
Abstract
Through stochastic simulations, accuracies of breeding values and response to selection were assessed under traditional pedigree-(BLUP) and genomic-based evaluation methods (GBLUP) in forest tree breeding. The latter provides a methodological foundation for genomic selection. We evaluated the impact of clonal replication in progeny testing on the response to selection realized in seed orchards under variable marker density and target effective population sizes. We found that clonal replication in progeny trials boosted selection accuracy, thus providing additional genetic gains under BLUP. While a similar trend was observed for GBLUP, however, the added gains did not surpass those under BLUP. Therefore, breeding programs deploying extensive progeny testing with clonal propagation might not benefit from the deployment of genomic information. These findings could be helpful in the context of operational breeding programs.
Similar content being viewed by others


Introduction
Genomic selection (GS) has garnered increased attention for its potential to deliver significant genetic gains per unit of time and cost. In traditional genetic evaluation, phenotypic performance is regressed on the identity-by-descent (IBD) probabilities inferred from pedigrees, producing the best linear unbiased predictions (BLUP) of additive genetic (breeding) values. However, when genetic covariance is estimated from DNA markers (e.g., SNP), genomic-based predictions (GBLUP) of true breeding values may provide additional benefits over that of the traditional BLUP, mainly when used in the context of GS.
In forest trees, recurrent GS is based on establishing training populations (i.e., marker-trait associations). These, in turn, eliminate, to a certain extent (loss of accuracy over time), the needed phenotypic evaluation phase of selection candidates. Using computer simulation, the theoretical potential of GS in tree breeding was demonstrated with the conclusion that it could radically increase breeding efficiency1,2. They assessed GS scenarios under variable narrow-sense heritability (h2), number of QTLs, marker density, and effective population size (Ne). Recent studies utilizing empirical data reported additional prediction accuracies3,4.
Unlike the studies mentioned above, the genetic response to selection was recently simulated in a seed production population, providing a more realistic basis for comparing actual genetic gains available in forest reproductive material5. In particular, they expressed the efficiency of GBLUP/BLUP protocols based on the respective ranking of selection candidates and genetic gains provided in forest reproductive material. Furthermore, a combination of low h2, high Ne, and dense marker coverage resulted in the maximum genomic prediction efficiency and added within-family selection accuracy (exploitation of the Mendelian sampling term). Adoption of GS in operational breeding programs is challenging in predominantly outbred forest tree species. These are characterized by: (1) long generation intervals, (2) sensitivity to inbreeding, i.e., breeding relying on high Ne, (3) fast gametic-phase disequilibrium decay, (4) considerable temporal and spatial environmental sensitivity, and (5) large genome sizes (especially in conifers), requiring dense SNP genotyping involving many individuals6. Regarding forest tree species' spatial and temporal sensitivity, excessive field experiments are required to assess genotype-by-environment interactions and age-age correlations on top of complicated covariance structure among multiple traits and its shift with selection. These issues are specific to populations, traits, ages, and environmental conditions and require testing hundreds of families in several environments over an excessive time scale7.
Genetically improved seed production predominantly relies on seed orchards, i.e., bulk seed from open-pollinated crosses, capturing additive genetic variance. Additionally, one can exploit non-additive genetic effects through the mass deployment of full-sib families (dominance) or clonal mixtures (dominance + epistasis). Apart from deploying improved forest reproductive material, one can clonally replicate selection candidates (offspring genotypes) in progeny trials, enhancing the precision of forward selection8,9. In Sweden, clonal replication in progeny testing has provided operational benefits in the Norway spruce breeding program by boosting the within-family response to selection while minimizing genetic diversity loss9,10. Under fixed progeny test size, a trade-off exists between the family size and the number of clonal propagules per genotype (NR), i.e., clonal size11.
Here, building on our earlier stochastic simulations5, we evaluated the impact of clonal replication in progeny testing on the efficiency of BLUP and GBLUP evaluation and the actual genetic response realized in seed orchards. Specifically, we assessed the combined effect of marker density, effective population size (Ne), family size, and NR.
Methods
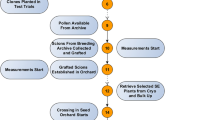
We utilized a stochastic simulation model developed in R12. We created parental and offspring populations5 using the function “glSim” implemented within the R package “adegenet”13 to generate allelic frequencies in a founder population. Linkage disequilibrium (LD) was set to reflect typical values in outcrossed forest trees6. We generated offspring populations (50 parents) of two different sizes using a single pair mating design (SPM) to evaluate the impact of family size (80/160), so the overall population size varied from 2050 to 4050 individuals.
