
Abstract
Prognosis of patients with lung cancer remains extremely poor; thus, we sought to unearth novel competing endogenous RNA (ceRNA) networks associated with the prognosis of lung adenocarcinoma (LUAD). Aberrant mRNAs were identified from the intersection of three Gene Expression Omnibus (GEO) datasets. A protein–protein interaction (PPI) network was constructed, and miRNAs and long noncoding RNAs (lncRNAs) upstream of mRNAs were predicted. In the present study, 402 upregulated and 638 downregulated genes in lung cancer tissues were identified. Functional analysis showed significant enrichment of cancer pathways. In these top hub genes, 10 upregulated and 7 downregulated genes had substantial prognostic values in LUAD. Thirty-seven miRNAs were predicted to target 17 key genes, and only five miRNAs exhibited prognostic correlation. Through stepwise reverse prediction and validation from miRNA to lncRNA, four key lncRNAs were identified using expression and survival analysis. Ultimately, the co-expression analysis identified LINC00665-miR-let-7b-CCNA2 as the key ceRNA network associated with the prognosis of LUAD. We successfully constructed a novel ceRNA network wherein each component was significantly associated with the prognosis of LUAD. Hence, we propose that this network may provide key biomarkers or potential therapeutic targets for LUAD prognosis.
Similar content being viewed by others

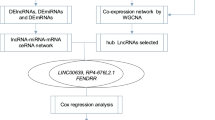

Introduction
Lung cancer remains the leading cause of cancer-related death worldwide, accounting for 19.4% of overall cancer mortality1. Notably, most lung cancer patients have a poor prognosis2. Primary lung cancer is generally divided into small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC). NSCLC accounts for about 80% of all primary lung cancers, of which adenocarcinoma, squamous cell carcinoma, and large cell carcinoma are the main histological subset3. Despite improvements and advancements in early diagnostic and treatment methods, the 5-year survival rate for primary lung cancer is still as low as 11–16%4,5. With a deeper understanding of the molecular biological mechanisms of lung cancer, targeted therapy has made great progress.
Whole-genome analysis of gene expression shows that exons account for less than 3% of the human genome, and an increase in the number of noncoding RNA (ncRNA) genes in introns reportedly regulates gene expression6. ncRNAs include three types: long ncRNAs (lncRNAs), mid-size ncRNAs, and short ncRNAs. Among short ncRNAs, miRNAs target gene regulation and can affect lung cancer treatment7. Salmena et al. proposed the competing endogenous RNA (ceRNA) hypothesis whereby mRNAs, miRNAs, and lncRNAs could cross-talk, forming a regulatory network8. Through miRNA response elements, lncRNAs could be sequestering RNA-binding proteins and microRNAs leading to changes in the miRNA-regulated mRNA levels. Increasing evidence indicates that this ceRNA network plays a pivotal role in a variety of human cancers, such as breast cancer9, gastric cancer10, liver cancer11, and pancreatic cancer12.
Generally, lncRNAs consist of more than 200 nucleotides with limited or no protein-coding capacity. To date, more than 3000 lncRNAs have been identified. Abnormal expression of various lncRNAs is related to carcinogenesis. In particular, it can lead to the occurrence, development, and metastasis of multiple cancers, such as breast cancer, hepatocellular carcinoma, and cardiac adenocarcinoma13,14,15. In physiological conditions, lncRNAs participate in multiple biological processes, including splicing, transcription, epigenetic gene expression, and chromatin modification16.
Research has shown that the expression of HOTAIR, MALAT1, HOTTIP, ANRIL, and ZXF2 were upregulated in lung cancer tissues, which is related to increased tumor lymph node metastasis rate, advanced lymph node metastasis, and decreased overall survival17,18,19,20,21. Considering their function in the development of lung cancer, they could become promising biomarkers for the diagnosis or prognosis of lung cancer22. However, the current understanding of the key lncRNA–miRNA–mRNA ceRNA networks that are significantly related to the prognosis of lung cancer remains unclear.
