
Abstract
Signaling through integral membrane G protein-coupled receptors (GPCRs) is influenced by lipid composition of cell membranes. By using novel high affinity ligands of human cannabinoid receptor CB2, we demonstrate that cholesterol increases basal activation levels of the receptor and alters the pharmacological categorization of these ligands. Our results revealed that (2-(6-chloro-2-((2,2,3,3-tetramethylcyclopropane-1-carbonyl)imino)benzo[d]thiazol-3(2H)-yl)ethyl acetate ligand (MRI-2646) acts as a partial agonist of CB2 in membranes devoid of cholesterol and as a neutral antagonist or a partial inverse agonist in cholesterol-containing membranes. The differential effects of a specific ligand on activation of CB2 in different types of membranes may have implications for screening of drug candidates in a search of modulators of GPCR activity. MD simulation suggests that cholesterol exerts an allosteric effect on the intracellular regions of the receptor that interact with the G-protein complex thereby altering the recruitment of G protein.
Similar content being viewed by others

Introduction
Lipid composition of membranes plays an important role in modulation of G protein-coupled receptors (GPCR)1,2,3,4. In particular, cholesterol content varies significantly among different types of cell membranes, and was shown to influence the ligand-induced signal transduction through GPCRs5,6,7,8. Early stage pharmacological characterization of GPCR ligands typically involves screening of prospective drug candidates in cell- and membrane-based assays9. Hence, it is important to understand how a physiological response of the target receptor to small molecule ligands is modulated by the lipid composition of membranes.
Membranes of mammalian cells typically used for expression of recombinant GPCR for cell- and membrane-based assays may contain significant amount of cholesterol10. Plasma membranes of human embryonic kidney (HEK293) and Chinese hamster ovary (CHO) cells were reported to contain as much as 25–40 mol% of cholesterol relative to phospholipid10,11,12. The lipid and cholesterol content of cultured mammalian cells fluctuates depending on nutrient composition of growth media, temperature of cultivation, age of cell culture and other growth parameters10. Other commonly used expression hosts such as Escherichia coli13,14, Spodoptera frugiperda (Sf9), and Trichoplusia ni (Tn) synthesize only trace amounts of cholesterol or do not produce it at all11.
A large proportion of pharmacological drugs, currently on the market or under development, target membrane proteins. Since pharmacological profiles of drug candidates could be influenced by the properties of cell membranes harboring these receptors, variations in composition of the lipid bilayer may result in inconsistencies of the receptor response15,16. This is especially true for hydrophobic ligands targeting GPCR since interaction between the drug and the lipid may influence the pharmacological characteristics of the compound such as its binding affinity and selectivity17,18,19. While it was demonstrated that lipids and cholesterol can shift the response of the receptor to ligand binding, it is not well known if the pharmacological categorization of ligands, i.e. agonism, antagonism or inverse agonism, depends on composition of the lipid matrix. Here, we sought to elucidate whether the functional response of the cannabinoid receptor CB2 to a series of novel specific ligands is affected by the cholesterol content of membranes.
Cholesterol is critical for the formation of lateral domains (clusters)20, and it may induce negative curvature elastic stress in lipid bilayers21. The cholesterol-dependent increase in membrane stiffness increases the decay length of protein-induced perturbations in the lipid matrix22. Furthermore, cholesterol may directly interact with GPCR at sites identified in several receptors23,24,25,26. Cholesterol was reported to negatively modulate the activity of type 1 cannabinoid receptor (CB1) in nerve cells27. Furthermore, it was proposed that the CB1 receptor possesses a specific cholesterol binding site28. However, the activity of the structurally close cannabinoid receptor CB2 was reported to be not influenced by changes in membrane cholesterol content29. The CB2 receptor is known for its role as a regulator of inflammation and is commonly found in tissues containing variable levels of cholesterol30. Therefore, it is important to understand if CB2 is sensitive to variation in cholesterol content.
Here, we examined the effects of cholesterol on CB2 by analyzing G protein activation by this receptor in response to novel, rationally designed CB2 ligands (MRI-2646, MRI-2654, MRI-2687, MRI-2653, MRI-2659). We will demonstrate that cholesterol increases the basal activation levels of CB2 receptor, thereby altering pharmacological classification of these ligands from inverse agonists to partial agonists. We also use MD simulation to probe structural effects of cholesterol and ligands.
Results
Based on the rational modification of the A-836339 thiazole scaffold, some of us previously reported that the synthetic compounds MRI-2687 and MRI-2594 show contrasting activity on the CB2 receptor31. The two compounds only differ in arm 1 featuring the extended central 6-methylbenzothiazole ring in MRI-2687 and 4,5-dimethylthiazole ring in MRI-2594, respectively. Unexpectedly, MRI-2687 behaved as an inverse agonist, whereas MRI-2594 acted as an agonist on CB2 (similar to A-836339)31,32. Detailed molecular docking analysis showed that these two ligands adopt similar binding poses within CB2 wherein the different central rings reside in the same position as arm 1 of the receptor bound AM10257 CB2 crystal structure31. The arm 1 of MRI-2687 along with its 6-Me substitution forms π–π interactions with the side chain of Trp2586.48 and confines its conformation to a similar rotamer as in the CB2-AM10257 structure. As opposed to MRI-2687, the lack of a large substituent on arm 1 of MRI-2594 prevents it from extending sufficiently deep to constrain the conformation of Trp2586.48 and allows its unrestrained movement and, thereby, leading to receptor activation. The proposed differential interactions with the toggle switch residue Trp2586.48 prompted us to investigate the effect of these novel ligands on CB2 receptor further in various cell-and membrane-based assays.
To assess the function of CB2 in vitro, we measured the activation of the receptor by quantifying the rates of nucleotide exchange on the Gα subunit of G protein that interacts with CB2. The full agonist CP-55,940 is not well suited for studying modulatory effects of the lipid bilayer on receptor function since its strong activating effect may mask moderate effects exerted by the lipid matrix. This is an important consideration since, unlike full synthetic agonists, many endogenous agonists of GPCR only partially activate these receptors. We hypothesized that our novel series of high-affinity ligands would allow detection of moderate modulating effects of the lipid matrix on CB2 activation. Specifically, we sought to investigate the role of different substitutions at the 6-position of the benzothiazole ring in this series of ligands. Elaborating on our earlier approach31, we synthesized structurally-related compounds (Fig. 1) and evaluated their effects on CB2 receptor activity.

Benzothiazole-based CB2 ligands. (A) Structure of benzothiazole-based CB2 ligands. (B) Synthesis scheme for ligands, Reagents and conditions: a. 2-bromoethanol 90 °C; b. BOP, 2,2,3,3-tetramethylcyclopropane carboxylic acid. c. Acetyl Chloride.
In this study, we used the recently developed cannabinoid ligands which were synthesized as shown in Fig. 1A. The R group at the 6-position of the benzothiazole were varied to include Me, OMe, OCF3, Cl, and Br, synthesized as shown in Fig. 1B.
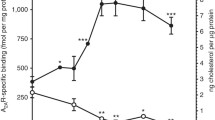
The affinity of the MRI ligands for CB2 receptor was determined to be in 0.053–0.1 nM range in hCB2-expressing-CHO cell membranes by a displacement binding experiment (Fig. 2a).

Affinity and functional effects of novel MRI ligands on CB2 receptor in CHO cell membranes. (a) binding affinities (nM) and Emax (% and nM) of [35S]-GTPγS binding to CHO membranes (PerkinElmer, Cat. No ES111-M400UA) expressing hCB2. (b) [35S]-GTPγS binding to membranes as a factor of ligand concentration. Binding of [35S]-GTPγS was determined as described in “Methods”. Non-specific binding was defined as 0% activity. The assay of GTPγS non-specific binding contains non-radioactive GTPγS.
Then, we assessed the functional effect of the compounds on G-protein signaling in [35S]-GTPγS binding assays using hCB2-expressing-CHO cell membranes obtained from PerkinElmer (see “Methods”). MRI-2646 had no effect on basal signaling (Fig. 2b) whereas MRI-2654, MRI-2687, MRI-2653, and MRI-2659 reduced basal signaling to 55% with high potency (Fig. 2a) as a function of inverse agonism. Unlike other structural analogues, MRI-2594 demonstrated full agonism with high affinity (hCB2R, Ki = 0.031 nM) and potency (EC50: 0.09 nM) (Fig. 2a). We were intrigued by the seemingly neutral antagonist activity of MRI-2646 in CHO cells even though its structural features were in line with the other benzothiazole ligands which behaved as inverse agonist. Hence, in the present work, we used this novel, high affinity cannabinoid ligand to explore the role of lipid environment in determining functional activity.
Based on our preliminary studies, we concluded that ligands such as MRI-2646 may act either as a weak partial agonist or a neutral antagonist, depending on the cell type used in the assay. To better understand the reason for these discrepancies, we assessed the activation of CB2 receptor by the in vitro G protein activation assay that measures the rates of nucleotide exchange on the Gα subunit of G protein (GEF assay) as described previously33,34. The assay reports the rates of formation of the complex of [35S]-GTPγS, a homolog of GTP, with the Gα subunit of G protein. Typical assay conditions require small (nanogram) quantities of the receptor protein either in cell membranes or reconstituted into lipid bilayers. The readout of the assay is the amount of the non-hydrolysable complex of Gα with [35S]-GTPγS which, under selected experimental conditions, is proportional to the amount of activated receptor in the assay.
For convenience, the results of the GEF assay are normalized such that activation of CB2 in the presence of CP-55,940 was set to 100%, and residual activity in the presence of saturating concentrations of the full inverse agonist SR-144,528 was set to 0%.
We first compared two different types of membranes: E. coli BL21 (DE3) and a commercially available preparation of CHO cell membranes expressing CB2 (Millipore EMD, Cat. No HTS020M) in the GEF assay (Fig. 3). Depending on the source of membranes, a significant difference in activation behavior of CB2 was observed. In this assay, the tested ligands (with the exception of the previously described full agonist MRI-259431 and strong inverse agonist MRI-2659) behaved as partial agonists of the CB2 receptor in E. coli membranes. However, these same ligands acted as partial inverse agonists on CB2 expressed in CHO cells (Fig. 3). The activities of the ligands relative to each other, which were roughly inversely correlated to the ligand substituent size, were not greatly changed, and correlated roughly to the R-group size.

