
Abstract
In order to explore the role of exosomal circRNAs in the occurrence and development of sepsis, we looked for potential diagnostic markers to accurately identify sepsis and to lay a molecular basis for precise treatment. Ultracentrifugation was used to extract exosomes from the serum of patients with sepsis and healthy individuals. Then, changes in circRNA expression in exosomes were studied by circRNA microarray analysis. Gene ontology (GO) analysis and Kyoto City Encyclopaedia of Genes and Genomes (KEGG) pathway analysis were used to annotate the biological functions and pathways of genes, and a circRNA-miRNA-mRNA regulatory network was constructed. In the microarray analysis, 132 circRNAs were significantly differentially expressed, including 80 and 52 that were upregulated and downregulated, respectively. RT-qPCR verified the results of microarray analysis: hsa_circRNA_104484 and hsa_circRNA_104670 were upregulated in sepsis serum exosomes. ROC analysis showed that hsa_circRNA_104484 and hsa_circRNA_104670 in serum exosomes have the potential to be used as diagnostic markers for sepsis. The circRNA-miRNA-mRNA network predicted the potential regulatory pathways of differentially expressed circRNAs. There are differences in the expression of circRNA in serum exosomes between patients with sepsis and healthy individuals, which may be involved in the occurrence and development of the disease. Among them, elevations in hsa_circRNA_104484 and hsa_circRNA_104670 could be used as novel diagnostic biomarkers and molecular therapeutic targets.
Similar content being viewed by others

Introduction
Sepsis is defined as life-threatening organ dysfunction and is not a specific disease, but rather a syndrome of physiological, pathological, and biochemical abnormalities caused by the host's unregulated response to infection1. Sepsis is a heterogeneous disease state that progresses rapidly, and its early diagnosis and intervention can significantly improve prognosis2. Our diagnosis of sepsis mainly relies on Sequential Organ Failure Assessment (SOFA) scoring system, which has certain limitations; currently, there is no ‘gold standard’ for laboratory diagnosis. With the development of high-throughput sequencing technology, genomics and metabolomics analyses have found that the levels of various genes and metabolites in sepsis have changed, and that the changes occur earlier than clinical symptoms3,4. Identifying these molecular changes in sepsis is highly valuable for understanding the course of the disease, and for predicting prognosis and response to treatment. Exploring the changes in sepsis at cellular and molecular levels is helpful to explore the nature of its pathogenesis and may help to identify the causes of heterogeneity in the body's response5. Individualised therapy targeting the core molecules of the disease can improve the efficiency of the treatment and reduce toxicity. Therefore, these differentially expressed molecules may serve as diagnostic markers for sepsis and may become targets for molecular targeted therapy.
Exosomes are small extracellular vesicles derived from the endosomal system, ranging from 40 to 160 nm (about 100 nm on average) in diameter6. In sepsis, exosomes are secreted by a variety of cells (including mesenchymal stem cells and macrophages, among others), and act on recipient cells (e.g., cardiomyocytes, macrophages, vascular endothelial cells) to promote inflammation, inhibit inflammation, or regulate immunity7,8,9. Their contents are rich and diverse, containing a variety of proteins, DNA, RNA (e.g., mRNA, miRNA, lncRNA, circRNA), amino acids, and metabolites6. The uptake of cytoplasmic components during exosomal biogenesis is not random, but is a highly regulated and selective process, which is very important for disease identification and diagnosis10. The cell-free RNA in the blood is easily inactivated by endogenous RNase, while RNA encapsulated in exosomes can be prevented from degradation by RNase and can exist stably11. In addition, the exosomes released to the outside of cells exist in a variety of body fluids and are easy to separate and extract11,12. These characteristics give exosomes diagnostic and therapeutic potential.
CircRNA is a large class of non-coding RNAs produced by reverse splicing events13. CircRNAs are produced in the nucleus and are then transported to the cytoplasm. They have the characteristics of tissue specificity, cell specificity, high stability, and species conservation14. Some can be distributed to exosomes, where they are enriched and stably exist15,16. In disease states, the expression level of exosome circRNA changes, and it plays a regulatory role in cell proliferation, tumour metastasis, and drug resistance, among other processes17. CircRNAs are involved in the occurrence and progression of various diseases through multiple mechanisms. For example, circRNAs act as miRNA sponges to regulate gene expression and participate in the occurrence and development of tumours13; they also act as a protein sponge to mediate the immune response during viral infection18.
Numerous studies have shown that the expression of exosomal circRNAs is different between patients and healthy people, and its detection can help to identify patients. Therefore, exosomal circRNAs may be used as novel disease diagnostic markers19. To date, there have been no reports on the expression or role of exosomal circRNAs in sepsis. This study aimed to detect circRNAs in serum exosomes of patients with sepsis and to explore their value in the diagnosis of sepsis and in molecular targeted therapy.