Bi-allelic marker data were simulated for the full-sib families using the function “genomesim” implemented within the R package “pedantics”14. We set the number of markers (SNPs) per centiMorgan (cM) to 1, 5, and 10 covering chromosome lengths of 120 cM. In total, two chromosomes (linkage groups) were simulated, comparably to the previous studies3, with the maximum number of markers equal to 2400. As the impact of traits' genetic architecture was evaluated earlier5, we modeled only a fixed QTL number (NQTL = 200). QTL effects were randomly assigned to selected loci and were sampled from a standardized normal distribution to emulate polygenic traits. We generated phenotypic data as the sum of allelic effects across all QTL loci with the addition of residual effects reflecting h2 = 0.2, which approximates growth traits in forest tree species. Clonal replicates were derived as the sum of a genotypic value and the average of NR independent samples of residual effects. We conducted 200 independent stochastic iterations of the above scenarios.
We conducted separate genetic evaluations in ASReml software V.315 for pedigree- (BLUP) and genomic-based (GBLUP) relationships to predict offspring breeding value (BV; i.e., forward selection) using the animal model in the REML framework16. The marker-based relationship matrix G was constructed as follows17:
where Z is M—P, M is the marker matrix containing genotypes coded as 0, 1, and 2 for the first allele homozygote, heterozygote, and second allele homozygote, and P is the vector of doubled frequencies of the second allele, p is the frequency of the second allele at the loci.
Breeding value (BV) accuracy was calculated as the correlation between their predicted (genetic evaluation of both BLUP and GBLUP strategies) and true values (as determined by the sum of simulated allelic effects). Next, the reported standard error of the overall accuracy across 200 iterations (calculated as the respective standard deviation divided by the square root of the iteration count). Following the genetic evaluation, a set of unrelated offspring with top breeding values (considered as parents in seed orchards) was chosen by mathematical programming18 to meet the predetermined effective population size (Ne = 5, 10, 20, and 25), thus maximizing the genetic response19. The method selects the best set of offspring individuals, maximizing the average additive genetic value (genetic gain) while meeting the declared effective population size (constraint). Relatedness among the selected trees was not permitted to avoid inbreeding in the seed orchard's crop. Optimization was conducted in Gurobi software 18. Details on the optimization algorithm are provided in Lstibůrek and Hodge 19.
Results
Accuracy of predicted breeding values
Table 1 provides BV's accuracies at variable family size, NR, and marker density. Under GBLUP, a steady increase in BV's accuracy is visible with higher marker density (mainly between 1 and 5 SNPs/cM). BV's accuracy under GBLUP was greater than BLUP under all investigated scenarios after marker density reached 5 SNPs/cM. However, under 1 SNP/cM, BV's accuracies of GBLUP were inferior to BLUP irrespective of NR and the family size.
Clonal replication boosted accuracies of both BLUP and GBLUP evaluations across all marker densities and family sizes. The difference is visible primarily between one to six clonal copies, while additional clonal replications (up to 12) provided lower increments. At low marker density (1 SNP/cM), the accuracy of GBLUP ranged from 62 to 74% of that under BLUP at both family sizes. Assuming the family size 80, clonal replication reduced relative accuracy of GBLUP over BLUP, e.g., reduction from 72% (NR = 1) to 66% (NR = 6) to 62% (NR = 12). The same observation was made under 5 SNPs/cM, namely the reduction from 112% (NR = 1) to 109% (NR = 6) to 105% (NR = 12). Similarly, under 10 SNPs/cM, relative accuracies dropped from 115% (NR = 1) to 111% (NR = 6) to 106% (NR = 12). A similar trend was observed under family size 160. As expected, absolute accuracies were boosted by family size under both BLUP and GBLUP except for the lowest marker coverage.
Selection response
In Table 2, we present differences in the standardized response to selection between GBLUP and BLUP. Relative differences are provided in Table S2. The impact of added marker density was most significant between 1 and 5 SNPs/cM, and it was diminishing towards the 10 SNPs/cM, primarily under the family size 80.