GEF of CB2 expressed in: (a) E. coli membranes and (b) CHO membranes (Millipore EMD). 2 μg of total protein per assay. Each point represents an average of four independent measurements (n = 4). Ligands were added at a concentration of 2 μM to ensure saturation of the receptor, and G protein was added as described in “Methods”. The dotted line indicates the rates of activation of G protein in the absence of a ligand. The rates of activation with CP-55,9040 are set to 100%, and rates of activation in the presence of SR-144,528 to 0%.
Intrigued by these observations, we compared several other available sources of membranes expressing CB2. MRI-2646 ligand was selected as a representative of a cohort of related benzothiazole ligands for all subsequent measurements since it activated CB2 in E. coli cell membranes the most while being a partial inverse agonist of CB2 in CHO membranes. Several preparations of membranes expressing CB2 were compared (Fig. 4). While MRI-2646 behaved as a neutral antagonist of CB2 in membranes from CHO cells obtained from PerkinElmer, it acted as a partial inverse agonist in two other commercial preparations of CB2 in CHO membranes procured from EMD Millipore and Applied Cell Sciences (CHO-K1 membranes). Likewise, MRI-2646 was a partial inverse agonist of CB2 expressed in membranes Expi293F and Expi293F GNTI- cells. On the other hand, this ligand was a partial agonist of CB2 expressed in baculovirus infected insect Sf9 cells and in E. coli BL21 (DE3) cells.

GEF on CB2 expressed in different cell lines. An amount of 2 μg of membrane protein per assay was used. Each data points represents an average of four independent measurements (n = 4) with error rates indicated by the bars. 100% represents full activation in the presence of 2 μM of CP-55,940, and 0%–with 2 μM SR-144,528.
Therefore, MRI-2646 exerts differential effects on CB2: in mammalian cell membranes this ligand acts as an inverse agonist or neutral antagonist while in bacterial- and insect-cell membranes it behaves as a partial agonist of CB2 receptor. Likely contributing factors to such discrepancies may include: (i) differences in lipid composition (in particular, cholesterol content) between mammalian, insect, and bacterial membranes; (ii) different pattern of post-translational modifications (palmitoylation, glycosylation) of receptor molecules expressed in different expression hosts; (iii) differences in expression levels of the receptor and densities of ligand-binding sites in membrane preparations from different sources, and (iv) composition of endogenous G proteins in membrane preparations. The endogenous membrane-associated G protein contained in preparations of CHO, HEK and Sf9 cell membranes expressing CB2 contributes to the GEF signal. E. coli cells do not produce G protein. The subtle role of the sterics and electronics of the 6-Cl substituent in this intricate modulatory mechanism cannot be ruled out either.
Effects of lipid composition
We considered the effects of lipid composition of CB2-containing membranes obtained from different expression hosts. Specifically, the CHO cell membranes are known to contain high concentrations of cholesterol, unlike the E. coli cell membranes that are devoid of cholesterol12. We quantified the relative content of lipids and cholesterol in several preparations of membranes of mammalian, insect, and E. coli cells (Supporting Fig. 1). Lipids were extracted from membranes, and their composition determined by 1H-NMR as described in Legend to Supporting Fig. 1. Consistent with the previously published data, cholesterol was not detected in membrane preparations from E. coli and from Sf9 cells11,35,36,37 while membrane preps obtained from CHO cells and suspension culture of HEK Expi293F expressing CB2 contained 39% and 26% cholesterol relative to phospholipids, respectively10,38. While the bacterial-, insect- and mammalian cell membranes differ significantly not only in content of cholesterol but also in composition of phospholipids10,11,13, a variability in cholesterol content between membranes from different expression cell lines correlates strongly with the signaling pattern of CB2 activated by the novel ligand. Therefore, we hypothesized that the cholesterol content of membranes affects the activation of the cannabinoid receptor CB2.
Endogenous vs. exogenous G protein
Besides lipids, the content of endogenous G proteins in cell membrane preparations from different sources may also affect the readout of the [35S]-GTPγS binding and the GEF G-protein activation assays used in this study (see “Methods”). The [35S]-GTPγS binding assay measures the binding of the radiolabeled nucleotide analogue to the endogenous G protein that is already present in membrane preparations while the GEF assay relies on the exogenous G protein subunits of Gαi1 and Gβ1γ2 added in large excess relative to receptor. Therefore, the GEF assay typically affords a good signal-to-noise ratio and enables comparison of multiple samples at standardized conditions. The addition of G protein is necessary to analyze the activation of CB2 in E. coli membranes since these membranes do not contain endogenous G protein. On the other hand, the membranes obtained from mammalian and insect cell cultures contain endogenous G proteins and, therefore in these membranes, the GEF assay reports on the rates of activation of a combined pool of endogenous as well as exogenous G protein.
To assess the contribution of endogenous G protein to the total GEF signal, we performed the GEF assay on membranes of Expi293F, CHO (Perkin Elmer) and Sf9, in the absence as well as in the presence of exogenous G protein (Fig. 5).

GEF analysis of three membrane preparations with or without supplementation with exogenous G protein. (a) Expi293F membranes; (b) CHO membranes; (c) Sf9 membranes. Bars represent an average of four independent measurements (n = 4). Dotted lines represent the rates of activation of G protein in the absence of a ligand.
As expected, in the absence of exogenous G protein the magnitude of the signal was significantly lower (three to tenfold) compared to the standard GEF assay. Yet, the pattern of activation of CB2 by the MRI-2646 was similar in assays performed with- and without addition of exogenous G protein. While in membrane preps from Expi293F cells, MRI-2646 acted as an inverse agonist of CB2 (Fig. 5a); in CHO cell membranes (PerkinElmer) it behaved as a neutral antagonist (Fig. 5b); and in membranes of Sf9 cells expressing CB2, it acted as a partial agonist (Fig. 5c).
Receptor density in membranes
We next examined the density of ligand binding sites (by saturation 3H-CP-55,940 radioligand binding assay, Supporting Fig. 2). The density of the ligand binding sites varied between ~ 22 and 50 pmol/ mg of membrane protein. However, there was no correlation between the density of the ligand binding sites and behavior of the MRI-2646 ligand as an inverse agonist, neutral antagonist or partial agonist of CB2. Thus, the differential pattern of activation of CB2 by the MRI-2646 cannot be explained by differences in expression levels and density of binding sites of receptor in membrane preparations.
The results above suggest that the functional effect of MRI-2646 on CB2 varies following the same pattern as the content of cholesterol in membranes of cell lines expressing CB2. While in membranes devoid of cholesterol, the MRI-2646 is a partial agonist, and, in cholesterol-containing membranes it acts either as an inverse agonist or a neutral antagonist of CB2.
Treatment with methyl-β-cyclodextrin
The content of cholesterol in cell membrane preparations can be altered by pre-treatment with methyl-β-cyclodextrin (MβCD)8,39 (Supporting Fig. 3). In membranes of Expi293F cells expressing CB2, treated with 20 mM MβCD, the rates of activation of G protein on CB2 decreased by about twofold although the levels of the CB2 receptor in these membranes were unchanged (Supporting Fig. 3b). In the MβCD-treated membranes, the activity of CB2 in the presence of MRI-2646 was higher than the basal signaling in the absence of ligands, indicating that MRI-2646 ligand acted as a partial agonist of CB2 receptor at these conditions.
MβCD can also be used as a carrier of cholesterol in order to enrich cell membranes with cholesterol40. To test the effect of the exogenously added cholesterol on activation of CB2, we pre-treated the E. coli membranes expressing CB2 with a solution of MβCD/cholesterol, and measured the rates of activation of G protein (Fig. 6a). In the MβCD/cholesterol-treated membranes of E. coli, the MRI-2646 reproducibly acted as a neutral antagonist of CB2, while in the untreated membranes it exhibited agonistic effects (Fig. 6b). There was no noticeable change in the levels of CB2 in membranes, and the density of ligand binding sites did not change upon treatment with MβCD/cholesterol (Supporting Fig. 4a,b) These results provide a more direct proof that the activation of CB2 bound to MRI-2646 is modulated by cholesterol content of membranes.