Materials and methods
Patient samples and ethics statement
In this study, a total of 25 patients with sepsis who underwent treatment at the Second Hospital of Jilin University from September 2018 to January 2019 were included, in addition to 22 healthy individuals. Sepsis was defined according to the Sepsis-3 criteria1. All study participant’s peripheral blood samples (4–5 mL) were collected in the early phase (within 24 h) of the diagnosis of sepsis and centrifuged at 3000 rpm for 10 min to obtain the serum, which was stored at − 80 °C after being labelled. The patients’ clinical and laboratory data are shown in Table 1. This study was approved by the Ethics Committee of the Second Hospital of Jilin University. All experiments were performed in accordance with relevant named guidelines and regulations. All participants signed an informed consent form.
Exosome collection
We used ultracentrifugation to extract exosomes from the serum, and the whole process was completed at 4 °C. First, the serum was centrifuged at 2000 × g for 30 min to remove dead cells and was then centrifuged at 10,000 × g for 30 min to remove cell debris and impurities. Then, the exosomes were preliminarily precipitated by centrifugation at 110,000 × g for 80 min. Phosphate buffer saline (PBS) solution was added to wash the soluble protein impurities, and then the sample was centrifuged again at 110,000 × g for 80 min to obtain pure exosomes. Finally, the pellet was resuspended in PBS solution (100μL PBS solution per 1 mL of serum) and was stored in a − 80 °C freezer.
Western blotting analysis
Exosomal marker proteins were detected by immunoblotting. Protein was extracted from the same volume of exosomes, and the protein concentration of exosomes was quantified using the BCA method (Beyotime, China). Then, 20 μg of exosomal protein was separated by electrophoresis on a 12% SDS-PAGE gel and was then transferred to a PVDF membrane (Millipore, USA). Immunoblotting was performed with anti-CD63 and anti-TSG101 antibodies (Affinity, USA) at 4 °C. The primary antibodies were then detected with a horseradish peroxidase-conjugated secondary antibody (#SA00001-1 or #SA00001-2; Proteintech Group, USA). Finally, the ECL chemiluminescence agent (Thermo Fisher Scientific, USA) was used to display protein bands, and the results were recorded with photos.
Electron microscopy
For electron microscopy, 5 μl of the exosome suspension was spotted on copper mesh and dried at room temperature. The sample was then negatively stained with 5 μl of 2% (w/v) phosphotungstic acid solution. The morphology of exosomes was observed at 80 kV under a transmission electron microscope (JEM-1400, JEOL, Japan), and the results were photographed.
RNA extraction and quality control
Total RNA was extracted from the exosome suspension using the TRI Reagent BD (Molecular Research Center, Inc., USA) according to the manufacturer’s protocol. The total RNA from each exosome sample was quantified and its purity was evaluated using a NanoDrop 2000 ultra-micro spectrophotometer (Thermo Fisher Scientific, USA).
circRNA microarray analysis
CircRNA microarray analysis was performed on serum exosomes from three people with sepsis and three healthy persons. According to the manufacturer's protocol (Arraystar Inc., USA), sample labelling and microarray hybridization were performed. First, RNA was fluorescently labelled. Rnase R reagent (Epicenter, Inc., USA) was used to digest total RNA to remove linear RNA and enrich circRNAs. The enriched circRNAs were then transcribed into fluorescently labelled cRNA using a random priming method (Arraystar Super RNA Labelling Kit; Arraystar, USA). The labelled cRNAs were purified using the RNeasy Mini Kit (Qiagen, Germany). Microarray hybridisation was then performed in an Agilent Hybridisation oven. The fluorescently labelled cRNAs were cleaved into fragments and were then hybridised on the circRNA expression microarray slide. After hybridisation was completed, the hybridised microarrays were washed, fixed, and scanned using the Agilent Scanner G2505C. Agilent Feature Extraction software was used to extract raw data from the scanned images. Quantile normalisation of raw data was performed using the limma package (version 3.48.0)20 in R, and the circRNAs labelled by the software were retained for subsequent difference analysis. A t-test was used to estimate the statistical significance of the difference. Fold changes and p-values were used to screen for significant differences in the expression of circRNAs between the two groups of samples. Volcano plots and heat maps were used to display differentially expressed circRNAs.
real-time quantitative PCR (RT-qPCR) analysis
Total RNA was extracted from serum exosomes of 25 sepsis patients and 22 controls. Real-time quantitative polymerase chain reaction (RT-qPCR) was used to verify the experiment. The sequences of the primers used in the experiment are shown in Table 2. Total RNA was reverse transcribed into complementary DNA (cDNA) using a PrimeScript RT reagent kit (Takara, Japan) according to the manufacturer’s protocol. Real-time quantitative PCR reactions were then carried out with a real-time PCR system (LightCycler480, Roche, Switzerland) using TB Green Premix Ex Taq II (Takara, Japan). The PCR conditions were 95 °C for 30 s, followed by 40 cycles at 95 °C for 10 s, and 60 °C for 60 s. β-actin was used as a reference gene, and all qPCR reactions were repeated three times. The 2-△△CT value reflects the relative expression level of circRNAs.