Under clonal replication (NR = 6–12), GBLUP yielded minor benefit in selection response under 5–10 SNPs/cM, but the difference was not statistically significant (alpha = 0.05). The impact of NR on the absolute selection response of both methods is prominent irrespective of the marker density, primarily between 1 and 6 clonal replicates (yet additional gain was generated under NR = 12). While the clonal replication improves gains of both evaluation methods, the major boost of selection response was observed under the BLUP. Under the lowest marker density, i.e., 1 SNP/cM, BLUP generated a higher selection response in the range of app. 0.4–1.3 standard deviations. Under NR = 1 (no cloning), GBLUP was superior to BLUP, primarily under higher marker densities and larger families (see Table 2, NR = 1). The above trends were generally true across the range of Ne, yet the added difference between the two methods was diluted at larger NR. Under NR = 1 and moderate marker density (5 SNP/cM), a larger family size (160) boosted the difference between the two methods. Note that values in Table 2 are differences among standardized genetic gains of BLUP and GBLUP. Thus, they are not reflecting baseline genetic gains, e.g., a significant drop of selection response with added Ne. Under NR = 1, low marker density (1 SNP/cM), Ne = 5, the absolute difference − 0.52 (Table 2) reflects standardized gains of 0.88 (GBLUP) and 1.4 (BLUP). Assuming the same parameters, but Ne = 25, the absolute difference − 0.38 reflects a significant drop in standardized gains due to lower selection intensity, i.e., 0.13 (GBLUP) and 0.51 (BLUP). For clarity, standardized genetic gains of all strategies are provided in Table S1. Relative genetic gains, i.e., ratios of the standardized genetic response of GBLUP/BLUP, are provided in Table S2.
Discussion
Here, we estimated the relative efficiency of the genomic evaluation protocol over the traditional phenotypic alternative. Our findings resemble animal and plant breeding studies, i.e., the added prediction accuracy and anticipated selection response under genomic evaluation. This relative superiority of GBLUP is conditional on dense marker coverage, lower narrow-sense heritabilities, and the presence of the population-wise linkage disequilibrium1,3,5,20,21. In operational tree breeding programs, additional factors contribute to the breeding efficiency, e.g., sizes of breeding and production populations, mating design, progeny test size and configuration, maximum acceptable inbreeding rate, extend of genotype by environment interactions, cost and time parameters of breeding activities, etc. (see7 for introduction to forest tree genetics and breeding).
The novelty of our comparison is attributed to the inclusion of clonal replication in progeny test trials as used in operational tree breeding programs to boost selection accuracy. The main added value of the GBLUP evaluation is its ability to capitalize on capturing within-family additive genetic variance and unmasking cryptic relatedness22,23. Under all investigated scenarios, the relative genetic gain efficiency of GBLUP decreased with added clonal propagules per offspring individual (NR). Under 1 SNP/cM, BLUP provided a significantly higher genetic response over the GBLUP across the whole range of Ne and family sizes. Both evaluations yielded comparable genetic gains with denser marker coverage (5–10 SNPs/cM); differences were not significant (alpha = 0.05). This finding implies that combining both cloning and genomic evaluation does not bring added genetic response. Thus, our results could inspire breeders to consider two broader alternatives. One involves investing resources into clonal test trials, the other one to genomic evaluation. In agreement with previous studies8,9, relatively low NR (6) provided sufficient accuracy. While both strategies benefited in prediction accuracies from the added NR, their relative efficiency was equalized by applying diversity constraint (Ne) in selection. This is a clear message to operational forest tree breeding programs. Without cloning, the superiority of GBLUP is limited to low h2, large family sizes, and higher marker coverage (5–10 SNPs/cM). Genetic response in smaller families is limited due to the model oversaturation under dense marker coverage23. On the contrary, large family sizes (160 offspring per cross) become impractical in many species, even though they are not prone to oversaturation and provide options for higher selection intensity (larger number of selection candidates). GBLUP provided no additional benefit over the BLUP alternative across the diversity range in production populations scenarios (seed orchards) with clonal replication.
There are practical scenarios under which GBLUP could become economically more feasible. These include programs with too costly or unavailable clonal propagation technology. In analogy, SNP genotyping platform has been developed and is currently operationally feasible in a limited number of forest tree species. As forest trees are long-lived perennials with generational intervals spanning decades, the principal added benefit of genomic selection is reducing the breeding cycle's length. Therefore, GBLUP is becoming a viable platform in this context. Our conclusions are relevant to full-scale operational tree breeding programs that capture general combining ability with repeated cycles of control crosses (single-pair mating, progeny testing, and selection). Adaptive genetic response in natural populations, in theory, could be enabled by the GBLUP evaluation based on the SNP chip platform. However, this is limited by the magnitude of genetic covariance, i.e., the product of genetic coancestry and the respective genetic variance in natural populations. Future research could investigate, by stochastic simulation, the genomic-based single-step model (HBLUP) augmented by clonal replication in progeny testing.
Data availability
In our study, we described a stochastic simulation model and compared hypothetical breeding strategies. No real-world data of any species have been used throughout the study. However, output data have been outlined and published in tables and figures included in the manuscript. The complete R code was submitted as a compressed folder within supplements (S3 file).
References
Grattapaglia, D. & Resende, M. D. V. Genomic selection in forest tree breeding. Tree Genet. Genomes 7, 241–255 (2010).