Effect of treatment with MβCD/Chol of E. coli membranes on activation of CB2 by synthetic ligands. (a) Membranes expressing CB2 were treated with 20 mM MβCD/Chol for 1 h at 4 °C, washed with PBS, and the activation of CB2 in the presence of ligands determined by GEF; (b) control, untreated E. coli membranes expressing CB2. Each data point represents an average of four independent measurements (n = 4) with standard deviation indicated by vertical bars.
Liposome-reconstituted CB 2 and post-translational modifications
To assess the possible role of post-translational modifications of CB2 on its activation by the MRI ligands, the recombinant CB2 receptor was isolated from two different expression cell lines, E. coli BL21 (DE3)41 and Expi293F GNTI-42. The protein was purified and reconstituted into lipid bilayers containing 1-palmitoyl-2-oleoyl-glycero-3-phosphocholine (POPC) and, 1-palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol) (POPG) at a molar ratio of 3/1, either supplemented or not supplemented with cholesterol, as described in “Methods”10,13. We reported previously that the presence of phospholipids with a negatively charged headgroup stabilizes CB2 protein in lipid bilayers43. Therefore, purified CB2 was reconstituted into POPC/POPG (3/1, mol/mol) liposomes containing 0, 20 and 40 mol% cholesterol (reported as total content of lipids) as described in “Methods”. In one case, the protein purified from E. coli cells was reconstituted into lipids extracted from brain tissue (Avanti Polar Lipids). The ratio of protein-to-lipid in the resulting samples was in the range of 1:850 to1:1100 (mol/mol). The levels of protein, and the density of ligand binding sites for receptor preparations reconstituted in liposomes with different content of cholesterol varied only slightly (Supporting Fig. 5a,b).
The results of the GEF assay performed on these liposome-reconstituted CB2 samples demonstrate that the MRI-2646 ligand acts as a partial agonist of bacterially expressed CB2 in liposomes devoid of cholesterol (Fig. 7a). However, in the lipid matrix containing 20% or 40% of cholesterol or in liposomes composed of lipids extracted from brain tissues (Fig. 7b–d), the basal (without ligand) activation of CB2 receptor was increased. In all cholesterol-containing liposomes, including those containing the brain lipid extract, the MRI-2646 ligand acted as a partial inverse agonist of the receptor.

Activation of G protein on CB2 receptor reconstituted in liposomes. Liposome composition: 75% POPC, 25% POPG and cholesterol content relative to total phospholipids as indicated, or total lipids extracted from bovine brain, as indicated. Proteins were reconstituted at a protein-to-lipid ratio in the range 1:850 to 1:1100 mol/mol. (a–d) CB2 purified from E. coli cells. (e,f) CB2 purified from Expi293F GNTI- cells. Average values of four independent measurements are plotted (n = 4). Dotted lines represent the rates of activation of G protein in the absence of a ligand.
For comparison, the activation behavior of CB2 isolated from the Expi293F GNTI- cells42 and reconstituted into liposomes was studied (Fig. 7e). In liposomes without cholesterol, the MRI-2646 acted as a partial agonist of CB2, similar to its action on the bacterially expressed protein. At the same time, MRI-2646 acted as a partial inverse agonist on HEK cell-expressed CB2 protein reconstituted into liposomes with 40% of cholesterol (Fig. 7f and Supporting Fig. 5, 6). These results corroborate the above described inverse agonism of MRI-2646 on CB2 in membrane preps containing cholesterol. Therefore, it can be concluded that cholesterol is involved in modulating the activation behavior of CB2. Post-translational modifications of CB2 do not seem to play a significant role in modulation the activation of the receptor by MRI-2646 in the presence of cholesterol.
Structural effects of cholesterol on CB 2 in molecular dynamics simulation
Using molecular dynamics (MD) simulations, we evaluated the effect of membrane cholesterol on three of the ligands’ interaction with the known toggle switch residue Trp2586.48as well as the displacement and fluctuation of the ICL3-TM6 region of CB2, which would interact with the G-protein upon its recruitment. We hypothesized there would be a significant effect of membrane cholesterol on these regions in free CB2.We anticipated three alternatives for the effect of ligand type on the simulation results: (1) no obvious effect of ligand; (2) effects specific to the experimentally observed ligand pharmacological category; or (3) effects correlating mainly to ligand size. MRI-2646. MRI-2659, and MRI-2594 were simulated, chosen for their diversity.
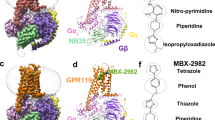
We conducted equilibrium all-atom molecular MD simulations of the cryo-EM structure of CB2 (PDB: 6PT0)44 with and without 40% membrane cholesterol and a POPC/POPG, 3/1, mol/mol ratio. The cholesterol molecules in the extracellular part of the TM5-6 region reported in the cryo-EM structure were retained, particularly since this is a known cholesterol binding site45,46. In both of these conditions, simulations were done with no bound ligand as well as with each of the three chosen ligands. Each ligand was placed respectively in the orthosteric site (Fig. 8) by analogy with the configuration of the structurally similar AM10257 ligand agonist present in the X-ray structure of CB231. There were eight total simulations.

Depictions of ligand binding site and preferentially affected residues. (a) Representative snapshot of MRI-2659 in orthosteric binding site. MRI-2646 and MRI-2659 share common backbone position; (b) Cryo-EM structure (PDB: 6PT0)44. Those residues whose simulation average RMSD from the cryo-EM structure changed by > 1 Å as a function of 0% vs 40 mol% cholesterol are highlighted by spheres. Such residues from free and ligand-bound conditions (MRI-2594, MRI-2646, MRI-2659) are all shown. Note proximity to Gα subunit (grey). A number of these residues reside in the extracellular loops; the functional relevance is not known.
The R-groups of the ligands were in close proximity to the toggle switch residue Trp258 6.48. In each simulation, the protein underwent an initial relaxation phase from the initial antagonist-bound cryo-EM conformation within 10 ns, as shown by evolution of whole protein RMSD over time (Supporting Fig. 7). Each ligand remained stable in its initial position throughout (Supporting Fig. 7). We discarded the first 50 ns of each production simulation, to include the relaxation phase, for an aggregated grand total of 2 µs of simulation trajectory analyzed across all conditions. Key binding site interactions are shown in Fig. 8a.
In GEF experiments, membrane cholesterol increased the constitutive activity of CB2, and the relative order of MRI-2594, MRI-2646, and MRI-2659 by activity was preserved, even though these ligands would be categorized differently relative to the benchmark of constitutive activity. Since G-protein recruitment is thought to depend on the toggle switch Trp2586.48, we hypothesized that the rotameric state of this residue would be differentially changed by ligand type, particularly since the ligand substituent is in close proximity to it.
We found that the distribution of Trp2586.48 side-chain χ1 rotamer angles (from the N–Cα–Cβ–Cγ dihedral) during the simulations varied by ligand arm 1 substituent size and presence of cholesterol. In ligand-free, cholesterol-free CB2, the distribution of side-chain angles had a peak at ~ 280° (and very small peak at roughly 180o which will not be discussed further, since it is unlikely to relate to experimental results). In each case the average magnitude was increased when cholesterol was present (Fig. 9a). The difference in mean χ1 angles between cholesterol conditions for each bound ligand are shown in Fig. 9a and plotted in Fig. 9b. As the ligand size grew, the effect of cholesterol on the rotamer angle decreased.

Difference in Trp2586.48 rotamer angle related to presence of cholesterol. (a) Trp2586.48 χ1 rotamer angle distributions with 0% and 40 mol% cholesterol. (b) Difference in mean Trp2586.48 χ1 rotamer angle between no cholesterol vs 40 mol% cholesterol conditions; green (negative change) dashed line. TM domains are annotated, with up and down arrows indicating direction of helix, with up from intracellular to extracellular.
Some amount of conformational change from the reference cryo-EM structure is to be expected in simulation, but we hypothesized that residues interfacing with G-protein, and therefore most likely to change the activation rate, would be preferentially affected as a function of the presence of cholesterol. To test this idea, we compared the average root-mean-square deviation (RMSD) of each Cα, with respect to the original cryo-EM structure at each simulation frame, between 0% cholesterol and 40% cholesterol conditions by subtraction. This comparison was made for free and each ligand-bound state. If a residue in the 40% cholesterol structure deviated more than the same residue in the 0% cholesterol structure, the resulting quantity would be positive, and if less, negative. Even though the reference cryo-EM structure is antagonist-bound, if there were no effect of cholesterol or ligand, the RMSD would be expected to be similar across conditions, resulting in zero RMSD difference. We found that residues that would interact with the G-protein complex, primarily in the ICL3 loop and the N-terminal side of TM6 (residues 222–236), were preferentially displaced in the presence of cholesterol across all ligand conditions (Fig. 10). The difference in average per-residue RMSD was relatively variable in this region compared to the rest of the structure, where this quantity was mostly close to zero (Fig. 10). The number of residues in this region with > 1 Å mean change in RMSD as a function of cholesterol presence was 9, 6, 2, and 3 in free, MRI-2594, MRI-2646, and MRI-2659-bound simulations respectively. This number decreased with ligand substituent size—though we cannot rule out other factors, particularly including the chemical composition of the substituent.

