LUAD may help in determining the potential prognostic characteristics of cancer and aid in the development of novel therapeutic methods.
In the current study, 23 m6A regulatory factors from m6a-related literature were collected, and then their expression levels, mutations, and prognosis in LUAD were explored. It was observed that ZC3H13 (writers) showed the highest mutation frequency, followed by FMR1 (readers). Upon comparing with normal tissues it was found that METTL3, VIRMA, RBM15, YTHDF1, YTHDF2, LRPPRC, HNRNPA2B1, IGFBP3, RBMX, FTO, and ALKBH5 were significantly up-regulated in LUAD tissues, while WTAP, METL16, METL14, and ZC3H13 were significantly down-regulated. WTAP, ZC3H13, RBM15, HNRNPC, LRPPRC, HNRNPA2B1, IGFBP1, and IGFBP3 were considered to be the risk factors for poor prognosis. In addition, based on 23 m6A regulatory variables, three distinct m6A methylation modification patterns in LUAD were discovered. The best prognosis among these three models was m6Acluster C, whereas, the worst prognosis was m6Acluster A. Interestingly, these three models varied TME cell infiltration characteristics and biological behaviors. Activated B cell, Activated CD4 T cell, Activated CD8 T cell, activated dendritic cell, CD56 bright natural killer cell, immature dendritic cell, MDSC, Macrophage, Neutrophil, Type 1 T helper cell, and Type 17 T helper cell were found to be prevalent in innate immune cell infiltration. However, patients with this m6A modification pattern showed the worst prognosis compared to the other two patterns. The results of GSVA analysis revealed that m6Acluster A is associated with immune-related pathways, such as primary immunodeficiency, autoimmune thyroid disease, allograft rejection, nod-like receptor signaling pathway, toll-like receptor signaling pathway, t-cell receptor signaling pathway, natural killer cell-mediated cytotoxicity, and jak stat signaling pathway. This might explain the poor prognosis of m6Acluster A. Some comprehensive pathways were enriched by m6Acluster B, and the tumor metabolism-related pathways were highly represented by m6Acluster C. Based on the above-mentioned m6A modification patterns in LUAD, the m6A-related transcriptional expression patterns in these modifications were further explored and it was identified that 15 m6A phenotypic DEGs were present. These genes were significantly associated with immune-related biological pathways, according to the results of GO and KEGG enrichment analyses. Following that, three genomic subtypes based on m6A characteristic genes were identified in this study. These subtypes were also linked to the matrix and immunological activation, demonstrating the importance of m6A modification in creating distinct TME landscapes. TME plays an important role in the initiation and progression of tumorigenesis. The function of m6A regulatory factors in the immune microenvironment of LUAD to modify TME should be investigated further for the discovery of novel potential therapeutic targets.
Different patients had varying levels of m6A modifications. Thus, a scoring system (m6Ascore) was established to quantify the m6A modification pattern of patient tumors. It was observed that the high m6Ascore group was related to a better prognosis and reduced mutation frequency. According to the tumor microenvironment analyses the high m6Ascore group revealed higher ImmuneScore, StromalScore, and ESTIMATEScore. It was worth noting that the high expression levels of ImmuneScore, StromalScore, and ESTIMATEScore in LUAD patients were all associated with a better prognosis. The immunological checkpoints cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and programmed death 1 (PD-1) have provided novel tools for immunotherapy. Immune checkpoint inhibitors (ICI) acted against these molecules by relieving inhibition of certain pathways, thereby strengthening the immune system to produce anti-tumor effects. Therefore, the efficacy of ICI is strongly linked to the host's immune system and tumor immune microenvironment (TIME). The immunotherapeutic response to ICI therapy represented by CTLA-4/PD-1 inhibitors in the high and low m6Ascore groups was evaluated in this study. Patients with a high m6Ascore had higher ICI scores in the anti-CTLA-4 therapy cohort alone, according to the findings. In both the anti-PD-1 therapy alone and the anti-CTLA-4/PD-1 treatment cohorts, patients with a low m6Ascore had higher ICI scores. Therefore, the m6Ascore could be used in the future to evaluate the efficacy of the clinical responses of patients to immunotherapy.
However, our study has few limitations which are as follow: our conclusions are mainly based on bioinformatics methods, so further experiments and clinical verification are needed; There is a certain overlap between the m6Acluster subtypes that we have identified, so this method has certain limitations for some patients, and further development of more complete methods is needed in the future.