Functional analysis
Arraystar microRNA prediction software was used to predict miRNAs downstream of differentially expressed circRNAs. Then, the interactions between circRNA-microRNAs are explained in detail. TargetScan (http://www.targetscan.org/vert_71/), miRDB (http://www.mirdb.org/), and miRTarBase (http://mirtarbase.mbc.nctu.edu.tw/php/index.php) were used to predict the potential targets of miRNAs. The common genes in the three databases were collected using Venn diagrams. The circRNA–miRNA–mRNA regulatory map was visualised using Cytoscape 3.8.0. Gene ontology (GO) analysis was used to annotate the biological functions of genes in the ceRNA network, including molecular functions (MF), biological pathways (BP), and cellular components (CC). Kyoto City Encyclopaedia of Genes and Genomes (KEGG) Enrichment Analysis was used to evaluate the biological pathways of genes21. The enrichment of MF, BP, CC, and pathways of genes were annotated with DAVID 6.8 (https://david.ncifcrf.gov/) which is an online biological tool.
Statistical analysis
SPSS software (version 23.0, IBM, Chicago, IL, USA) was used for statistical analysis. If the data of continuous variables were distributed normally, the data were analysed using t-tests; results are expressed as the mean ± standard deviation. If data were non-normal, the Mann–Whitney U test was used, and the data are expressed in percentile form. Data of categorical variables between groups were tested using the Chi-square test. A p value of < 0.05 means that the difference is statistically significant. The receiver operating characteristic (ROC) curve was constructed to evaluate the diagnostic ability of exosomal circRNAs for sepsis. The area under the ROC curve (AUC) was used to evaluate the diagnostic efficacy of circRNA. The Youden Index was used to determine the optimal cut-off value, sensitivity and specificity (Youden Index = Sensitivity + Specificity-1). The highest Youden index corresponds to the optimal cut-off value, sensitivity and specificity.
Ethics approval and consent to participate
This study was approved by the Ethics Committee of the second hospital of Jilin University. All participants were informed and willing to sign informed consent.
Consent for publication
All the authors read and consented to the publication of the manuscript.
Results
Characterization of circulating serum exosomes
The serum exosome was confirmed by transmission electron microscopy (TEM) and WB for CD63 or TSG101 (Fig. 1a). The exosomes are round or oval ‘cup-shaped’, with a diameter in the range of 40–160 nm. CD63 and TSG101 showed positive expression in WB (Fig. 1b).

(a) Electron micrographs and (b) WB results of serum exosomes.
Identification of differentially expressed circRNAs
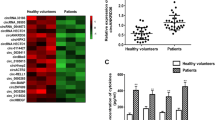
We used circRNA microarray technology to detect changes in the circRNA expression profile of serum exosomes in sepsis. After scanning the fluorescent signal of circRNA microarray hybridisation, a total of six scanning pictures of the sepsis and control groups were obtained (Fig. 2a). The box plot shows the results of the quality control analysis of the microarray data (Fig. 2b). Volcano plots and scatter plots were used to visually show the differences in circRNA expression between the two groups. In the volcano map (Fig. 2c), the vertical lines represent 1.5 times up and down, and the horizontal lines represent p ≤ 0.05. Red dots indicate circRNAs that are significantly differently expressed, and grey dots indicate circRNAs that are not significantly differently expressed. In the scatter plot (Fig. 2d), the X-axis and Y-axis represent the normalised signal values of the two groups of samples, respectively, and the green line is the fold line. Plots distributed above the upper green line and below the lower green line represent significantly differently expressed circRNAs.


(a) The probe fluorescence signal displayed in each microarray scanning picture was uniform and clear. (A, B, C: sepsis group, a, b, c: control group). (b) Box plot: The abscissa represents each sample, and the ordinate represents the normalized intensity value. The expression of circRNAs in each sample was almost the same after normalization. (c) Volcano map: Differentially expressed circRNAs between sepsis and healthy human serum exosomes. (d) Scatter plot: Changes of circRNAs expression levels between sepsis and healthy human serum exosomes. (e) Cluster analysis: the distinguishable circRNA expression profile between sepsis and healthy human serum exosomes. The quantile normalisation and difference analysis were performed using limma package (version 3.48.0) in R. The Volcano map and Scatter plot were performed using python (version 2.7). Cluster analysis was performed using gplots package (version 3.1.1) in R.