Difference in per-residue root-mean-square deviation (RMSD) between simulation and antagonist-bound cryo-EM structure. Trp2586.48 highlighted in red, residues with > 1 Å difference highlighted in orange (positive change) or green (negative change) dashed line. TM domains are annotated, with up and down arrows indicating direction of helix, with up from intracellular to extracellular.
Given that RMSD change as a function of the presence of cholesterol was related to substituent size, we hypothesized that the root-mean-square fluctuation (RMSF) of each residue with respect to the average structure would also be affected in a systematic way (Supporting Fig. 8). A decrease in RMSF of specific residues would suggest a localized decrease in entropy. The ICL3 and first part of TM6 (residue 221–236) exhibited a relatively high RMSF (mean 2.58–3.80 Å with no cholesterol, 2.85–4.65 Å with 40% cholesterol). The magnitude of the RMSF was inversely correlated to the size of the ligand substituent. In unbound CB2, the RMSF in this region in the 40% cholesterol condition was substantially higher than in the 0% cholesterol condition (4.65 Å vs 2.58 Å). While relatively small in magnitude, the mean of absolute per-residue difference in RMSF as a function of the presence of cholesterol also decreased as ligand substituent size increased (1.01, 0.58, and 0.40 Å for MRI-2594, MRI-2646, and MRI-2659 respectively, shown in Supporting Fig. 8).
Discussion
Here we demonstrated that cholesterol increases the basal activation levels of CB2 receptor, thereby altering the pharmacological classification of novel synthetic cannabinoid ligands (Fig. 11). While in membranes devoid of cholesterol the MRI ligands act as partial agonist of CB2, in cholesterol-enriched membranes that same ligands became either inverse agonists or neutral antagonists of this receptor.

Membrane cholesterol dependent protean agonism.
Some of us have previously reported the stabilizing effect of anionic lipids such as PG and PS on purified CB2 protein reconstituted in liposomes43,47. It was also shown that the negatively charged cholesterol derivative, cholesteryl hemisuccinate (CHS), stabilizes the recombinant CB2 protein in detergent micelles and liposomes43. At the same time, it was reported that cholesterol did not affect the activation of CB2 by the full agonist CP-55,94047. Since these observations were made on a CB2 receptor activated by the high affinity full agonist CP-55,940, the relatively moderate effects of the lipid matrix on receptor activation may have been masked.
Cholesterol is the major sterol found in higher eukaryotes48. The rigid planar structure of cholesterol modulates fluidity, thickness, curvature and permeability of membranes49,50,51. The presence of cholesterol in membranes increases ordering of lipid acyl chains. Physicochemical parameters of membranes have been implicated in the regulation of function of integral membrane proteins49,52,53. Lateral compression and hydrophobic matching between the lipid bilayer and transmembrane domains of the protein are affected by cholesterol content. These parameters are important for the structural stability of embedded membrane proteins51.
It was previously reported that cholesterol rafts in human immune cell membranes modulate the activity of CB1 receptor but do not affect the activation behavior of CB2 receptor29. That conclusion was reached by quantifying several signaling pathways in cells treated with methyl-β-cyclodextrin (MβCD). However, the effects of cholesterol/ MβCD were assessed in the presence of the full agonist of CB2, CP-55,940 that may have masked moderate effects on activation of CB2 receptor by the lipid bilayer. Here, by using a novel synthetic ligand MRI-2646 we demonstrated that the content of cholesterol in lipid bilayers modulates CB2 activation.
We have demonstrated that cholesterol increases the constitutive activity of the CB2 receptor. This effect was first shown in membrane preparations of several types of cells expressing CB2. One can argue that the difference between the membranes of mammalian cells (HEK, CHO) on the one hand, and insect and bacterial cells—on the other hand is not only in the content of cholesterol but in many other parameters including composition of phospholipids. Indeed, it has been reported that the content of phosphocholine (PC) lipids in HEK cell membranes is about 33% of total lipids, about two-fold higher compared to Sf9 membranes which have higher phosphoethanolamine (PE) (almost 40% of total lipids)10. The difference in lipid composition is even more pronounced when E. coli membranes are considered: they consist predominantly (almost 75%) of zwitterionic PE with the remainder consisting of anionic lipids (PG) and cardiolipin54. To prove that the presence or absence of cholesterol plays a major role in modulating the spontaneous signaling by CB2, we reconstituted the purified receptor into lipid bilayers of defined composition, containing POPC/POPG, 3/1, mol/mol and cholesterol in the range of 0–40% of total lipids. Indeed, the presence of cholesterol in these artificial bilayers resulted in an increase of spontaneous signaling by CB2. This effect was observed for the recombinant receptor isolated from E. coli cells as well as from the HEK Expi293F cells, providing additional evidence for an increase of basal activity of the receptor by cholesterol.
What are the mechanisms by which cholesterol modulates function of GPCR? There are several examples of GPCR that exhibit a certain affinity for cholesterol, and whose activities are regulated in response to the content of cholesterol in membranes. This includes the β2-adrenergic receptor and the μ-opioid receptor for which specific high affinity cholesterol binding sites have been reported near the transmembrane helices7,45.
A putative cholesterol binding sequence (CRAC) was reported for transmembrane helix 7 of human CB1 receptor28. This sequence was proposed to be involved in directing the interaction of CB1 receptor with cholesterol-rich microdomains of cell membranes. Moreover, the presence of a cholesterol molecule was recently reported in a crystal structure55 and a cryo-EM structure56 of CB1 receptor. At the same time, there was no evidence of a specific retention of cholesterol in a recently published CB2 crystal31 structure. There is cholesterol included in PDB 6PT0, a cryo-EM structure of CB244, although its origin is unclear since the protein was expressed in Sf9 cells that produce very little if any cholesterol10. Also, any specific interaction of cholesterol with certain sites on the receptor may not explain why modulation of receptor function occurs at relatively high cholesterol concentrations in the lipid matrix surrounding the receptor.
Using MD simulations, we searched for structural and dynamical correlates of the experimental results. In simulations of free CB2, the ICL3-TM6 region, known to interact with Gα, deviated significantly in RMSD/RMSF from the antagonist-bound conformation in the presence of cholesterol. Since the antagonist-bound conformation would be less likely to recruit G protein by definition, deviating from it is consistent with the large experimentally observed increase in constitutive activity. By contrast, the MRI-2659-bound structure showed relatively little deviation in this region, consistent with the strong antagonist activity of MRI-2659 in both cholesterol conditions.
The situation is less clear with the other two simulated ligands. We observed that the size of the ligand is inversely correlated with the effect of cholesterol on both Trp258 and ICL3-TM6. Yet this correlation does not map precisely to the corresponding changes in categorizing the ligands’ actions. When cholesterol is included in the membrane, the strong agonist MRI-2594 remains a (less-strong) agonist, the weaker agonist MRI-2646 becomes an inverse agonist, and the strong inverse agonist MRI-2659 remains as such (Fig. 3). The ligands are categorized relative to the baseline of ligand-free constitutive activity. The relative activity rank order of the MRI ligand series ligands (Fig. 3, y-axes) is preserved in both cholesterol conditions. We consider that the simulation-observed changes in the binding and ICL3-TM6 (i.e. G-protein binding) sites may be independent components contributing to the overall experimentally-observed effects, rather than ligand-specific effects that directly correlate with the pharmacological categories of ligands, since the simulation results correlated with the ligand sterics rather than their pharmacological categories. This is consistent with the hypothesis that the primary effect of cholesterol is to modify the baseline constitutive activity that defines how the tested ligands are categorized.
Our simulation cannot elucidate the allosteric pathway from binding pocket to G protein that would be responsible for the data—such an endeavor is well outside the scope of this work. While the chemical identity of the ligand is presumably important, we have not specifically addressed the effects of specific ligands beyond sterics. Future work might include simulating the CB2-G-protein complex and estimating the binding energy difference as a function of cholesterol and ligand, but this is an exceedingly large task.
The data do not explain how the chloro- substituent at the 6-position of the benzothiazole arm may regulate effects of cholesterol on CB2 activation. While the toggle switch Trp2586.48 functions as an important molecular determinant in activation or deactivation of the receptor, it sheds limited light on differential interactions leading to neutral vs. inverse agonism. That the compound MRI-2654 with a bromo-substituent still behaves as an inverse agonist attests to the subtle difference in size and electronegativity of the chloro-group in influencing the molecular dynamics and signaling processes resulting in modulation of receptor function.
By using MRI-2646 ligand we demonstrate that cholesterol increases the constitutive activity of CB2 receptor. The content of cholesterol in preparations of cell membranes expressing CB2 correlates with an increase in basal signaling through CB2, which could be reversed by depletion of cholesterol using cyclodextrin. These results suggest that the pharmacological properties of synthetic ligands can be influenced by the cholesterol composition of cell membranes harboring cannabinoid receptor. Such a regulatory mechanism may contribute for well documented tissue- and cell-specific differences in the efficacy of partial CB2 agonists, such as the endocannabinoid anandamide or the plant-derived cannabinoid Δ9-tetrahydrocannabinol57. For example, THC acted as a full CB2 agonist in suppressing interferon-γ-induced activation of microglia58, whereas it had no CB2 agonist activity and acted as a CB2 antagonist by blocking 2-AG-induced migration of natural killer cells59, which have high levels of membrane lipid, including cholesterol60.
Methods
Materials
Chromatographic resin Ni–NTA was purchased from Qiagen (Germantown, MD). Streptactin XT was from IBA Life Sciences (Goettingen, Germany). The detergents CHAPS (3-[(3-Cholamidopropyl)-Dimethylammonio]-1-Propane Sulfonate] • N,N-Dimethyl-3-Sulfo-N-[3-[[3α,5β,7α,12α)-3,7,12-Trihydroxy-24-Oxocholan-24-yl]Amino]propyl]-1-Propanaminium Hydroxide, Inner Salt) , LMNG (Lauryl Maltose Neopentyl Glycol) and DDM (Dodecyl-β-D-Maltoside) were from Anatrace (Maumwee, OH). CHS-Tris salt was from Anatrace. The detergent Façade-TEG (3a,7a,12a-tri-((O-b-D-glucopyranosyl)ethyloxy)-cholane) and lipids POPC, POPG, brain lipid extract and cholesterol were from Avanti Polar Lipids (Alabaster, AL).
The potent non-selective CB2 agonist CP-55,940 ((-)-cis-3[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxtpropyl) cyclohexanol, the high affinity selective CB2 inverse agonist SR-144,528 5-(4-Chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl]-1H-pyrazole-3-carboxamide were from Cayman Chemical (Ann Arbor, MI). 3H-labeled CP-55,940 was from Perkin Elmer Life Sciences (Akron, OH). All other reagents were from Sigma-Aldrich (USA).
Preparations of CHO cell membranes expressing CB2 were obtained from the following sources: PerkinElmer (Cat. No ES111-M400UA, Billerica, MA), Millipore ChemiScreen (Cat. No P34972, Burlington, MA) and Applied Cell Sciences (Cat. No A318, Rockville, MD). Preparations of Sf9 cell membranes expressing CB2 were from Signal Screen (Cat. No 6110130, Rockville, MD).
Expi293F HEK cells were obtained from ThermoFisher Scientific (Cat. No: A14527), and CB2 expressed and membranes obtained in house as described elsewhere42. The E. coli cells BL21 (DE3) were obtained from EMD Millipore-Sigma (Cat. No. 69450), and CB2 expressed and membranes were obtained in house as described elsewhere.34,61.
Chemistry
Commercially available regents were purchased and used as is. Proton (1H NMR) spectra were recorded on a Varian 400 or Bruker 500 MHz spectrometer in solvents indicated with the values given in ppm (TMS as internal standard) and J (Hz) assignments of proton resonance coupling. Mass spectra (HRMS) were recorded on a JEOL SX102a mass spectrometer. Thin layer chromatography (TLC) analyses were carried out on 5 cm × 10 cm silica gel GHLF 0.25 mm plates using various gradients of EtOAc:n-hexane with visualization under UV light. Flash column chromatography was performed on Combiflash system. Product yields are reported as un-optimized. Study compounds had ≥ 95% purity. Purity and structural characterization was done by a combination of TLC, 1H-NMR, and LC/MS. LC–MS detection was carried out on Agilent 1200 using two different methods/columns: Luna C18 3 um (3 × 75 mm) where the mobile phase was 4% to 100% acetonitrile (0.05% TFA) standard gradient and EC18, 2.7 um (3 × 50 mm) where the method was 50% acetonitrile in water (0.1% formic acid) for 3 min ramping up to 98% acetonitrile over 7.5 min. The LC–MS chromatogram showed the correct molecular (MH+) ion as well as a single peak at UV (254 nm).
Synthesis and characterization of MRI-268728 and MRI-259431,32,62
Synthesis and characterization of MRI-2646, MRI-2654, MRI-2653 and MRI-2659 were carried as outlined in Li et al.28
N-(6-methyl-3-(2-hydroxyethyl)benzo[d]thiazol-2(3H)-ylidene)-2,2,3,3-tetramethylcyclopropane-1-carboxamide (4a)28.
N-(6-chloro-3-(2-hydroxyethyl)benzo[d]thiazol-2(3H)-ylidene)-2,2,3,3-tetramethylcyclopropane-1-carboxamide (4b).