Conclusion
In conclusion, this study elucidated several extensive regulatory mechanisms underlying m6A methylation modification in LUAD. An m6A-scoring signature was created to identify m6A modification patterns in individual tumors. The heterogeneity of m6A modification patterns was highlighted, and the findings may enhance the understanding of the characterization of the tumor microenvironment and guide the development of effective immunotherapeutic strategies in the future.
Data availability
The data sets analysed during the current study are available in the TCGA (https://portal.gdc.cancer.gov/) and GEO repository (https://www.ncbi.nlm.nih.gov/geo/).
Abbreviations
- LUAD:
-
Lung adenocarcinoma
- m6A:
-
N6-methyladenosine
- TCGA:
-
The Cancer Genome Atlas
- GEO:
-
Gene-Expression Omnibus
- FPKM:
-
Fragments per kilobase million
- TPM:
-
Transcripts per million
- TCIA:
-
The Cancer Immunome Database
- ICI:
-
Immune checkpoint inhibitor
- TIME:
-
Tumor immune microenvironment
References
Hirsch, F. R. et al. Lung cancer: Current therapies and new targeted treatments. Lancet 389(10066), 299–311. https://doi.org/10.1016/S0140-6736(16)30958-8 (2017).
Schabath, M. B. & Cote, M. L. Cancer progress and priorities: Lung cancer. Cancer Epidemiol. Biomark. Prev. 28(10), 1563–1579. https://doi.org/10.1158/1055-9965 (2019).
Torre, L. A., Siegel, R. L. & Jemal, A. Lung cancer statistics. Adv. Exp. Med. Biol. 893, 1–19. https://doi.org/10.1007/978-3-319-24223-1_1 (2016).
Nasim, F., Sabath, B. F. & Eapen, G. A. Lung cancer. Med. Clin. North Am. 103(3), 463–473. https://doi.org/10.1016/j.mcna.2018.12.006 (2019).
Duma, N., Santana-Davila, R. & Molina, J. R. Non-small cell lung cancer: Epidemiology, screening, diagnosis, and treatment. Mayo Clin Proc. 94(8), 1623–1640. https://doi.org/10.1016/j.mayocp.2019.01.013 (2019).
Duruisseaux, M. & Esteller, M. Lung cancer epigenetics: From knowledge to applications. Semin. Cancer Biol. 51, 116–128. https://doi.org/10.1016/j.semcancer.2017.09.005 (2018).
Denisenko, T. V., Budkevich, I. N. & Zhivotovsky, B. Cell death-based treatment of lung adenocarcinoma. Cell Death Dis. 9(2), 117. https://doi.org/10.1038/s41419-017-0063-y (2018).
Chen, Z. et al. Molecular features of lung adenocarcinoma in young patients. BMC Cancer 19(1), 777. https://doi.org/10.1186/s12885-019-5978-5 (2019).
Shao, X., Niu, R., Jiang, Z., Shao, X. & Wang, Y. Role of PET/CT in management of early lung adenocarcinoma. AJR Am. J. Roentgenol. 214(2), 437–445. https://doi.org/10.2214/AJR.19.21585 (2020).
Jonna, S. & Subramaniam, D. S. Molecular diagnostics and targeted therapies in non-small cell lung cancer (NSCLC): An update. Discov. Med. 27(148), 167–170 (2019).
Evison, M. & Limited, A. U. K. The current treatment landscape in the UK for stage III NSCLC. Br. J. Cance. 123(Suppl 1), 3–9. https://doi.org/10.1038/s41416-020-01069-z (2020).
Wang, T., Kong, S., Tao, M. & Ju, S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 19(1), 88. https://doi.org/10.1186/s12943-020-01204-7 (2020).
Dai, D., Wang, H., Zhu, L., Jin, H. & Wang, X. N6-methyladenosine links RNA metabolism to cancer progression. Cell Death Dis. 9(2), 124. https://doi.org/10.1038/s41419-017-0129-x (2018).
Zhou, Z. et al. Mechanism of RNA modification N6-methyladenosine in human cancer. Mol. Cancer. 19(1), 104. https://doi.org/10.1186/s12943-020-01216-3 (2020).
Ma, Z. & Ji, J. N6-methyladenosine (m6A) RNA modification in cancer stem cells. Stem Cells. https://doi.org/10.1002/stem.3279 (2020).
Wu, L., Wu, D., Ning, J., Liu, W. & Zhang, D. Changes of N6-methyladenosine modulators promote breast cancer progression. BMC Cancer 19(1), 326. https://doi.org/10.1186/s12885-019-5538-z (2019).