In our work, we screened mRNAs that were differentially expressed (DE-mRNAs) in lung cancer tissues compared to healthy tissues by mining three Gene Expression Omnibus (GEO) datasets (GSE18842, GSE19188, and GSE33532). The common aberrantly expressed mRNAs were listed, and their functional enrichment analysis was carried out. Subsequently, we conducted protein–protein interaction (PPI) analysis using the STRING database and identified several up- and downregulated hub genes. Then, miRTarBase was used to predict the upstream miRNAs. Taking miRNA expression and prognostic values into consideration, candidate miRNAs were further selected to predict the potential upstream lncRNAs. Furthermore, the prognostic value of these potential lncRNAs was assessed, and the co-expression analysis between mRNAs, miRNAs, and lncRNAs was evaluated using the ceRNA hypothesis. Finally, we successfully established a novel ceRNA regulatory network, which was significantly associated with the prognosis of patients with LUAD. Our research sheds light on the onset of LUAD and identifies potential diagnostic biomarkers or therapeutic targets.
Results
Identification of significant differentially expressed genes (DEGs) in LUAD
Searching for gene expression microarrays with regard to LUAD from the GEO database, three datasets (GSE18842, GSE19188, and GSE33532) were selected, and information of three GEO data sets were shown in Table 1. Next, DEGs analysis was performed using GEO2R (|log2FC|> 1 and adj. p-value < 0.05), and significant DEGs in each dataset were identified (Fig. 1A–C). In the GSE18842 dataset, a total of 815 upregulated and 1034 downregulated mRNAs were screened. For the GSE19188 dataset, there were 557 upregulated and 911 downregulated genes in lung cancer tissues compared to those in normal control samples. Finally, in the GSE33532 dataset, 866 upregulated and 1263 downregulated genes were identified. After the upregulated or downregulated mRNAs from each dataset were intersected, we identified a total of 402 upregulated and 638 downregulated DE-mRNAs shared among the three datasets and selected them for subsequent analyses (Fig. 1D, E). The detailed DE-mRNAs are listed in Supplementary Excel 1–3.

Identification of differentially expressed mRNAs between lung adenocarcinoma (LUAD) and normal samples in three Gene Expression Omnibus (GEO) datasets. (A–C) Volcano plots of DE-mRNAs in GSE18842, GSE19188, and GSE33532. Horizontal axis represents the − log10 (adjusted p-value) and vertical axis represents the log2 (fold change) of gene expression. Green dots and red dots represent significantly downregulated and upregulated genes, respectively. Black dots represent genes with no significant difference. (D, E) The intersection of upregulated and downregulated genes in three datasets, respectively. DEG; DE-mRNA: Differentially expressed mRNA.
Functional analysis of the DE-mRNAs
To predict the biological functions of the identified DE-mRNAs, we performed Gene Ontology (GO) enrichment and Kyoto Encyclopaedia of Genes and Genomes (KEGG) pathway analysis. Predicting the biological function of identified DE-mRNAs, we performed GO term enrichment and KEGG pathway analysis. Analysis of biological processes was mainly performed in the GO annotation. For upregulated DE-mRNAs, our results indicated that these genes were notably enriched in processes associated with cell division, DNA replication, and cytoskeletal movements (Fig. 2A).Additionally, cell cycle and malignancy-related pathways, such as the p53 signaling pathway and amino acid metabolism, were observed in the KEGG pathway analysis (Fig. 2B). Notably, positive regulation of angiogenesis was observed, and the biological process analysis also indicated several cancer-related GO terms, such as cell adhesion and extracellular region/space (Fig. 2C). Furthermore, the KEGG pathway analysis also found that some downregulated genes were significantly enriched in well-known cancer-associated pathways, including cell adhesion molecules, TNF, PPAR, and chemokine signaling pathways (Fig. 2D). Altogether, these results indicated that both up- and downregulated DE-mRNAs were closely related to lung cancer.

Functional and protein–protein interaction (PPI) networks analysis for the intersected differentially expressed mRNAs. (A, C) Enriched GO pathways of the significantly upregulated and downregulated genes, respectively. (B, D) Enriched KEGG pathways of the significantly upregulated and downregulated genes, respectively. BP biological process; CC cellular component; MF molecular function; GO Gene Ontology; KEGG Kyoto Encyclopaedia of Genes and Genome.
Construction of the PPI network and identification of hub genes
STRING-based database analysis, Supplementary Figure 1a, c show significantly different PPI networks related to up- and downregulated DEGs, respectively. Based on the degree of each node, we identified 20 hub genes among these significant differences. For better visualization, we used the Cytoscape software to reconstruct the top 20 interacting genes of up- (Supplementary Figure 1B) and downregulated (Supplementary Figure 1D) hub genes. The top 20 hub genes that were upregulated and the top 20 hub genes that were downregulated were selected for subsequent analyses.