2-amino-6-chlorobenzothiazole (1.84 g, 10.0 mmol) gave compound 4b (700 mg, 20%) as a white powder over two steps. Mp 147–149 °C;1H-NMR (400 MHz, CDCl3): δ 7.56 (s, 1H), 7.36 (d, J = 8.7 Hz, 1H), 7.22 (d, J = 8.7 Hz, 1H), 4.49 (t, J = 4.6 Hz, 3H), 4.06 (t, J = 4.6 Hz, 3H), 1.58 (s, 2H), 1.32 (s, 8H), 1.22 (s, 8H). LCMS [M + H]+: 353.2.
N-(6-bromo-3-(2-hydroxyethyl)benzo[d]thiazol-2(3H)-ylidene)-2,2,3,3-tetramethylcyclopropane-1-carboxamide (4c).

2-amino-6-bromobenothiazole (2.29 g, 10.0 mmol) gave compound 4c (950 mg, 24%) as a white powder over two steps. Mp 173–175 °C;1H-NMR (400 MHz, CDCl3): δ 7.70 (s, 1H), 7.50 (d, J = 8.6 Hz, 1H), 7.17 (d, J = 8.7 Hz, 1H), 4.49 (t, J = 4.5 Hz, 2H), 4.17 (s, 1H), 4.06 (s, 2H), 1.58 (s, 1H), 1.32 (s, 6H), 1.22 (s, 6H). LCMS [M + H]+: 397.1.
N-(3-(2-hydroxyethyl)-6-methoxybenzo[d]thiazol-2(3H)-ylidene)-2,2,3,3-tetramethylcyclopropane-1-carboxamide (4d).

2-amino-6-methoxybenothiazole (1.8 g, 10.0 mmol) gave compound 4d (920 mg, 26%) as a white powder over two steps. Mp 164–166 °C;1H-NMR (400 MHz, CDCl3): δ 7.18 (d, J = 8.9 Hz, 1H), 7.11 (s, 1H), 6.99–6.96 (m, 1H), 4.82 (s, 1H), 4.48 (t, J = 4.5 Hz, 2H), 4.05 (d, J = 3.5 Hz, 2H), 3.83 (s, 3H), 1.55 (s, 4H), 1.32 (s, 6H), 1.21 (s, 6H). LCMS [M + H]+: 349.2.
N-(3-(2-hydroxyethyl)-6-(trifluoromethoxy)benzo[d]thiazol-2(3H)-ylidene)-2,2,3,3-tetramethylcyclopropane-1-carboxamide (4e).

2-amino-6-trifluoromethoxybenothiazole (1.5 g, 6.4 mmol) gave compound 4 (610 mg, 24%) as a white powder over two steps. Mp 149–151 °C; 1H-NMR (400 MHz, CDCl3): δ 7.46 (s, 1H), 7.30 (t, J = 7.6 Hz, 2H), 4.51 (s, 2H), 4.07 (s, 3H), 1.59 (s, 1H), 1.32 (s, 6H), 1.22 (s, 6H). LCMS [M + H]+: 403.2.
2-(6-methyl-2-((2,2,3,3-tetramethylcyclopropane-1-carbonyl)imino)benzo[d]thiazol-3(2H)-yl)ethyl acetate (MRI-2687) (2a)28.
2-(6-Chloro-2-((2,2,3,3-tetramethylcyclopropane-1-carbonyl)imino)benzo[d]thiazol-3(2H)-yl)ethyl acetate (MRI-2646) (2b).

Compound 4b (300 mg, 0.85 mmol) was used as a starting material to give compound 2b (195 mg, 58%) as a white powder. Mp 123–125 °C;1H-NMR (400 MHz, CDCl3): δ 7.54 (s, 1H), 7.34 (d, J = 8.8 Hz, 1H), 7.23 (s, 1H), 4.56 (t, J = 5.2 Hz, 2H), 4.46 (t, J = 5.3 Hz, 2H), 1.95 (s, 3H), 1.64 (s, 1H), 1.33 (s, 6H), 1.23 (s, 6H). LCMS [M + H]+: 395.2.
2-(6-Bromo-2-((2,2,3,3-tetramethylcyclopropane-1-carbonyl)imino)benzo[d]thiazol-3(2H)-yl)ethyl acetate (MRI-2654) (2c).

Compound 4c (300 mg, 0.75 mmol) was used as a starting material to give compound 2c (140 mg, 42%) as a white powder. Mp 127–129 °C;1H-NMR (400 MHz, CDCl3): δ 7.68 (s, 1H), 7.48 (d, J = 8.3 Hz, 1H), 7.19 (d, J = 8.6 Hz, 1H), 4.55 (d, J = 5.3 Hz, 2H), 4.47 (d, J = 5.2 Hz, 2H), 1.95 (s, 3H), 1.64 (s, 1H), 1.57–1.49 (m, 7H), 1.33 (s, 6H), 1.23 (s, 6H). LCMS [M + H]+: 439.2.
2-(6-Methoxy-2-((2,2,3,3-tetramethylcyclopropane-1-carbonyl)imino)benzo[d]thiazol-3(2H)-yl)ethyl acetate (MRI-2653) (2d).