A total of 13228 circRNAs were detected by circRNA microarray analysis, of which 6247 were upregulated and 6981 were downregulated. Among them, 132 circRNAs were differentially expressed (p < 0.05, fold change > 1.5), including 80 upregulated and 52 downregulated circRNAs. Specific details are shown in Tables 3 and 4. Then, cluster analysis was performed on the significantly differentially expressed circRNAs to visually display the differentially expressed circRNAs and to test their rationality and accuracy. As shown in the heat map (Fig. 2e), red represents highly expressed circRNAs and green represents low-expressed circRNAs. The results showed distinguishable circRNA expression profiles between the two groups of samples.
RT-qPCR validation of the differentially expressed circRNAs
RT-qPCR was used to verify the differentially expressed circRNAs in sepsis. We selected five circRNAs that are most likely to be related to sepsis for verification based on the fold changes in microarray analysis: hsa_circRNA_406194, hsa_circRNA_104670, hsa_circRNA_104484, hsa_circRNA_103864, and hsa_circRNA_101491. Because the microarray analysis may contain false positive results, we first verified in 3 sepsis patients and 3 healthy volunteers that had been tested by microarray to confirm the accurate expression of circRNAs. The expression levels of hsa_circRNA_406194 (0.95 ± 0.32 to 1.05 ± 0.37; p = 0.751), hsa_circRNA_104670 (2.37 ± 0.19 to 1.02 ± 0.23; p = 0.001), hsa_circRNA_104484 (1.98 ± 0.08 to 1.01 ± 0.15; p = 0.001), hsa_circRNA_103864 (1.62 ± 0.68 to 1.04 ± 0.36; p = 0.265), and hsa_circRNA_101491 (1.18 ± 0.55 to 1.03 ± 0.28; p = 0.699) (Fig. 3). Among these five circRNAs, only hsa_circRNA_104484 and hsa_circRNA_104670 were significantly increased.

RT-qPCR verification of five circRNAs in microarray samples. The drawings were performed using GraphPad Prism software (version 8.0, https://www.graphpad.com/scientific-software/prism/).
We further verified the expression levels of hsa_circRNA_104484 and hsa_circRNA_104670 in the serum exosomes of 22 patients with sepsis and 19 controls collected subsequently. As shown in Fig. 4, the expression of hsa_circRNA_104484 (1.829 ± 0.718 to 1.124 ± 0.506; p = 0.005) and hsa_circRNA_104670 (2.045 [1.319–3.049] to 0.948 [0.684–1.639]; p = 0.003) in serum exosomes of patients with sepsis increased, and the expression differences were statistically significant, which was consistent with the results of microarray analysis.

Expression of hsa_circRNA_104484 and hsa_circRNA_104670 in the serum exosomes of 22 patients with sepsis and 19 controls.
ROC analysis of serum exosomal hsa_circRNA_104484 and hsa_circRNA_104670 in sepsis
The results of qPCR were used to construct the ROC curve to evaluate the diagnostic value of exosomal hsa_circRNA_104484 and hsa_circRNA_104670 in sepsis (Fig. 5). Compared with healthy subjects, the AUC of hsa_circRNA_104484 in sepsis exosomes was 0.782 (95% confidence interval [CI]: 0.643–0.921; p < 0.05), the sensitivity and specificity were 0.545 and 0.947, respectively. The highest Youden index was 0.492 and the corresponding optimal cut-off value was 31.901. The AUC of hsa_circRNA_104670 was 0.775 (95% CI: 0.632–0.919; p < 0.05), and the sensitivity and specificity were 0.591 and 0.895, respectively. The highest Youden index was 0.486 and the corresponding optimal cut-off value was 1.357. The results indicate that hsa_circRNA_104484 and hsa_circRNA_104670 have a medium diagnostic value and have the potential to be used as diagnostic markers in sepsis.

ROC curve for hsa_circRNA_104484 and hsa_circRNA_104670.
Identification of circRNA‐targeting miRNAs and construction of circRNA‐miRNA‐mRNA networks
Arraystar microRNA prediction software was used to predict the miRNAs targeted by hsa_circRNA_104484 and hsa_circRNA_104670. The results showed that the miRNAs targeted by hsa_circRNA_104484 were hsa-miR-34b-5p, hsa-miR-508-3p, hsa-miR-378a-3p, hsa-miR-378d, and hsa-miR-30c-2-3p. Further, the miRNAs targeted by hsa_circRNA_104670 were hsa-miR-17-3p, hsa-miR-433-3p, hsa-miR-367-5p, hsa-miR-335-3p, and hsa-miR-642a-5p. The interaction between circRNA-microRNA is annotated in detail, and the results are shown in Fig. 6a. The ceRNA network was used to visually show the relationship between hsa_circRNA_104484 and hsa_circRNA_104670, miRNAs, and target genes (Fig. 6b).


