
Abstract
Shortening the juvenile stage in citrus and inducing early flowering has been the focus of several citrus genetic improvement programs. FLOWERING LOCUS T (FT) is a small phloem-translocated protein that regulates precocious flowering. In this study, two populations of transgenic Carrizo citrange rootstocks expressing either Citrus clementina FT1 or FT3 genes under the control of the Arabidopsis thaliana phloem specific SUCROSE SYNTHASE 2 (AtSUC2) promoter were developed. The transgenic plants were morphologically similar to the non-transgenic controls (non-transgenic Carrizo citrange), however, only AtSUC2-CcFT3 was capable of inducing precocious flowers. The transgenic lines produced flowers 16 months after transformation and flower buds appeared 30–40 days on juvenile immature scions grafted onto transgenic rootstock. Gene expression analysis revealed that the expression of SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1) and APETALA1 (AP1) were enhanced in the transgenics. Transcriptome profiling of a selected transgenic line showed the induction of genes in different groups including: genes from the flowering induction pathway, APETALA2/ETHYLENE RESPONSE FACTOR (AP2/ERF) family genes, and jasmonic acid (JA) pathway genes. Altogether, our results suggested that ectopic expression of CcFT3 in phloem tissues of Carrizo citrange triggered the expression of several genes to mediate early flowering.
Similar content being viewed by others

Introduction
In the genetic improvement of citrus by conventional breeding or biotechnology, the long juvenile phase is one of the major challenges encountered towards rapid evaluation of fruit quality1. Shortening this phase has been the focus of several studies2,3,4. In citrus, the transition from juvenility to maturity can range from as little as two years for Hong Kong kumquat, to over 12 years for some pummelo cultivars2,5,6. After this phase the plant transitions from the vegetative to the reproductive phase7. The process of flower development occurs in the shoot apical meristem (SAM), which transitions from producing vegetative structures to developing floral structures8.
Flowering is governed by a complex and finely tuned regulated network of signals. In Arabidopsis, four major regulatory pathways for flowering have been discovered including: the photoperiod pathway, autonomous floral initiation, the vernalization pathway, and those regulated by gibberellins. These pathways can be negatively or positively regulated by transcription factors, repressors, endogenous signal cascades, and/or environmental signals, which converge to regulate floral meristem identity genes that promote flowering9,10,11. FLOWERING LOCUS T (FT) is a key component of these flowering pathways. FT is a small protein synthesized in phloem companion cells which is upregulated by the transcription factor CONSTANS (CO). The FT protein translocates through the phloem to the SAM to induce floral primordia formation via the activation of meristem identity genes such as APETALA1 (AP1)12,13 and LEAFY (LFY)14,15, converting the vegetative SAM into inflorescence SAM which later on starts producing flowers10,16,17 (Fig. 1).