Validation of gene expression of hub genes and survival analysis
Aiming to determine the expression of key genes in lung cancer, we used gene expression profiling analysis (GEPIA) and the Kaplan–Meier plotter database to analyze the expression and prognostic value of the top 10 up- and downregulated hub genes, respectively. Combining expression patterns and survival analysis, we found that 10 upregulated hub genes (CDK1, CCNB1, CCNA2, TOP2A, AURKA, MAD2L1, CDC20, CCNB2, AURKB, and KIF11) were not only significantly upregulated in lung cancer but also significantly correlated with poor prognosis of lung cancer patients (p < 0.05; Fig. 3A–K). Conversely, seven of the downregulated hub genes (TLR4, PECAM1, SELP, CXCL12, VWF, KDR, and CD34) showed both low expression and good prognosis in lung cancer patients (p < 0.05; Fig. 4A–H). These 17 key genes, which met the criteria of converse expression pattern and survival prognosis, were selected for the next analyses.

Screening the key upregulated genes in LUAD. (A) Identification of the key genes with high expression and poor prognosis values among significantly upregulated hub genes. (B–K) Representative expression and prognostic value of hub genes validated with Gene Expression Profiling Interactive Analysis (GEPIA) and Kaplan–Meier plotter databases, respectively.

Screening the key downregulated genes in LUAD. (A) Identification of key genes with low expression and good prognosis values in significantly downregulated hub genes. (B–H) Representative expression and prognostic value of hub genes validated with GEPIA and Kaplan–Meier plotter databases, respectively.
Prediction and validation of key miRNAs upstream of critical genes
In order to identify key miRNAs that regulate the pivotal hub genes, we predicted their upstream miRNAs by using the miRTarBase database, which is the experimentally validated miRNA-target interaction. Finally, we identified 37 miRNAs that may regulate the expression of key pivot genes, as shown in Fig. 5A. We downloaded the miRNA expression profile and survival information of lung adenocarcinoma patients from TCGA project. Then, we selected 36 out of 37 miRNAs obtained from TCGA project to construct a risk signature using multivariate Cox regression analysis. The risk score of each patient was based on a linear combination of miRNA expression level (X) multiplied by the regression coefficient (α) from the multivariate Cox regression analysis. The formula is as follows: risk score = X1α1 + X2α2 + X3α3 + ⋯ + Xnαn. Twelve miRNAs comprise the risk signature. The results showed that high-risk patients had a shorter survival time than patients with lung adenocarcinoma, while the risk signature was not a good prediction model for prognosis of lung adenocarcinoma patients (Supplementary Figure 2a, b). According to the negative regulatory relationship between miRNA and its target genes, we further evaluated the prognostic value of miRNA for overall survival in patients with LUAD using the Kaplan–Meier plotter database. Survival analysis showed that high expression of two downregulated miRNAs, miR-548b and miR-let-7b, functioned as better prognostic biomarkers in patients with LUAD (Fig. 5B, C). By contrast, three upregulated miRNAs, miR-17, miR-137, and miR-23a, displayed negative prognostic function in these patients (Fig. 5D–F). These miRNAs were identified as key regulators and were analyzed further.

Construction of miRNA-gene network using Cytoscape software. (A) The rectangle in the network represents miRNA. The ellipse in the network represents hub genes. (B–F) Representative prognostic value of key miRNAs validated using Kaplan–Meier plotter databases.
Prediction and validation of upstream key lncRNAs
lncRNAs can regulate gene expression by influencing the transcription, mRNA turnover, and translation by sequestering RNA binding proteins and microRNAs23. We further used the online database, miRNet, to predict the lncRNAs that may bind to 5 key miRNAs (miR-548b, miR-let-7b, miR-17, miR-137, and miR-23a). A total of 198 lncRNAs were discovered in the database for the two upregulated miRNAs, while 426 lncRNAs were found to be able to potentially regulate the three downregulated miRNAs (Supplementary Excel 4, 5). The ceRNA hypothesis assumes that lncRNA can attenuate miRNA activity through chelation, thereby upregulating the expression of miRNA-target genes. Therefore, the eligible lncRNA should negatively correlate with miRNA while positively correlating with target mRNA. Therefore, the predicted expression level and prognostic value of lncRNA were verified using GEPIA and Kaplan–Meier plotter databases, respectively. Only LINC00665 was notably upregulated in LUAD samples when compared to healthy controls (Fig. 6A). Subsequent survival analysis showed that patients with high expression of LINC00665 had poor prognosis (Fig. 6B). By contrast, three lncRNAs (LINC01140, NEAT1, and PCAT19) were significantly downregulated compared to those of healthy controls and displayed a good prognosis (Fig. 6C–F). Therefore, we defined these four as key lncRNAs in the ceRNA network.