Prediction of circRNA-miRNA-mRNA regulatory relationship. (a) Annotation of detailed regulatory relationship between hsa_circRNA_104484, hsa_circRNA_104670 and miRNAs. (b) circRNA‐miRNA‐mRNA network established using hsa_circRNA_104484 and hsa_circRNA_104670.
Prediction of the potential functions of target genes
GO analysis results showed that the biological process and molecular functions of target genes were concentrated in several aspects, such as ‘negative regulation of transcription from the RNA polymerase II promoter’, ‘transcription’, ‘positive regulation of transcription’, ‘negative regulation of transcription’, ‘positive regulation of transcription from the RNA polymerase II promoter’, ‘protein binding’, ‘DNA binding’, ‘transcriptional activator activity’, ‘RNA polymerase II transcription factor activity’, ‘transcription factor activity’, and ‘transcriptional repressor activity’ (Fig. 7a). Most of them were related to the transcriptional regulation of gene expression. Therefore, hsa_circRNA_104484 and hsa_circRNA_104670 might participate in the process of sepsis by regulating transcription.

Functional analysis of circRNA. (a) Gene Ontology Analysis. (b) KEGG pathway Enrichment Analysis. The drawings were performed using Microsoft Excel (version 16.43, https://www.microsoft.com/zh-cn/microsoft-365/excel).
KEGG pathway analysis results show that the target gene-related signalling pathways are the PI3K-Akt signalling pathway, signalling pathways regulating the pluripotency of stem cells, the MAPK signalling pathway, hepatitis B, viral carcinogenesis, osteoclast differentiation, hepatitis C, HTLV-I infection, TNF signalling pathway, and the insulin signalling pathway, among others (Fig. 7b). Among them, the PI3K-Akt signalling pathway22, MAPK signalling pathway23, and the TNF signalling pathway have been confirmed by several studies to be related to sepsis.
Discussion
In recent years, despite significant advances in antimicrobial treatment and organ support technologies, sepsis remains the leading cause of death in patients with severe infections24. This may be related to the lack of specificity of clinical manifestations, the complexity of pathophysiological processes, and the heterogeneity of sepsis5. Unfortunately, despite the continuous exploration of its mechanism, our understanding of it is still far from being sufficient. In fact, there are currently no laboratory testing methods to accurately identify sepsis and there are no individualised therapies to cure it. Therefore, researchers are committed to developing a precision medicine method that aims to classify patients into different types based on transcriptomic signatures and other biological and clinical data, thus providing a molecular basis for precision targeted therapy. Improving the identification and diagnosis of sepsis, exploring its pathogenesis, classification, and individualised therapy can maximise the efficacy and improve prognosis.
In recent years, exosomes have been extensively studied as a new form of intercellular signal transduction. Studies have shown that circRNAs are specifically enriched and stable in exosomes and can be detected in a variety of bodily fluids17. This means that exosomal circRNA has the potential to diagnose diseases as a biomarker5,19. They are also involved in the pathogenesis of various diseases, such as tumours25,26, cardiovascular diseases27,28,29, neurological disorders30,31,32, infections, and immune-related diseases30,33,34, indicating that they may be used as targets for precise treatment. To date, the expression and function of exosomal circRNAs in sepsis have not been reported. In order to clarify their regulatory role in the pathophysiology of sepsis, it is necessary to explore the changes in circRNA expression levels in serum exosomes and their regulatory pathways.
By comparing and analysing the results of microarrays, molecules with fold changes > 1.5 and p values < 0.05 were considered statistically significant. Then, we selected five circRNA molecules for experimental verification, including hsa_circRNA_101491, hsa_circRNA_103864, hsa_circRNA_104484, hsa_circRNA_104670, and hsa_circRNA_406194. These circRNA molecules were then verified by RT-qPCR among the 3 septic patients and 3 healthy volunteers that had been tested by microarray to determine the reliability of the microarray results. Among these five circRNA molecules, the expression of two circRNA molecules (hsa_circRNA_104484 and hsa_circRNA_104670) were significantly upregulated, consistent with the microarray results, but the other three circRNA molecules (hsa_circRNA_101491, hsa_circRNA_103864, and hsa_circRNA_406194) were not significantly different between the two groups. This indicates that microarray results contain false positives, thus, only differential circRNA molecules qualified by RT-qPCR are considered reliable. We continued to verify hsa_circRNA_104484 and hsa_circRNA_104670 in small clinical samples, and the results are consistent with those of previous studies. To the best of our knowledge, this study is the first report the expression of hsa_circRNA_104484 and hsa_circRNA_104670 in sepsis serum exosomes.