Compound 4d (400 mg, 1.1 mmol) was used as a starting material to give compound 2d (230 mg, 51%) as a white powder. Mp 154–156 °C;1H-NMR (400 MHz, CDCl3): δ 7.21 (s, 1H), 7.09 (s, 1H), 6.96 (d, J = 9.3 Hz, 1H), 4.55 (d, J = 5.3 Hz, 2H), 4.47 (d, J = 5.2 Hz, 2H), 7.24–1.22 (m, 82H), 3.83 (s, 3H), 1.95 (s, 3H), 1.62 (s, 1H), 1.34 (s, 6H), 1.22 (s, 6H). LCMS [M + H]+: 391.2.
2-(2-((2,2,3,3-tetramethylcyclopropane-1-carbonyl)imino)-6-(trifluoromethoxy)benzo[d]thiazol-3(2H)-yl)ethyl acetate (MRI-2659) (2e).

Compound 4e (300 mg, 0.75 mmol) was used as a starting material to give compound 2e (190 mg, 57%) as a white powder. Mp 81–83 °C; 1H-NMR (400 MHz, CDCl3): δ 7.44 (s, 1H), 7.32 (d, J = 8.8 Hz, 1H), 7.26 (s, 2H), 4.57 (d, J = 5.4 Hz, 2H), 4.47 (t, J = 5.4 Hz, 2H), 1.95 (s, 3H), 1.64 (s, 1H), 1.33 (s, 6H), 1.23 (s, 6H). LCMS [M + H]+: 445.3.
CB 2 expression in E. coli and purification
CB2 was expressed as a fusion with the maltose binding protein (MBP) in BL21 (DE3) E. coli culture and purified on milligram-scale as previously described41,63. In brief, 10 L of 2xYT media containing 0.2% glucose supplemented with ampicillin was inoculated with an overnight culture of E. coli. After reaching an optical density of 0.4, CB2 expression was induced by addition of 0.5 mM IPTG and 2.5 μM CB2 agonist CP-55,490. Expression was conducted for additional 42 h at 20 °C. After expression, cells were harvested by centrifugation, washed with cold PBS, and lysed in a cell homogenizer (Avestin). Receptor was solubilized for 1 h at 4 °C under continuous stirring by addition of concentrated detergent to final concentrations of (0.1% CHS, 1.0% DDM, 0.5% CHAPS, all w/v). The insoluble material was removed by centrifugation at 170,000 × g for 1 h and the solubilized receptor was then purified by the affinity chromatography in 50 mM Tris pH 7.5, 150 mM NaCl, 0.1% CHS, 0.1% DDM, 0.5% CHAPS, all w/v; 30% glycerol (v/v) and 10 μM CP-55,490 (buffer A) on Ni–NTA (Qiagen). MBP fusion partner was then removed upon incubation with the tobacco etch virus (TEV) protease for 4 h at 4 °C, and the released receptor was further purified by chromatography on StrepTactin XT (IBA Biosciences) and eluted in buffer A supplemented with 50 mM biotin as described previously41.
CB 2 expression in HEK cells and purification
Biomass from 3 L of Expi293F GNTI- cells expressing CB2 containing N-terminal twin-Streptag and C-terminal His10 tag was obtained according to manufacturer’s protocol (ThermoFisher Scientific). Protein was solubilized in detergents and purified by the two successive rounds of affinity chromatography on Ni–NTA resin and StrepTactin XT resin as described41.
Removal of the CHS and ligand from purified CB 2
An amount of 2 mg of purified CB2 was bound to 1200 μL of HisPur Co2+ (ThermoFisher) resin in buffer A and incubated under shaking for 2 h at 4 °C. The protein sample was then transferred to a disposable gravity column and washed with 40 column volumes (CV) of 0.5% CHAPS/0.1% DDM in 50 mM Tris–HCl, pH 7.5, 150 mM NaCl. Protein was then eluted with 6xCV of the same buffer supplemented with 250 mM imidazole; combined eluates concentrated on 30 kDa MWCO spin concentrator and washed 3 times to remove imidazole. Concentrated protein was supplemented with 15% glycerol and aliquots stored at − 80 °C until further use. To confirm that both ligand and CHS have been removed from the sample, 20 μL aliquot was mixed with 300 μL of chloroform–methanol mixture (1:1 v/v) and 1H-NMR spectra acquired.
Reconstitution of CB 2 into liposomes
Reconstitution of the purified CB2 into liposomes was performed as described earlier43. Briefly, 200 μg of the purified protein was mixed with 2 mg of lipid mixture (POPC:POPG, 3:1, mol/mol without or with addition or 20 mol% or 40 mol% of cholesterol) solubilized in 1% CHAPS at a concentration of 5 mg lipid/mL, and incubated on ice for 30 min. The detergents were then removed on 4 mL Detergent Removal spin column (Pierce), according to manufacturer’s instructions. The combined filtrate containing proteoliposomes was collected, and aliquots frozen in liquid nitrogen. Frozen liposomes were stored at − 80 °C until further use. Content of protein in proteoliposomes was determined by BioRad DC assay.
Ligand-binding assay in hCB 2 -CHO-K1 cell membranes
The assay was performed as described previously64. Briefly, binding affinity of the compounds to CB2R was determined by radioligand displacement assays using 0.2 nM of [3H] CP-55,940 as the radioligand. Plasma membranes were from cultured CHO-K1 cells stably transfected with human CB2R (Perkin Elmer). Two microgram plasma membrane protein was used in a 1 mL reaction mixture. Ki values were derived by computerized curve fitting and using the Cheng-Prusoff equation to account for the affinity of the radioligand, using the GraphPad Prism 8 program (GraphPad Prism Software Inc.).
[ 35 S] GTPγS binding assay in hCB 2 -CHO-K1 cell membranes 65
[35S] GTPγS binding was assayed as described earlier65 with slight modifications. Briefly, hCB2-CHO-K1 cell membranes (4 µg) were incubated with 0.05 nM [35S] GTPγS, and the indicated concentrations of ligands in TEM buffer (50 mM Tris–HCl, 0.2 mM EGTA, and 9 mM MgCl2, pH 7.4) containing 100 µM GDP, 150 mM NaCl, and 0.1% (w/v) bovine serum albumin in a total volume of 1 ml for 60 min at 30 °C.
[ 35 S] GTP nucleotide exchange (GEF) assays
The subunits of G protein were expressed and purified as described previously63. The nucleotide exchange assay was performed as previously described66.
Molecular dynamics simulations
The CHARMM36 force field67 was used. Before ligand parameterization, each ligand was geometry optimized using the B3LYP/6-31G** quantum mechanics level of theory and basis set using Gaussian0968. Ligand parameters were derived from CGenFF69; these high affinity ligands were not expected to explore the extremes of their conformational space.
The CB2 structure starting point was the cryo-EM structure previously described (Protein Data Bank: 6PT0). This was oriented and placed in a lipid membrane using the Orientations of Proteins in Membranes (OPM) database70 using the CHARMM-GUI71 input generator. In the with-cholesterol condition, the lipid membrane consisted of 40% cholesterol, and POPC:POPG in a 3:1 ratio. Sodium and chloride atoms were added to 0.15 M with excess for electroneutrality. The cholesterol molecules present in the cryo-EM structure were retained, particularly since the extracellular cholesterol conformations were analogous to those observed in the structurally similar mu opioid receptor. The system was minimized and equilibrated with side chain and backbone restraints which were subsequently released, and production simulations were run in the isothermic-isobaric ensemble at 303.15 K using NAMD 2.13 with GPU extensions. Particle Mesh Ewald summation of long-range interactions was used, as were the Langevin barostat and thermostat.
Abbreviations
- GPCR:
-
G protein-coupled receptor
- ICL:
-
Intracellular loop (in GPCR)
- TM:
-
Transmembrane domain (in GPCR)
- HEK293:
-
Human embryonic kidney
- CHO:
-
Chinese hamster ovary
- E. coli :
-
Escherichia coli
- Sf9 cells:
-
A clonal isolate of Spodoptera frugiperda baculovirus infected insect cell
- MD:
-
Molecular dynamics
- hCB2 :
-
Human cannabinoid receptor CB2
- hCB1 :
-
Human cannabinoid receptor CB1
- MBP:
-
Maltose binding protein of E. coli
- CP-55,940:
-
Synthetic cannabinoid agonist 2-[(1R,2R,5R)-5-Hydroxy-2-(3-hydroxypropyl)cyclohexyl]-5-(2-methyloctan-2-yl)phenol
- [3H]-CP55,940:
-
Radiolabeled synthetic ligand CP-55,940
- SR-144,528:
-
Synthetic cannabinoid inverse agonist 5-(4-Chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N-[(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]heptan-2-yl]-1H-pyrazole-3-carboxamide
- [35S]-GTPγS:
-
Radiolabeled synthetic analog of guanosine triphosphate
- MβCD:
-
Methyl-β-cyclodextrin
- Gα i1 and Gβ 1 γ 2 :
-
Subunits of heterotrimeric G protein
- GEF assay:
-