Screening the key lncRNAs in LUAD. (A, B) Identification of the key long noncoding RNAs (lncRNAs) with high expression and poor prognosis values among the predicted candidate lncRNAs by combining expression and prognosis analyses using GEPIA and Kaplan–Meier-plotter databases, respectively. (C–F) Identification of key lncRNAs with low expression and better prognosis values among predicted candidate lncRNAs by combining expression and prognosis analyses using GEPIA and Kaplan–Meier-plotter databases, respectively.
Construction of the lncRNA–miRNA–mRNA regulatory network in LUAD
Based on the previous prediction and validation, a vital lncRNA–miRNA–mRNA ceRNA network in patients with LUAD was established. Following the ceRNA hypothesis mentioned above, miRNA expression was inversely correlated with mRNA and lncRNA, whereas lncRNA expression was positively correlated with mRNA. Thus, we further assessed the correlation of interactions between all these RNA pairs in the network by using the starBase database. As shown in Fig. 7A–D, only the LINC00665-miR-let-7b-5p-CCNA2 regulatory network could fit within the ceRNA mechanism. Finally, we constructed a new three sub-networks of mRNA-miRNA-lncRNA, which significantly correlated with the prognosis of LUAD (Fig. 7E). This sub-network can also be used to identify promising diagnostic biomarkers or to develop therapeutic targets for LUAD.

Construction of the lncRNA–miRNA–mRNA regulatory network in LUAD. Correlations within mRNA, miRNA, and lncRNA using the starBase database (A–D). (E) Novel mRNA–miRNA–lncRNA competing endogenous RNA (ceRNA), triple-regulatory network associated with the prognosis of LUAD.
Discussion
Lung cancer is known for its poor prognosis and high mortality; its metastatic mechanism is complex and remains unclear24,25. Poor prognosis for patients with lung cancer prompts us to formulate effective treatment measures and find more effective prognostic indicators26. Therefore, revealing molecular switches that control the malignant transformation of lung cancer is of considerable significance, which may also reveal new prognostic indicators. Recent research suggests that ncRNAs, including miRNAs and lncRNAs, play pivotal roles in the occurrence or development of cancer27,28. There is increasing evidence of a close, complex relationship between miRNAs and lncRNAs in cancer29,30. Based on the ceRNA hypothesis first proposed by Salmena et al.8, active research on ceRNAs in human cancer has been implemented, which has shown that they participate in various pathological processes, including tumorigenesis31,32. Liu et al. reported that lncRNA-XIST as ceRNA negatively regulates the expression of miR-34a and drives thyroid cancer proliferation and growth via the MET-PI3K-AKT signaling pathway33. lncRNA XLOC-006390 also has the function of ceRNA and negatively regulates the expression of miR-331-3p and miR-338-3p, thereby promoting tumorigenesis and metastasis of cervical cancer34. Moreover, Gao et al. determined that lncRNA ZEB2-AS1 promotes pancreatic cancer cell growth and invasion by regulating miR-20435.
Regarding lung cancer, Schmidt et al. identified the lncRNA, MALAT1, as a prognostic marker for metastasis and patient survival in NSCLC36. In addition, Jen and colleagues revealed a new mechanism whereby Oct4 transcriptionally activates lncRNAs, NEAT1 and MALAT1, via promoter and enhancer-binding, respectively, to promote tumor cell proliferation and metastasis, leading to lung tumorigenesis and poor prognosis37. Functional validation shows that the largest differentially expressed lncRNA in lung cancer, LCAL1 (lung cancer-associated lncRNA 1), contributes to tumor cell proliferation38. Furthermore, siRNA-mediated downregulation of lncRNA and MVIH (microvascular invasion of HCC) can inhibit cell growth by regulating the expression of MMP-2 and MMP-9 proteins in NSCLC39.