At present, ceRNA is the most common circRNA regulation mechanism. CircRNA targets miRNAs and indirectly regulates the expression of miRNA target genes and plays an important role in the occurrence and development of diseases35. Studies have found that circulating miRNAs are differentially expressed in inflammation-related diseases and can target the tumour necrosis factor pathway (TLR/NF-κB signalling pathway), acting as inflammation regulators36,37. Therefore, we speculate that circRNA may indirectly regulate the expression of inflammation-related genes by targeting miRNAs in sepsis. The annotation of the circRNA-miRNA regulatory axis and the construction of the ceRNA network showed that five miRNAs and several targeted mRNAs interacted with hsa_circRNA_104484 and hsa_circRNA_104670, respectively.
Among them, hsa_circRNA_104484 is a sponge molecule of hsa-miR-378a-3p/hsa-miR-378d. In recent experimental studies, miR-378 has been found to act directly or indirectly as a regulator of inflammation and participates in the processes of inflammation and immune regulation. Platelet-derived exosomal miR-378a-3p directly targets PDK1, resulting in the inhibition of the Akt/mTOR pathway and promoting the formation of neutrophil extracellular traps (NET) in sepsis38. A study by Caserta et al.36 showed that miR-378a-3p is differentially expressed in systemic inflammatory response syndrome (SIRS) and correlated with its severity. miR-378a can directly target ZBTB20, which plays a role in cell growth and apoptosis39. ZBTB20 is a transcriptional repressor that inhibits the transcription of the IκBα gene and positively regulates the activation of NF-κB, triggering an innate immune response40,41. This is consistent with the results of the GO analysis. In addition, miR-378 negatively regulates nuclear respiratory factor-1 (NRF-1), AMP-activated protein kinase γ2 (AMPKγ2), and phosphoinositide 3-kinase (PI3K), inhibits energy metabolism processes, and activates the NF-κB-TNFα pathway, which may be related to SIRS and sepsis42,43,44. Similarly, hsa_circRNA_104670 is a sponge molecule of hsa-miR-17-3p. Jiang and Li et al.45 found that lipopolysaccharide (LPS) and TNF-α can regulate the expression of miR-17-3p. miR-17-3p directly targets intercellular adhesion molecule 1 (ICAM-1) and inhibits its expression in LPS-induced acute lung injury (ALI)46. ICAM-1 is an important inflammatory mediator, and its expression is upregulated in sepsis, which enhances inflammatory cell infiltration and organ damage47,48. Therefore, we speculate that hsa_circRNA_104484 and hsa_circRNA_104670 may be involved in the pathogenesis of sepsis.
Conclusions
Our study compared the differences in the expression levels of circRNAs in serum exosomes between sepsis and healthy people, and initially evaluated the clinical application value of hsa_circRNA_104484 and hsa_circRNA_104670. The results provide a basis for mechanistic research. However, our research sample is relatively small; in the future, the sample size will be enlarged. We will further explore the biological functions of hsa_circRNA_104484 and hsa_circRNA_104670 through cell and animal experiments. Currently, the pathogenesis of sepsis is still unclear. As such, there is no effective therapeutic intervention; the exploration of the circRNA regulatory mechanism in sepsis will have great clinical translation research value.
Abbreviations
- RT-qPCR:
-
Real-time quantitative polymerase chain reaction
- ROC:
-
Receiver operating characteristic
- GO:
-
Gene ontology
- KEGG:
-
Kyoto City Encyclopaedia of Genes and Genomes
- circRNAs:
-
Circular RNA
- miRNA:
-
MicroRNA
- PBS:
-
Phosphate buffer saline
- cDNA:
-
Complementary DNA
- MF:
-
Molecular functions
- BP:
-
Biological pathways
- CC:
-
Cellular components
- AUC:
-
Area under the ROC curve
- TEM:
-
Transmission electron microscopy
- CI:
-
Confidence interval
- NET:
-
Neutrophil extracellular traps
- SIRS:
-
Systemic inflammatory response syndrome
- NRF-1:
-
Nuclear respiratory factor-1
- AMPKγ2:
-
AMP-activated protein kinase γ2
- PI3K:
-
Phosphoinositide 3-kinase
- LPS:
-
Lipopolysaccharide
- ICAM-1:
-
Intercellular adhesion molecule 1
- ALI:
-
Acute lung injury
- SOFA:
-
Sequential organ failure assessment
References
Singer, M. et al. The third international consensus definitions for sepsis and septic shock (Sepsis-3). JAMA 315(8), 801–810 (2016).