Iwata, H., Hayashi, T. & Tsumura, Y. Prospects for genomic selection in conifer breeding: A simulation study of Cryptomeria japonica. Tree Genet. Genomes 7, 747–758 (2011).
Denis, M. & Bouvet, J. M. Efficiency of genomic selection with models including dominance effect in the context of Eucalyptus breeding. Tree Genet. Genomes 9, 37–51 (2013).
Id, Y. L. & Dungey, H. S. Expected benefit of genomic selection over forward selection in conifer breeding and deployment. 13, 1–21 (2018).
Stejskal, J., Lstibůrek, M., Klápště, J., Čepl, J. & El-Kassaby, Y. A. Effect of genomic prediction on response to selection in forest tree breeding. Tree Genet. Genomes 14, 1–9 (2018).
Neale, D. B. & Savolainen, O. Association genetics of complex traits in conifers. Trends Plant Sci. 9, 325–330 (2004).
White, T. L., Adams, W. T. & Neale, D. B. Forest genetics. (Cabi, 2007).
Russell, J. H. & Libby, W. J. Clonal testing efficiency: The trade-offs between clones tested and ramets per clone. Can. J. For. Res. 16, 925–930 (1986).
Rosvall, O., Lindgren, D. & Mullin, T. J. Sustainability robustness and efficiency of a multi-generation breeding strategy based on within-family clonal selection. Silvae Genet. 47, 307–321 (1998).
Lindgren, D., Danusevicius, D. & Rosvall, O. Unequal deployment of clones to seed orchards by considering genetic gain, relatedness and gene diversity. Forestry 82, 17–28 (2009).
Russell, J. H. & Loo-Dinkins, J. A. Distribution of testing effort in cloned genetic tests. Silvae Genet. 42, 98 (1993).
R Core Team, Rf. R: A language and environment for statistical computing. (2013).
Jombart, T. & Ahmed, I. adegenet 1.3–1: New tools for the analysis of genome-wide SNP data. Bioinformatics 27, 3070–3071 (2011).
Morrissey, M. B. & Wilson, A. J. Pedantics: An r package for pedigree-based genetic simulation and pedigree manipulation, characterization and viewing. Mol. Ecol. Resour. 10, 711–719 (2010).
Gilmour, A. R., Gogel, B. J., Cullis, B. R., Welham, S. J. & Thompson, R. ASReml user guide release 4.1 functional specification. Hemel Hempstead VSN Int. Ltd (2015).
Mrode, R. A. Linear models for the prediction of animal breeding values. (Cabi, 2014).
VanRaden, P. M. Efficient methods to compute genomic predictions. J. Dairy Sci. 91, 4414–4423 (2008).
Gurobi Optimization, I. Gurobi optimizer reference manual, version 6.0. http//www.gurobi.com. Retrieved (2014).
Lstibůrek, M., Hodge, G. R. & Lachout, P. Uncovering genetic information from commercial forest plantations: Making up for lost time using “Breeding without Breeding”. Tree Genet. Genomes 11, 55 (2015).
Henryon, M. et al. Pedigree relationships to control inbreeding in optimum-contribution selection realise more genetic gain than genomic relationships. Genet. Sel. Evol. 51, 1–12 (2019).
Jighly, A. et al. Boosting genetic gain in allogamous crops via speed breeding and genomic selection. Front. Plant Sci. 10, 1364 (2019).
Habier, D., Fernando, R. L. & Dekkers, J. C. M. The impact of genetic relationship information on genome-assisted breeding values. Genetics 177, 2389–2397 (2007).
Habier, D., Fernando, R. L. & Garrick, D. J. Genomic BLUP decoded: A look into the black box of genomic prediction. Genetics 194, 597–607 (2013).
Acknowledgements
Funding received from: The project FORGENRES benefits from a 835 000 € grant from the Norway Grants and Technology Agency of the Czech Republic within the KAPPA Programme; the project EXTEMIT-K: “Building up an excellent scientific team and its spatiotechnical background focused on mitigation of the impact of climatic changes to forests from the level of a gene to the level of a landscape at the FFWS CULS Prague”, Grant No. CZ.02.1.01/0.0/0.0/15_003/0000433 financed by OP RDE.
Author information
Authors and Affiliations
Contributions
J.S. and M.L. coordinated the research activities. J.K., J.Č., J.S., and M.L. were involved in the coding of stochastic simulations. J.S., M.L., J.K., and Y.K. wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Stejskal, J., Klápště, J., Čepl, J. et al. Effect of clonal testing on the efficiency of genomic evaluation in forest tree breeding. Sci Rep 12, 3033 (2022). https://doi.org/10.1038/s41598-022-06952-8
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-022-06952-8
- Springer Nature Limited