In our research, we identified two important subsets, including 402 upregulated genes and 638 downregulated genes from three GEO datasets, GSE18842, GSE19188, and GSE3353240. GO analysis of these significant DEGs demonstrated that they were significantly enriched in GO terms associated with cancer biological behavior, including cell division41,42, cell adhesion43, and positive regulation of angiogenesis44,45. KEGG pathway enrichment analysis shows that multiple pathways are enriched, mainly p53 signaling pathways and other important regulatory pathways in cancer46 and cell cycle-related pathways47. More importantly, we found that the deregulation of amino acid metabolism, such as that of pyrimidine, purine, and glutathione, plays vital roles in the reprogramming of cellular metabolism and is essential for tumorigenesis48,49. Therefore, these important DEGs may be involved in regulating the occurrence and metastasis of lung cancer.
We hypothesized that these screened DE-mRNAs might interact with each other. Thus, two PPI networks were constructed separately using the STRING database, which displayed complex associations among these DE-mRNAs, especially in the upregulated group. Subsequently, we selected the top 20 upregulated and downregulated hub genes for further expression validation and survival analyses. Convincingly, nearly all hub genes were well-validated in the GEPIA database and proved to be significantly associated with prognosis, implying that they may function as key genes in lung cancer.
Interestingly, some of the hub genes have been widely reported to be involved in cancer. For instance, overexpression of CCNB1 and CDK1 induces tumor growth and metastasis in human breast cancer50. CCNA2 modulates CDK6 and MET-mediated cell cycle pathway, and epithelial-mesenchymal transition progression is blocked by miR-381-3p in bladder cancer51.
In order to systematically explore potential ceRNAs that regulate the above-mentioned central genes, we first predicted their upstream miRNA based on an experimentally verified miRNA-target interaction database miRTarBase. According to the ceRNA hypothesis and combined with expression and survival analysis, 17 candidate miRNAs were identified as key miRNA, of which two (miR-548b and miR-let-7b) had a good prognosis, and three (miR-17, miR-137 and miR- 23a) had a poor prognosis.
Furthermore, lncRNAs regulate downstream genes by sequestering miRNA, according to the ceRNA mechanism. Therefore, we further predicted the upstream lncRNAs for the key miRNAs. Similarly, after strict expression validation and survival analysis, only four (LINC00665, LINC01140, NEAT1, and PCAT19) out of 624 lncRNAs were screened as key lncRNAs. Interestingly, some interactions found in this network were identified in previous studies. Z, C. et al. determined that LINC00665 expression is significantly upregulated in lung cancer tissues and exerts its oncogenic role by competing with miR-98, and subsequently activating downstream AKR1B10-ERK signaling pathway52. Moreover, LINC00665 is important for NSCLC to develop drug resistance, and in cells with acquired gefitinib resistance, downregulation of the LINC00665 gene reverses gefitinib sensitivity in vitro and in vivo53.
In conclusion, the ceRNA regulatory network is becoming a research focus in the field of noncoding RNA. Employing stepwise reverse prediction from mRNA to lncRNA, we successfully constructed a potential mRNA-miRNA-lncRNA regulatory network in LUAD. Each component of the ceRNA network is significantly related to the prognosis of patients with lung cancer. More importantly, LINC00665- miR-let-7b- CCNA2 was identified as a novel key ceRNA network for its possible oncogenic function in lung cancer.
Our findings may provide new insights into the pathogenesis of lung cancer. However, further experimental validation is required. In particular, future work should focus on clarifying the function of LINC00665- miR-let-7b- CCNA2 axis in LUAD with in vitro and in vivo studies. Additional experiments and large-scale clinical trials are required in the future. We have successfully constructed a silencing plasmid for LINC00665 and plan to investigate the tumor biology effect of the intervention of LINC00665 on lung cancer cells. Parallel silencing of LINC00665 in animal tumor models was performed to investigate whether tumor growth could be inhibited. Further, we clarified the direct regulation effect of LINC00665 on miR-let-7b through luciferase reporting and ChIP experiments. Finally, we used a large number of clinical samples to verify that the LINC00665-miR-let-7b-CCNA2 axis plays an important regulatory role in LUAD patients. We believe that identifying ceRNA networks associated with metastasis or staging of LUAD will be relevant for clinical research and is worthy of further investigation and development.
Methods
Datasets collection
We searched for the datasets of lung cancer tissues from the GEO database (http://www.ncbi.nlm.nih.gov/geo/). The clinical characteristics of selected datasets were extracted for further analysis. Finally, three datasets (GSE18842, GSE19188, and GSE33532), were selected for subsequent analyses. To increase the reliability of the differential mRNA screening results, we added the TCGA datasets to the discovery set.