Maity, A. & Das, B. N6-methyladenosine modification in mRNA: Machinery, function and implications for health and diseases. FEBS J. 283(9), 1607–1630. https://doi.org/10.1111/febs.13614 (2016).
Zhang, C. et al. Reduced m6A modification predicts malignant phenotypes and augmented Wnt/PI3K-Akt signaling in gastric cancer. Cancer Med. 8(10), 4766–4781. https://doi.org/10.1002/cam4.2360 (2019).
Li, J. et al. The m6A demethylase FTO promotes the growth of lung cancer cells by regulating the m6A level of USP7 mRNA. Biochem. Biophys. Res. Commun. 512(3), 479–485. https://doi.org/10.1016/j.bbrc.2019.03.093 (2019).
Xu, J. et al. m6A methyltransferase METTL3 maintains colon cancer tumorigenicity by suppressing SOCS2 to promote cell proliferation. Oncol. Rep. 44(3), 973–986. https://doi.org/10.3892/or.2020.7665 (2020).
Manfredi, M. et al. Mining cancer biology through bioinformatic analysis of proteomic data. Expert Rev. Proteom. 16(9), 733–747. https://doi.org/10.1080/14789450.2019.1654862 (2019).
Tao, Z. et al. Microarray bioinformatics in cancer—A review. J. Buon. 1, 838–843 (2017).
Milan, T. & Wilhelm, B. T. Mining cancer transcriptomes: Bioinformatic tools and the remaining challenges. Mol. Diagn. Ther. 21(3), 249–258. https://doi.org/10.1007/s40291-017-0264-1 (2017).
Zhang, B. et al. m6A regulator-mediated methylation modification patterns and tumor microenvironment infiltration characterization in gastric cancer. Mol. Cancer. 19(1), 53. https://doi.org/10.1186/s12943-020-01170-0 (2020).
Zhang, X. et al. m6A regulator-mediated RNA methylation modification patterns are involved in immune microenvironment regulation of periodontitis. J. Cell Mol. Med. 25(7), 3634–3645. https://doi.org/10.1111/jcmm.16469 (2021).
Leek, J. T. svaseq: Removing batch effects and other unwanted noise from sequencing data. Nucl. Acids Res. 42(21), e161. https://doi.org/10.1093/nar/gku864 (2014).
Leek, J. T., Johnson, W. E., Parker, H. S., Jaffe, A. E. & Storey, J. D. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics 28(6), 882–883. https://doi.org/10.1093/bioinformatics/bts034 (2012).
Zhang, Y. et al. Expression and prognostic significance of m6A-related genes in lung adenocarcinoma. Med. Sci. Monit. 22(26), e919644. https://doi.org/10.12659/MSM.919644 (2020).
Zhou, J. et al. Gene signatures and prognostic values of m6A regulators in clear cell renal cell carcinoma—A retrospective study using TCGA database. Aging (Albany NY). 11(6), 1633–1647. https://doi.org/10.18632/aging.101856 (2019).
Shi, H. et al. Retrospective study of gene signatures and prognostic value of m6A regulatory factor in non-small cell lung cancer using TCGA database and the verification of FTO. Aging (Albany NY). 12(17), 17022–17037. https://doi.org/10.18632/aging.103622 (2020).
Wang, X. et al. An individualized immune prognostic index is a superior predictor of survival of hepatocellular carcinoma. Med. Sci. Monit. 31(26), e921786. https://doi.org/10.12659/MSM.921786 (2020).
Wang, Y., Zhao, W., Liu, X., Guan, G. & Zhuang, M. ARL3 is downregulated and acts as a prognostic biomarker in glioma. J. Transl. Med. 17(1), 210. https://doi.org/10.1186/s12967-019-1914-3.PMID:31234870;PMCID:PMC6591946 (2019).
Hänzelmann, S., Castelo, R. & Guinney, J. GSVA: Gene set variation analysis for microarray and RNA-seq data. BMC Bioinf. 16(14), 7. https://doi.org/10.1186/1471-2105-14-7 (2013).
Zhang, J. et al. Establishing and validating a pathway prognostic signature in pancreatic cancer based on miRNA and mRNA sets using GSVA. Aging (Albany NY). 12(22), 22840–22858. https://doi.org/10.18632/aging.103965 (2020).
Yi, M., Nissley, D. V., McCormick, F. & Stephens, R. M. ssGSEA score-based Ras dependency indexes derived from gene expression data reveal potential Ras addiction mechanisms with possible clinical implications. Sci. Rep. 10(1), 10258. https://doi.org/10.1038/s41598-020-66986-8 (2020).