Determines rates of nucleotide exchange on the Gα subunit of G protein
- POPC:
-
1-Palmitoyl-2-oleoyl-glycero-3-phosphocholine
- POPG:
-
1-Palmitoyl-2-oleoyl-sn-glycero-3-phospho-(1′-rac-glycerol)
- CHS:
-
Cholesteryl hemisuccinate
- RMSD:
-
Root-mean-square deviation
- RMSF:
-
Root mean square fluctuation
References
Jastrzebska, B., Debinski, A., Filipek, S. & Palczewski, K. Role of membrane integrity on G protein-coupled receptors: Rhodopsin stability and function. Prog. Lipid Res. 50, 267–277. https://doi.org/10.1016/j.plipres.2011.03.002 (2011).
Marino, K. A., Prada-Gracia, D., Provasi, D. & Filizola, M. Impact of lipid composition and receptor conformation on the spatio-temporal organization of mu-opioid receptors in a multi-component plasma membrane model. PLoS Comput. Biol. 12, e1005240. https://doi.org/10.1371/journal.pcbi.1005240 (2016).
Salas-Estrada, L. A., Leioatts, N., Romo, T. D. & Grossfield, A. Lipids alter rhodopsin function via ligand-like and solvent-like interactions. Biophys. J. 114, 355–367. https://doi.org/10.1016/j.bpj.2017.11.021 (2018).
Escriba, P. V. et al. Role of lipid polymorphism in G protein-membrane interactions: Nonlamellar-prone phospholipids and peripheral protein binding to membranes. Proc. Natl. Acad. Sci. U.S.A. 94, 11375–11380. https://doi.org/10.1073/pnas.94.21.11375 (1997).
Maxfield, F. R. & van Meer, G. Cholesterol, the central lipid of mammalian cells. Curr. Opin. Cell. Biol. 22, 422–429. https://doi.org/10.1016/j.ceb.2010.05.004 (2010).
Chini, B. & Parenti, M. G-protein-coupled receptors, cholesterol and palmitoylation: Facts about fats. J. Mol. Endocrinol. 42, 371–379. https://doi.org/10.1677/JME-08-0114 (2009).
Manna, M. et al. Mechanism of allosteric regulation of beta2-adrenergic receptor by cholesterol. Elife https://doi.org/10.7554/eLife.18432 (2016).
Guixa-Gonzalez, R. et al. Membrane cholesterol access into a G-protein-coupled receptor. Nat. Commun. 8, 14505. https://doi.org/10.1038/ncomms14505 (2017).
Hefti, F. F. Requirements for a lead compound to become a clinical candidate. BMC Neurosci. 9, S7. https://doi.org/10.1186/1471-2202-9-S3-S7 (2008).
Dawaliby, R. et al. Phosphatidylethanolamine is a key regulator of membrane fluidity in eukaryotic cells. J. Biol. Chem. 291, 3658–3667. https://doi.org/10.1074/jbc.M115.706523 (2016).
Marheineke, K., Grunewald, S., Christie, W. & Reilander, H. Lipid composition of Spodoptera frugiperda (Sf9) and Trichoplusia ni (Tn) insect cells used for baculovirus infection. FEBS Lett. 441, 49–52 (1998).
Warnock, D. E. et al. Determination of plasma membrane lipid mass and composition in cultured Chinese hamster ovary cells using high gradient magnetic affinity chromatography. J. Biol. Chem. 268, 10145–10153 (1993).
Oursel, D. et al. Lipid composition of membranes of Escherichia coli by liquid chromatography/tandem mass spectrometry using negative electrospray ionization. Rapid Commun. Mass Spectrom. 21, 1721–1728. https://doi.org/10.1002/rcm.3013 (2007).
Morein, S., Andersson, A. S., Rilfors, L. & Lindblom, G. Wild-type Escherichia coli cells regulate the membrane lipid composition in a “window” between gel and non-lamellar structures. J. Biol. Chem. 271, 6801–6809. https://doi.org/10.1074/jbc.271.12.6801 (1996).
Vauquelin, G. Cell membranes and how long drugs may exert beneficial pharmacological activity in vivo. Br. J. Clin. Pharmacol. 82, 673–682. https://doi.org/10.1111/bcp.12996 (2016).
Jacobson, K. A. New paradigms in GPCR drug discovery. Biochem. Pharmacol. 98, 541–555. https://doi.org/10.1016/j.bcp.2015.08.085 (2015).
Overington, J. P., Al-Lazikani, B. & Hopkins, A. L. How many drug targets are there?. Nat. Rev. Drug Discov. 5, 993–996. https://doi.org/10.1038/nrd2199 (2006).
Yin, H. & Flynn, A. D. Drugging membrane protein interactions. Annu. Rev. Biomed. Eng. 18, 51–76. https://doi.org/10.1146/annurev-bioeng-092115-025322 (2016).
Talmont, F., Lebrun, C. & Zajac, J. M. Agonist binding of human mu opioid receptors expressed in the yeast Pichia pastoris: Effect of cholesterol complementation. Neurochem. Int. 132, 104588. https://doi.org/10.1016/j.neuint.2019.104588 (2020).
Veatch, S. L., Soubias, O., Keller, S. L. & Gawrisch, K. Critical fluctuations in domain-forming lipid mixtures. Proc. Natl. Acad. Sci. U.S.A. 104, 17650–17655. https://doi.org/10.1073/pnas.0703513104 (2007).
Chen, Z. & Rand, R. P. The influence of cholesterol on phospholipid membrane curvature and bending elasticity. Biophys. J. 73, 267–276. https://doi.org/10.1016/S0006-3495(97)78067-6 (1997).
Pan, J., Tristram-Nagle, S. & Nagle, J. F. Effect of cholesterol on structural and mechanical properties of membranes depends on lipid chain saturation. Phys. Rev. E Stat. Nonlin. Soft Matter. Phys. 80, 021931. https://doi.org/10.1103/PhysRevE.80.021931 (2009).
Rouviere, E., Arnarez, C., Yang, L. & Lyman, E. Identification of two new cholesterol interaction sites on the A2A adenosine receptor. Biophys. J. 113, 2415–2424. https://doi.org/10.1016/j.bpj.2017.09.027 (2017).
Gimpl, G. Interaction of G protein coupled receptors and cholesterol. Chem. Phys. Lipids 199, 61–73. https://doi.org/10.1016/j.chemphyslip.2016.04.006 (2016).
Sengupta, D., Prasanna, X., Mohole, M. & Chattopadhyay, A. Exploring GPCR-lipid interactions by molecular dynamics simulations: Excitements, challenges, and the way forward. J. Phys. Chem. B 122, 5727–5737. https://doi.org/10.1021/acs.jpcb.8b01657 (2018).
Legler, D. F. et al. Modulation of chemokine receptor function by cholesterol: New prospects for pharmacological intervention. Mol. Pharmacol. 91, 331–338. https://doi.org/10.1124/mol.116.107151 (2017).
Bari, M., Paradisi, A., Pasquariello, N. & Maccarrone, M. Cholesterol-dependent modulation of type 1 cannabinoid receptors in nerve cells. J. Neurosci. Res. 81, 275–283. https://doi.org/10.1002/jnr.20546 (2005).
Oddi, S. et al. Functional characterization of putative cholesterol binding sequence (CRAC) in human type-1 cannabinoid receptor. J Neurochem. 116, 858–865. https://doi.org/10.1111/j.1471-4159.2010.07041.x (2011).
Bari, M. et al. Effect of lipid rafts on Cb2 receptor signaling and 2-arachidonoyl-glycerol metabolism in human immune cells. J. Immunol. 177, 4971–4980 (2006).
Turcotte, C., Blanchet, M. R., Laviolette, M. & Flamand, N. The CB2 receptor and its role as a regulator of inflammation. Cell. Mol. Life Sci. 73, 4449–4470. https://doi.org/10.1007/s00018-016-2300-4 (2016).
Li, X. et al. Crystal structure of the human cannabinoid receptor CB2. Cell 176, 459–467. https://doi.org/10.1016/j.cell.2018.12.011 (2019).
Yao, B. B. et al. Characterization of a cannabinoid CB2 receptor-selective agonist, A-836339 [2,2,3,3-tetramethyl-cyclopropanecarboxylic acid [3-(2-methoxy-ethyl)-4,5-dimethyl-3H-thiazol-(2Z)-ylidene]-amide], using in vitro pharmacological assays, in vivo pain models, and pharmacological magnetic resonance imaging. J. Pharmacol. Exp. Ther. 328, 141–151. https://doi.org/10.1124/jpet.108.145011 (2009).
Yeliseev, A. A. Methods for recombinant expression and functional characterization of human cannabinoid receptor Cb2. Comput. Struct. Biotechnol. J. 2, 2. https://doi.org/10.5936/csbj.201303011 (2013).
Yeliseev, A. A., Wong, K. K., Soubias, O. & Gawrisch, K. Expression of human peripheral cannabinoid receptor for structural studies. Protein Sci. 14, 2638–2653. https://doi.org/10.1110/ps.051550305 (2005).
Parathath, S. et al. Changes in plasma membrane properties and phosphatidylcholine subspecies of insect Sf9 cells due to expression of scavenger receptor class B, type I, and CD36. J. Biol. Chem. 279, 41310–41318. https://doi.org/10.1074/jbc.M404952200 (2004).
Gimpl, G., Klein, U., Reilander, H. & Fahrenholz, F. Expression of the human oxytocin receptor in baculovirus-infected insect cells: High-affinity binding is induced by a cholesterol-cyclodextrin complex. Biochemistry 34, 13794–13801. https://doi.org/10.1021/bi00042a010 (1995).
Huang, Z. & London, E. Cholesterol lipids and cholesterol-containing lipid rafts in bacteria. Chem. Phys. Lipids 199, 11–16. https://doi.org/10.1016/j.chemphyslip.2016.03.002 (2016).