Differential expression analysis
Three matrix and associated platform annotation files of the aforementioned three GEO datasets were first downloaded from the GEO database. The software package “limma” was used in the R software to identify DEGs from three data sets, taking |log2FC|> 1 and an adjusted p-value < 0.05 as the cut-off criterion for differential expression analysis. In addition, VENNY 2.1.0 (http://bioinfogp.cnb) was used to draw the Venn diagram. Common DEGs in the GSE18842, GSE19188, and GSE33532 data sets were redefined as significant DEGs, including significantly up- and downregulated DEGs.
Functional enrichment analysis
These DE-mRNAs were introduced for GO function annotation and the KEGG pathway enrichment analysis to explore the related functions. The website, DAVID, was used for enriched GO terms and KEGG pathways enrichment analysis. A p-value < 0.05 was considered statistically significant. The R software ggplot2 software package was used to visualize the first 15 enriched GO terms and KEGG pathways.
Construction of PPI network
Protein–protein interactions were constructed separately for DE-mRNAs using the STRING (Search Tool for Retrieval of Interacting Genes/Proteins) database. PPIs with a combined confidence score > 0.4 were used to construct the PPI network. The images were displayed on a high-resolution monitor during the experiment and were downloaded from the webpage.
Prediction of miRNA
We used the miRTarbase database to predict upstream miRNAs of key genes, and the database was verified experimentally. We also used the Kaplan–Meier plotter database to further evaluate the prognostic value of these predicted miRNAs.
Prediction of lncRNA
The upstream potential lncRNAs were predicted using the miRNet database, which provides an integrated tool to comprehensively analyze the interaction between miRNA and its targeted lncRNA. Additionally, we evaluated the prognostic value of these predicted lncRNAs using the Kaplan–Meier plotter database.
Correlation analysis
Correlation analysis of mRNA/miRNA/lncRNA pairs in lung cancer was performed using the starBase database. Statistical significance was set at p-value < 0.05.
Data availability
The data used to support the findings of this study are available from the corresponding author upon reasonable request.
Abbreviations
- ceRNA:
-
Competing endogenous RNA
- lncRNAs:
-
Long noncoding RNAs
- GEO:
-
Gene Expression Omnibus
- PPI:
-
Protein–protein interaction
- LUAD:
-
Lung adenocarcinoma
References
Ferlay, J. et al. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 136, E359–E386 (2015).
Zhao, K., Li, Z. & Tian, H. Twenty-gene-based prognostic model predicts lung adenocarcinoma survival. OncoTargets Ther. 11, 3415 (2018).
Davidson, M. R., Gazdar, A. F. & Clarke, B. E. The pivotal role of pathology in the management of lung cancer. J. Thoracic Dis. 5, S463 (2013).
Verdecchia, A. et al. Recent cancer survival in Europe: a 2000–02 period analysis of EUROCARE-4 data. Lancet Oncol. 8, 784–796 (2007).
Chheang, S. & Brown, K. in Seminars in interventional radiology. 099–113 (Thieme Medical Publishers).
Hangauer, M. J., Vaughn, I. W. & McManus, M. T. Pervasive transcription of the human genome produces thousands of previously unidentified long intergenic noncoding RNAs. PLoS Genet. 9, e1003569 (2013).
Berindan-Neagoe, I., Braicu, C., Gulei, D., Tomuleasa, C. & Calin, G. A. Noncoding RNAs in lung cancer angiogenesis. In Physiologic and pathologic angiogenesis-signaling mechanisms and targeted therapy (eds Simionescu, D. & Simionescu, A.) (InTech, Rijeka, 2017).
Salmena, L., Poliseno, L., Tay, Y., Kats, L. & Pandolfi, P. P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language?. Cell 146, 353–358 (2011).
Fan, C.-N., Ma, L. & Liu, N. Systematic analysis of lncRNA–miRNA–mRNA competing endogenous RNA network identifies four-lncRNA signature as a prognostic biomarker for breast cancer. J. Transl. Med. 16, 264 (2018).
Wang, P. et al. A novel LncRNA–miRNA–mRNA triple network identifies LncRNA RP11-363E7. 4 as an important regulator of miRNA and gene expression in gastric cancer. Cell. Physiol. Biochem. 47, 1025–1041 (2018).