Ferrer, R. et al. Empiric antibiotic treatment reduces mortality in severe sepsis and septic shock from the first hour: results from a guideline-based performance improvement program. Crit. Care Med. 42(8), 1749–1755 (2014).
Xu, D. et al. Metabolomics coupled with transcriptomics approach deciphering age relevance in sepsis. Aging Dis. 10(4), 854–870 (2019).
Almansa, R. et al. Transcriptomic correlates of organ failure extent in sepsis. J. Infect. 70(5), 445–456 (2015).
Leligdowicz, A. & Matthay, M. A. Heterogeneity in sepsis: new biological evidence with clinical applications. Crit. Care 23(1), 80 (2019).
Kalluri, R. & LeBleu, V. S. The biology, function, and biomedical applications of exosomes. Science 367(6478), eaau6977 (2020).
Wang, X. et al. Exosomal miR-223 contributes to mesenchymal stem cell-elicited cardioprotection in polymicrobial sepsis. Sci. Rep. 5, 13721 (2015).
Song, Y. et al. Exosomal miR-146a contributes to the enhanced therapeutic efficacy of interleukin-1beta-primed mesenchymal stem cells against sepsis. Stem Cells 35(5), 1208–1221 (2017).
Zhang, Y. et al. Secreted monocytic miR-150 enhances targeted endothelial cell migration. Mol. Cell 39(1), 133–144 (2010).
Jeppesen, D. K. et al. Reassessment of exosome composition. Cell 177(2), 428–45.e18 (2019).
Ibsen, S. D. et al. Rapid isolation and detection of exosomes and associated biomarkers from plasma. ACS Nano 11(7), 6641–6651 (2017).
Kourembanas, S. Exosomes: vehicles of intercellular signaling, biomarkers, and vectors of cell therapy. Annu. Rev. Physiol. 77, 13–27 (2015).
Kristensen, L. S. et al. The biogenesis, biology and characterization of circular RNAs. Nat. Rev. Genet. 20(11), 675–691 (2019).
Wang, P. L. et al. Circular RNA is expressed across the eukaryotic tree of life. PLoS ONE 9(6), e90859 (2014).
Lasda, E. & Parker, R. Circular RNAs co-precipitate with extracellular vesicles: a possible mechanism for circRNA clearance. PLoS ONE 11(2), e0148407 (2016).
Li, Y. et al. Circular RNA is enriched and stable in exosomes: a promising biomarker for cancer diagnosis. Cell Res. 25(8), 981–984 (2015).
Wang, Y. et al. Exosomal circRNAs: biogenesis, effect and application in human diseases. Mol. Cancer 18(1), 116 (2019).
Li, X. et al. Coordinated circRNA biogenesis and function with NF90/NF110 in viral infection. Mol. Cell 67(2), 214–227 (2017).
Fanale, D. et al. Circular RNA in exosomes. Adv. Exp. Med. Biol. 1087, 109–117 (2018).
Ritchie, M. E. et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucl. Acids Res. 43(7), e47 (2015).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucl. Acids Res. 28(1), 27–30 (2000).
Xie, W. J. et al. Astaxanthin suppresses lipopolysaccharide-induced myocardial injury by regulating MAPK and PI3K/AKT/mTOR/GSK3β signaling. Mol. Med. Rep. 22(4), 3338–3346 (2020).
Wang, F. et al. Maresin1 ameliorates sepsis-associated lung injury by inhibiting the activation of the JAK2/STAT3 and MAPK/NF-κB signaling pathways. Microb. Pathog. 148, 104468 (2020).
Rudd, K. E. et al. Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet 395(10219), 200–211 (2020).
Shang, A. et al. Exosomal circPACRGL promotes progression of colorectal cancer via the miR-142-3p/miR-506-3p- TGF-β1 axis. Mol. Cancer 19(1), 117 (2020).
Huang, X. Y. et al. Exosomal circRNA-100338 promotes hepatocellular carcinoma metastasis via enhancing invasiveness and angiogenesis. J. Exp. Clin. Cancer Res. 39(1), 20 (2020).
Wang, W. et al. Circular RNAs as potential biomarkers and therapeutics for cardiovascular disease. PeerJ 7, e6831 (2019).
Wang, Y. et al. Exosomal circHIPK3 released from hypoxia-pretreated cardiomyocytes regulates oxidative damage in cardiac microvascular endothelial cells via the miR-29a/IGF-1 pathway. Oxid. Med. Cell Longev 2019, 7954657 (2019).
Han, J. et al. Circular RNA-expression profiling reveals a potential role of Hsa_circ_0097435 in heart failure via sponging multiple microRNAs. Front. Genet 11, 212 (2020).
He, J. et al. Exosomal circular RNA as a biomarker platform for the early diagnosis of immune-mediated demyelinating disease. Front. Genet 10, 860 (2019).