Liu, S. L. et al. Orthogonal lipid sensors identify transbilayer asymmetry of plasma membrane cholesterol. Nat. Chem. Biol. 13, 268–274. https://doi.org/10.1038/nchembio.2268 (2017).
Mahammad, S. & Parmryd, I. Cholesterol depletion using methyl-beta-cyclodextrin. Methods Mol. Biol. 1232, 91–102. https://doi.org/10.1007/978-1-4939-1752-5_8 (2015).
Zidovetzki, R. & Levitan, I. Use of cyclodextrins to manipulate plasma membrane cholesterol content: Evidence, misconceptions and control strategies. Biochim. Biophys. Acta 1768, 1311–1324. https://doi.org/10.1016/j.bbamem.2007.03.026 (2007).
Yeliseev, A., Zoubak, L. & Schmidt, T. G. M. Application of Strep-Tactin XT for affinity purification of Twin-Strep-tagged CB2, a G protein-coupled cannabinoid receptor. Protein Expr. Purif. 131, 109–118. https://doi.org/10.1016/j.pep.2016.11.006 (2017).
Yeliseev, A. et al. Thermostability of a recombinant G protein-coupled receptor expressed at high level in mammalian cell culture. Sci. Rep. 10, 16805. https://doi.org/10.1038/s41598-020-73813-7 (2020).
Vukoti, K., Kimura, T., Macke, L., Gawrisch, K. & Yeliseev, A. Stabilization of functional recombinant cannabinoid receptor CB2 in detergent micelles and lipid bilayers. PLoS ONE 7, e46290. https://doi.org/10.1371/journal.pone.0046290 (2012).
Xing, C. et al. Cryo-EM structure of the human cannabinoid receptor CB2-Gi signaling complex. Cell https://doi.org/10.1016/j.cell.2020.01.007 (2020).
Huang, W. et al. Structural insights into micro-opioid receptor activation. Nature 524, 315–321. https://doi.org/10.1038/nature14886 (2015).
Salari, R., Joseph, T., Lohia, R., Henin, J. & Brannigan, G. A streamlined, general approach for computing ligand binding free energies and its application to GPCR-bound cholesterol. J. Chem. Theory Comput. 14, 6560–6573. https://doi.org/10.1021/acs.jctc.8b00447 (2018).
Kimura, T. et al. Recombinant cannabinoid type 2 receptor in liposome model activates g protein in response to anionic lipid constituents. J. Biol. Chem. 287, 4076–4087. https://doi.org/10.1074/jbc.M111.268425 (2012).
Mouritsen, O. G. & Zuckermann, M. J. What’s so special about cholesterol?. Lipids 39, 1101–1113. https://doi.org/10.1007/s11745-004-1336-x (2004).
Niu, S. L., Mitchell, D. C. & Litman, B. J. Manipulation of cholesterol levels in rod disk membranes by methyl-beta-cyclodextrin: Effects on receptor activation. J. Biol. Chem. 277, 20139–20145. https://doi.org/10.1074/jbc.M200594200 (2002).
Cantor, R. S. The lateral pressure profile in membranes: A physical mechanism of general anesthesia. Biochemistry 36, 2339–2344. https://doi.org/10.1021/bi9627323 (1997).
Andersen, O. S. & Koeppe, R. E. Bilayer thickness and membrane protein function: An energetic perspective. Annu. Rev. Biophys. Biomol. Struct. 36, 107–130. https://doi.org/10.1146/annurev.biophys.36.040306.132643 (2007).
Yeagle, P. L. Modulation of membrane function by cholesterol. Biochimie 73, 1303–1310. https://doi.org/10.1016/0300-9084(91)90093-g (1991).
Pucadyil, T. J. & Chattopadhyay, A. Role of cholesterol in the function and organization of G-protein coupled receptors. Prog. Lipid Res. 45, 295–333. https://doi.org/10.1016/j.plipres.2006.02.002 (2006).
Sohlenkamp, C. & Geiger, O. Bacterial membrane lipids: Diversity in structures and pathways. FEMS Microbiol. Rev. 40, 133–159. https://doi.org/10.1093/femsre/fuv008 (2016).
Hua, T. et al. Crystal structure of the human cannabinoid receptor CB1. Cell 167, 750–762. https://doi.org/10.1016/j.cell.2016.10.004 (2016).
Krishna Kumar, K. et al. Structure of a signaling cannabinoid receptor 1-G protein complex. Cell 176, 448–458. https://doi.org/10.1016/j.cell.2018.11.040 (2019).
Reggio, P. H. Endocannabinoid binding to the cannabinoid receptors: What is known and what remains unknown. Curr. Med. Chem. 17, 1468–1486. https://doi.org/10.2174/092986710790980005 (2010).
Ehrhart, J. et al. Stimulation of cannabinoid receptor 2 (CB2) suppresses microglial activation. J. Neuroinflam. 2, 29. https://doi.org/10.1186/1742-2094-2-29 (2005).
Kishimoto, S. et al. Endogenous cannabinoid receptor ligand induces the migration of human natural killer cells. J. Biochem. 137, 217–223. https://doi.org/10.1093/jb/mvi021 (2005).
Rudd-Schmidt, J. A. et al. Lipid order and charge protect killer T cells from accidental death. Nat. Commun. 10, 5396. https://doi.org/10.1038/s41467-019-13385-x (2019).
Yeliseev, A. A. & Vukoti, K. in Production of membrane proteins (ed A.S. Robinson) 219–248 (Wiley-VCH, 2011).
Horti, A. G. et al. Synthesis and biodistribution of [11C]A-836339, a new potential radioligand for PET imaging of cannabinoid type 2 receptors (CB2). Bioorg. Med. Chem. 18, 5202–5207. https://doi.org/10.1016/j.bmc.2010.05.058 (2010).
Yeliseev, A. Expression and preparation of a G-protein-coupled cannabinoid receptor CB2 for NMR structural studies. Curr. Protoc. Protein Sci. 96, e83. https://doi.org/10.1002/cpps.83 (2019).
Cinar, R. et al. Hybrid inhibitor of peripheral cannabinoid-1 receptors and inducible nitric oxide synthase mitigates liver fibrosis. JCI Insight https://doi.org/10.1172/jci.insight.87336 (2016).
Iyer, M. R. et al. Structural basis of species-dependent differential affinity of 6-alkoxy-5-aryl-3-pyridinecarboxamide cannabinoid-1 receptor antagonists. Mol. Pharmacol. 88, 238–244 (2015).
Beckner, R. L., Zoubak, L., Hines, K. G., Gawrisch, K. & Yeliseev, A. A. Probing thermostability of detergent-solubilized CB2 receptor by parallel G protein-activation and ligand-binding assays. J. Biol. Chem. 295, 181–190. https://doi.org/10.1074/jbc.RA119.010696 (2020).
Huang, J. & MacKerell, A. D. Jr. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 34, 2135–2145. https://doi.org/10.1002/jcc.23354 (2013).
68M. J. Frisch, G. W. T., H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery, Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman, and D. J. Fox. Gaussian 09, Revision A02. Gaussian, Inc., (2016).
Vanommeslaeghe, K. et al. CHARMM general force field: A force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 31, 671–690. https://doi.org/10.1002/jcc.21367 (2010).
Lomize, M. A., Pogozheva, I. D., Joo, H., Mosberg, H. I. & Lomize, A. L. OPM database and PPM web server: Resources for positioning of proteins in membranes. Nucleic Acids Res. 40, D370-376. https://doi.org/10.1093/nar/gkr703 (2012).
Lee, J. et al. CHARMM-GUI input generator for NAMD, GROMACS, AMBER, OpenMM, and CHARMM/OpenMM simulations using the CHARMM36 additive force field. J. Chem. Theory Comput. 12, 405–413. https://doi.org/10.1021/acs.jctc.5b00935 (2016).
Acknowledgements
This work was supported by the Intramural Research Program of the NIAAA, NIH. TTJ was supported by the Foundation for Anesthesia Education and Research (MRTG-BS-Joseph). The Fig. 11 was generated in Biorender.
Author information
Authors and Affiliations
Contributions
A.Y., M.R.I., R.C. conceived the project and designed the experimental procedures. M.R.I. performed synthesis of cannabinoid ligands; L.Z. and A.Y. purified the protein and performed GEF experiments; R.C. and N.J.C. performed ligand binding and GTP-binding experiments; A.Y. prepared the proteoliposomes; T.T.J. performed the MD simulation; K.G. and G.K. supervised the work and provided funding. A.Y. and M.R.I. drafted the paper with contributions from R.C., T.T.J., K.G. and G.K. All authors reviewed the final paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Yeliseev, A., Iyer, M.R., Joseph, T.T. et al. Cholesterol as a modulator of cannabinoid receptor CB2 signaling. Sci Rep 11, 3706 (2021). https://doi.org/10.1038/s41598-021-83245-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-021-83245-6
- Springer Nature Limited
This article is cited by
-
Membrane protein Bcsdr2 mediates biofilm integrity, hyphal growth and virulence of Botrytis cinerea
Applied Microbiology and Biotechnology (2024)
-
Filling of a water-free void explains the allosteric regulation of the β1-adrenergic receptor by cholesterol
Nature Chemistry (2022)