Yan, H. et al. Identification of potential transcription factors, long noncoding RNAs, and microRNAs associated with hepatocellular carcinoma. J. Cancer Res. Ther. 14, 622 (2018).
Sun, Y. et al. LncRNA H19/miRer research and therapeuticsfactors, long noncoding Rmigration of pancreatic cancer. J. Cell. Biochem. 120, 3874–3886 (2019).
Wang, Y. et al. Microarray expression profile analysis of long non-coding RNAs in human gastric cardiac adenocarcinoma. Cell. Physiol. Biochem. 33, 1225–1238 (2014).
Chen, J., Lin, C., Yong, W., Ye, Y. & Huang, Z. Calycosin and genistein induce apoptosis by inactivation of HOTAIR/p-Akt signaling pathway in human breast cancer MCF-7 cells. Cell. Physiol. Biochem. 35, 722–728 (2015).
Li, C. et al. Progress and prospects of long noncoding RNAs (lncRNAs) in hepatocellular carcinoma. Cell. Physiol. Biochem. 36, 423–434 (2015).
Wang, K. C. & Chang, H. Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914 (2011).
Wu, T. et al. Roles of noncoding RNAs in metastasis of nonsmall cell lung cancer: a mini review. J. Cancer Res. Ther. 11, 7 (2015).
Loewen, G., Jayawickramarajah, J., Zhuo, Y. & Shan, B. Functions of lncRNA HOTAIR in lung cancer. J. Hematol. Oncol. 7, 90 (2014).
Zhang, J., Zhang, B., Wang, T. & Wang, H. LncRNA MALAT1 overexpression is an unfavorable prognostic factor in human cancer: evidence from a meta-analysis. Int. J. Clin. Exp. Med. 8, 5499 (2015).
Nie, F.-Q. et al. Long noncoding RNA ANRIL promotes non-small cell lung cancer cell proliferation and inhibits apoptosis by silencing KLF2 and P21 expression. Mol. Cancer Ther. 14, 268–277 (2015).
Yang, Z.-T. et al. Overexpression of long non-coding RNA ZXF2 promotes lung adenocarcinoma progression through c-Myc pathway. Cell. Physiol. Biochem. 35, 2360–2370 (2015).
Sang, H., Liu, H., Xiong, P. & Zhu, M. Long non-coding RNA functions in lung cancer. Tumor Biol. 36, 4027–4037 (2015).
Thomson, D. W. & Dinger, M. E. Endogenous microRNA sponges: evidence and controversy. Nat. Rev. Genet. 17, 272 (2016).
Balkwill, F. Cancer and the chemokine network. Nat. Rev. Cancer 4, 540 (2004).
Gay, L. J. & Felding-Habermann, B. Contribution of platelets to tumour metastasis. Nat. Rev. Cancer 11, 123 (2011).
Miao, Z. et al. CXCR7 (RDC1) promotes breast and lung tumor growth in vivo and is expressed on tumor-associated vasculature. Proc. Natl. Acad. Sci. 104, 15735–15740 (2007).
Zhang, H. et al. Long non-coding RNA: a new player in cancer. J. Hematol. Oncol. 6, 37 (2013).
Qiu, M.-T., Hu, J.-W., Yin, R. & Xu, L. Long noncoding RNA: an emerging paradigm of cancer research. Tumor Biol. 34, 613–620 (2013).
Esteller, M. Non-coding RNAs in human disease. Nat. Rev. Genet. 12, 861 (2011).
Chen, X., Xie, D., Zhao, Q. & You, Z.-H. MicroRNAs and complex diseases: from experimental results to computational models. Brief. Bioinform. 20, 515–539 (2017).
Wang, G., Li, X. & Wang, Z. APD3: the antimicrobial peptide database as a tool for research and education. Nucleic Acids Res. 44, D1087–D1093 (2015).
Luo, J. et al. Long non-coding RNA CARLo-5 is a negative prognostic factor and exhibits tumor pro-oncogenic activity in non-small cell lung cancer. Tumor Biol. 35, 11541–11549 (2014).
Liu, H., Deng, H., Zhao, Y., Li, C. & Liang, Y. LncRNA XIST/miR-34a axis modulates the cell proliferation and tumor growth of thyroid cancer through MET-PI3K-AKT signaling. J. Exp. Clin. Cancer Res. 37, 279 (2018).
Luan, X. & Wang, Y. LncRNA XLOC_006390 facilitates cervical cancer tumorigenesis and metastasis as a ceRNA against miR-331-3p and miR-338-3p. J. Gynecol. Oncol. https://doi.org/10.3802/jgo.2018.29.e95 (2018).