Xiao, B. et al. Identification and verification of immune-related gene prognostic signature based on ssGSEA for osteosarcoma. Front. Oncol. 15(10), 607622. https://doi.org/10.3389/fonc.2020.607622 (2020).
Li, L. et al. Exploration of immune-related genes in high and low tumor mutation burden groups of chromophobe renal cell carcinoma. Biosci. Rep. 40(7), 1. https://doi.org/10.1042/BSR20201491 (2020).
Xue, Y. et al. Tumor-infiltrating M2 macrophages driven by specific genomic alterations are associated with prognosis in bladder cancer. Oncol. Rep. 42(2), 581–594. https://doi.org/10.3892/or.2019.7196 (2019).
Shen, X. et al. The m6A methylation landscape stratifies hepatocellular carcinoma into 3 subtypes with distinct metabolic characteristics. Cancer Biol Med. 17(4), 937–952. https://doi.org/10.20892/j.issn.2095-3941.2020.0402 (2020).
Du, J. et al. m6A regulator-mediated methylation modification patterns and characteristics of immunity and stemness in low-grade glioma. Brief Bioinform. https://doi.org/10.1093/bib/bbab013 (2021).
Yang, L. et al. Identification of prognosis-related genes in the cervical cancer immune microenvironment. Gene 15(766), 145119. https://doi.org/10.1016/j.gene.2020.145119 (2021).
Luo, J. et al. Comprehensive insights on pivotal prognostic signature involved in clear cell renal cell carcinoma microenvironment using the ESTIMATE algorithm. Cancer Med. 9(12), 4310–4323. https://doi.org/10.1002/cam4.2983 (2020).
Liu, J. et al. Exploration of a novel prognostic risk signatures and immune checkpoint molecules in endometrial carcinoma microenvironment. Genomics 112(5), 3117–3134. https://doi.org/10.1016/j.ygeno.2020.05.022 (2020).
Zaretsky, J. M. et al. Mutations associated with acquired resistance to PD-1 blockade in melanoma. N. Engl. J. Med. 375(9), 819–829. https://doi.org/10.1056/NEJMoa1604958 (2016).
Deng, J. L., Xu, Y. H. & Wang, G. Identification of potential crucial genes and key pathways in breast cancer using bioinformatic analysis. Front. Genet. 2(10), 695. https://doi.org/10.3389/fgene.2019.00695 (2019).
Chen, Y. et al. WTAP facilitates progression of hepatocellular carcinoma via m6A-HuR-dependent epigenetic silencing of ETS1. Mol. Cancer. 18(1), 127. https://doi.org/10.1186/s12943-019-1053-8 (2019).
Chen, R. X. et al. N6-methyladenosine modification of circNSUN2 facilitates cytoplasmic export and stabilizes HMGA2 to promote colorectal liver metastasis. Nat. Commun. 10(1), 4695. https://doi.org/10.1038/s41467-019-12651-2 (2019).
Tong, J., Flavell, R. A. & Li, H. B. RNA m6A modification and its function in diseases. Front. Med. 12(4), 481–489. https://doi.org/10.1007/s11684-018-0654-8 (2018).
Ito, K. & Murphy, D. Application of ggplot2 to Pharmacometric Graphics. CPT Pharmacomet. Syst. Pharmacol. 2(10), e79. https://doi.org/10.1038/psp.2013.56.PMID:24132163;PMCID:PMC3817376 (2013).
Yu, G., Wang, L. G., Han, Y. & He, Q. Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS 16(5), 284–287. https://doi.org/10.1089/omi.2011.0118 (2012).
Kanehisa, M., Sato, Y., Kawashima, M., Furumichi, M. & Tanabe, M. KEGG as a reference resource for gene and protein annotation. Nucl. Acids Res. 44(D1), D457–D462. https://doi.org/10.1093/nar/gkv1070 (2016).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28(1), 27–30. https://doi.org/10.1093/nar/28.1.27.PMID:10592173;PMCID:PMC102409 (2000).
Author information
Authors and Affiliations
Contributions
Q.Z. and L.Y. designed study, analyzed data, and wrote the manuscript. Q.S., W.S., J.L., Y.L., and K.Z. analyzed data and contributed in writing the manuscript. K.Z. and L.Y. supervised research, analyzed data, and wrote the manuscript. All the authors have approved the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zeng, QC., Sun, Q., Su, WJ. et al. Analysis of m6A modulator-mediated methylation modification patterns and the tumor microenvironment in lung adenocarcinoma. Sci Rep 12, 20684 (2022). https://doi.org/10.1038/s41598-022-20730-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-20730-6
- Springer Nature Limited