Hosaka, T. et al. Extracellular RNAs as biomarkers of sporadic amyotrophic lateral sclerosis and other neurodegenerative diseases. Int. J. Mol. Sci. 20(13), 3148 (2019).
Zhao, R. T. et al. Circular ribonucleic acid expression alteration in exosomes from the brain extracellular space after traumatic brain injury in mice. J. Neurotrauma 35(17), 2056–2066 (2018).
Chen, X. et al. Circular RNAs in immune responses and immune diseases. Theranostics 9(2), 588–607 (2019).
Xie, R. et al. The role of circular RNAs in immune-related diseases. Front. Immunol. 11, 545 (2020).
Salmena, L. et al. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?. Cell 146(3), 353–358 (2011).
Caserta, S. et al. Severity of systemic inflammatory response syndrome affects the blood levels of circulating inflammatory-relevant microRNAs. Front. Immunol. 2017, 8 (1977).
Benz, F. et al. Circulating microRNAs as biomarkers for sepsis. Int. J. Mol. Sci. 17(1), 93 (2016).
Jiao, Y. et al. Platelet-derived exosomes promote neutrophil extracellular trap formation during septic shock. Crit. Care 24(1), 380 (2020).
Wang, J., Liu, Z. H. & Yu, L. J. Long non-coding RNA LINC00641 promotes cell growth and migration through modulating miR-378a/ZBTB20 axis in acute myeloid leukemia. Eur. Rev. Med. Pharmacol. Sci. 23(17), 7498–7509 (2019).
Liu, X. et al. Zinc finger protein ZBTB20 promotes Toll-like receptor-triggered innate immune responses by repressing IκBα gene transcription. Proc. Natl. Acad. Sci. USA 110(27), 11097–11102 (2013).
Qiu, J. et al. ZBTB20-mediated titanium particle-induced peri-implant osteolysis by promoting macrophage inflammatory responses. Biomater. Sci. 8(11), 3147–3163 (2020).
Jeon, T. I. et al. Fisetin protects against hepatosteatosis in mice by inhibiting miR-378. Mol. Nutr. Food Res. 57(11), 1931–1937 (2013).
Zhang, T. et al. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-κB-TNFα pathway. J. Hepatol. 70(1), 87–96 (2019).
Liu, W. et al. Hepatic miR-378 targets p110alpha and controls glucose and lipid homeostasis by modulating hepatic insulin signalling. Nat. Commun. 5, 5684 (2014).
Jiang, X. & Li, N. Induction of MiR-17-3p and MiR-106a [corrected] by TNFα and LPS. Cell. Biochem. Funct. 29(2), 164–170 (2011).
Suárez, Y. et al. Cutting edge: TNF-induced microRNAs regulate TNF-induced expression of E-selectin and intercellular adhesion molecule-1 on human endothelial cells: feedback control of inflammation. J. Immunol. 184(1), 21–25 (2010).
Laudes, I. J. et al. Disturbed homeostasis of lung intercellular adhesion molecule-1 and vascular cell adhesion molecule-1 during sepsis. Am. J. Pathol. 164(4), 1435–1445 (2004).
Hildebrand, F. et al. Role of adhesion molecule ICAM in the pathogenesis of polymicrobial sepsis. Exp. Toxicol. Pathol. 56(4–5), 281–290 (2005).
Acknowledgements
We thank all patients and volunteers who participated in this study.
Funding
This study was supported by the Department of Finance of Jilin Province (Grant ZXWSTZXEY019), the Department of Science and Technology of Jilin Province (Grant 20200708083YY, Grant 20191102012YY, Grant 20191313162SF).
Author information
Authors and Affiliations
Contributions
C.T. and K.W. conceived and performed the study. J.Y.L. and M.Z. participated in the collection of blood samples and patient characteristics. S.C. analysed the data and made the pictures and graphs. C.T. drafted and revised the manuscript. X.D. revised the manuscript. All the authors have read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Tian, C., Liu, J., Di, X. et al. Exosomal hsa_circRNA_104484 and hsa_circRNA_104670 may serve as potential novel biomarkers and therapeutic targets for sepsis. Sci Rep 11, 14141 (2021). https://doi.org/10.1038/s41598-021-93246-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-93246-0
- Springer Nature Limited
This article is cited by
-
Circ_0001714 knockdown alleviates lipopolysaccharide-induced apoptosis and inflammation in renal tubular epithelial cells via miR-129-5p/TRAF6 axis in septic acute kidney injury
Journal of Bioenergetics and Biomembranes (2023)
-
Roles of circRNAs in hematological malignancies
Biomarker Research (2022)
-
Emerging role of exosomes in cancer progression and tumor microenvironment remodeling
Journal of Hematology & Oncology (2022)