Gao, H. et al. LncRNA ZEB2-AS1 promotes pancreatic cancer cell growth and invasion through regulating the miR-204/HMGB1 axis. Int. J. Biol. Macromol. 116, 545–551 (2018).
Liu, X.-H. et al. The long non-coding RNA HOTAIR indicates a poor prognosis and promotes metastasis in non-small cell lung cancer. BMC Cancer 13, 464 (2013).
Jen, J. et al. Oct4 transcriptionally regulates the expression of long non-coding RNAs NEAT1 and MALAT1 to promote lung cancer progression. Mol. Cancer 16, 104 (2017).
White, N. M. et al. Transcriptome sequencing reveals altered long intergenic non-coding RNAs in lung cancer. Genome Biol. 15, 429 (2014).
Nie, F.-Q. et al. Long non-coding RNA MVIH indicates a poor prognosis for non-small cell lung cancer and promotes cell proliferation and invasion. Tumor Biol. 35, 7587–7594 (2014).
Yi, X., Du, Z. & Su, Z. PlantGSEA: a gene set enrichment analysis toolkit for plant community. Nucleic Acids Res. 41, W98–W103 (2013).
López-Lázaro, M. Stem cell division theory of cancer. Cell Cycle 14, 2547 (2015).
Wu, H. et al. Arginase-1-dependent promotion of TH17 differentiation and disease progression by MDSCs in systemic lupus erythematosus. Sci. Transl. Med. 8, 331ra340-331ra340 (2016).
Zantek, N. D. et al. MCF-10A-NeoST: a new cell system for studying cell-ECM and cell-cell interactions in breast cancer. Clin. Cancer Res. 7, 3640–3648 (2001).
Kalluri, R. Angiogenesis: basement membranes: structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 3, 422 (2003).
Adams, R. H. & Alitalo, K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat. Rev. Mol. Cell Biol. 8, 464 (2007).
Sherr, C. J. & McCormick, F. The RB and p53 pathways in cancer. Cancer Cell 2, 103–112 (2002).
Nakayama, K. I. & Nakayama, K. Ubiquitin ligases: cell-cycle control and cancer. Nat. Rev. Cancer 6, 369 (2006).
Wang, Y., Shi, J., Chai, K., Ying, X. & Zhou, B. The role of snail in EMT and tumorigenesis. Curr. Cancer Drug Targets 13, 963–972 (2013).
Vander Heiden, M. G., Cantley, L. C. & Thompson, C. B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033 (2009).
Li, Y. et al. Association study of germline variants in CCNB1 and CDK1 with breast cancer susceptibility, progression, and survival among Chinese Han women. PLoS ONE 8, e84489 (2013).
Li, J. et al. Dual regulatory role of CCNA2 in modulating CDK6 and MET-mediated cell-cycle pathway and EMT progression is blocked by miR-381-3p in bladder cancer. FASEB J. 33, 1374–1388 (2018).
Cong, Z. et al. Long non-coding RNA linc00665 promotes lung adenocarcinoma progression and functions as ceRNA to regulate AKR1B10-ERK signaling by sponging miR-98. Cell Death Dis. 10, 84 (2019).
Liu, X. et al. LINC00665 induces acquired resistance to Gefitinib through recruiting EZH2 and activating PI3K/AKT pathway in NSCLC. Mol. Ther. Nucleic Acids 16, 155–161 (2019).
Funding
This research was funded by the National Natural Science Foundation of China (No. 81700512) and Natural Science Foundation of Guangdong Province of China (No. 2016A030310252).
Author information
Authors and Affiliations
Contributions
Conceptualisation, Y.H., L.Z., K.N., and Y.L.; methodology, Y.H., L.Z., and K.N.; sofware, L.Z., S.S., L.L., and K.N.; data curation, L.Z., F.L., and K.N.; writing and original draft preparation, D. C..; writing, review, and editing, L.Z., K.N., Y.H., and Y.L.; funding acquisition, Y.L.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Huang, Y., Zhong, L., Nie, K. et al. Identification of LINC00665-miR-let-7b-CCNA2 competing endogenous RNA network associated with prognosis of lung adenocarcinoma. Sci Rep 11, 4434 (2021). https://doi.org/10.1038/s41598-020-80662-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-80662-x
- Springer Nature Limited