Schematic view of the flowering pathway in tropical and sub-tropical trees (adapted from Wilkie et al., 2008). Environmental stimuli promote the stabilization of the CO transcription factor, which upregulates FT protein. The FT protein is synthesized mainly in leaf tissue and transported by the phloem to the shoot apical meristem (SAM). In the SAM, FT induces flowering and TFL1 represses genes activated by FT. Downstream of FT, SOC1 up-regulates floral meristem identity genes AP1 and LFY. FT may also act independently of SOC1 by inducing AP1 and LFY gene expression. Up-regulation is represented by arrows and down-regulation or inhibition is represented by ‘an inverted T’.
Several studies have suggested that orthologs of the A. thaliana FT (AtFT) protein are a primary component of the flowering signal cascade in different plants3,16,18,19,20,21,22,23. When the tomato FT, also known as SFT, were overexpressed in either tomato or tobacco, an early flowering phenotype was observed. Subsequently, grafting experiments in tomatoes demonstrated that the signal was transmissible from transgenic plants expressing SFT under 35S promoter to non-transgenic sft mutant plants24.
In citrus, at least three loci encoding FT homolog proteins CiFT1, CiFT2 and CiFT3 have been identified3,25, however, two of them (CiFT1 and CiFT2) appear to be encoded by the same gene26. A shortened juvenile stage was observed when one of the homologs was ectopically expressed under the control of the Cauliflower mosaic virus (CaMV 35S) promoter in trifoliate orange (Poncirus trifoliata L. Raf.)3. In addition to the induction of early flowering phenotype, the overexpression of CiFT also induced abnormal morphological changes in the transgenic plants. Plants were dwarfed and their thorns replaced by flowers. This indicated that there were deleterious effects resulting from expression of FT gene under a strong constitutive promoter3. Similarly, the induction of flowering within four to six months was mediated by infection of citrus plants with a citrus leaf blotch virus-based vector expressing the FT protein (CiFT and AtFT)4. The virus infected plants showed no modification in plant, leaf, flower, or fruit morphology, suggesting that CiFT likely encodes a mobile floral signal and its expression does not lead to drastic phenotypic changes4.
In this study, we evaluated the early flowering phenotype using a FT citrus homolog cloned from Citrus clementina (CcFT3). A transgenic population of Carrizo citrange rootstocks (Citrus sinensis Osb. × Poncirus trifoliata L. Raf.) transformed with the AtSUC2-CcFT3 construct was generated, which showed normal morphological characteristics and exhibited normal vigor. Flowering occurred 16 months after transformation and clonally propagated transgenic rootstocks were able to induce precocious flowering in budded juvenile scions, demonstrating that the gene can induce earlier flowering in citrus.
Results
The AtSUC2 promoter efficiently drives expression of the CcFT3 transgene to induce early flowering in juvenile citrus
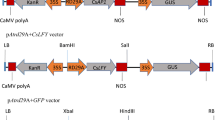
Three FT homologs have been identified in the satsuma mandarin (Citrus unshiu). Among the three, the FT1 and FT2 are considered alleles at the same locus26,27, and therefore, only FT1 and FT3, was used in this study (Supplementary Fig. S1). In this study, we identified FT1 and FT3 from the clementine mandarin. Then, a phylogenetic tree was constructed based in the predicted CcFT protein sequences and compared them with several other FT sequences identified previously from the satsuma mandarin, other fruit crops and Arabidopsis. The phylogenetic tree was produced by the maximum-likelihood method. When compared to other methods such as the maximum-parsimony or the neighbor-joining, results were almost identical (data not presented). Both the CcFT1 and CcFT3 sequences were homologous to the CiFT1 and CiFT3, were closely located and in the same clade, along with the CiFT3 (Fig. 2). The CcFT1 and CcFT3 cDNA sequences were cloned into binary vectors for Agrobacterium-mediated transformation of Carrizo citrange. We generated different populations of transgenic citrus plants for evaluation. Several promoters were examined to efficiently express the CcFT transgene. These included two constitutively expressed promoters (the strong 35S promoter derived from Cauliflower Mosaic Virus (CaMV) and a weaker NOPALINE SYNTHASE (NOS) promoter), the A. thaliana SUCROSE SYNTHASE 2 (AtSUC2) that localizes to vascular tissue28, and the A. thaliana HEAT SHOCK PROTEIN 18.2 (AtHSP18.2) heat inducible promoter29,30,31,32 (Supplementary Fig. S2). Non-transformed Carrizo citrange rootstocks that originate from control in vitro plates were used as non-transgenic controls.

Phylogenetic analysis of FT protein sequences of multiple plant species. Sequences evaluated include: Arabidopsis thaliana FT (AAF03936), Citrus clementina FT1 (MT707614), Citrus clementina FT3 (MT602515), Citrus unshiu FT1 (BAA77836), Citrus unshiu FT3 (BAF96645), Fragaria vesca FT (CBY25183), Malus domestica FT1 (BAD08340), Malus domestica FT2 (ADP69290), Prunus mume FT (CBY25181), Prunus persica FT (AEO72030), Pyrus communis FT (AJC01933) and Vitis vinifera FT (ABF56526).
None of the transgenic CcFT1 overexpressing lines flowered even after three years of transformation, heat treatment (to induce the AtHSP18.2 promoter) and plant regeneration (Table 1). In contrast, CcFT3 overexpression induced precocious flowering in many transgenic lines (Fig. 3A). Therefore, we focused on the evaluation of a CcFT3 transgenic line.

AtSUC2-CcFT3 transgenic rootstock flowering phenotype. (A) Precocious flowering from a 16-month old transgenic AtSUC2-CcFT3 plant. (B) Closeup of fruit set following self-pollination of the flowers. (C) A view of the whole plant. (D) Precocious flowering in a self-pollinated AtSUC2-CcFT3 F1 seedling within a year of seed germination. (E) Precocious in vitro flowering within a month of transformation using a 35S-CcFT3 construct and (F) relative CcFT3 gene expression of transgenic lines transformed with the different constructs. The error bars represent the means of different transgenic lines. Different letters represent a significant difference at P < 0.05 using Student's t-Test.
The transcripts levels were evaluated using CcFT3-specific primers to compare the expression of transgenic plants to the control plants (Supplementary Table S1). When the in vitro derived CcFT3 transcript levels were compared, transgenic lines expressing the 35S-CcFT3 construct (Fig. 3E) had on average over a 12,000-fold higher transgene expression than the AtSUC2-CcFT3 transgenic lines (Fig. 3F). The expression of the transgene under the 35S promoter also resulted in precocious flowering in the apical meristems in vitro, however, the explant was unable to survive after soil transplantation (Fig. 3E).
One transgenic NOS-CcFT3 line flowered within 18 months after transformation but did not flower in subsequent years. Forty lines expressing CcFT3 under control of AtHSP18.2 and 21 lines expressing CcFT3 under control of AtSUC2 promoter were regenerated and later transplanted to soil (Table 1). None of the AtHSP18.2-CcFT3 plants flowered during the time span of this study.
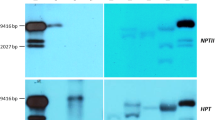
Interestingly, seven out of 21 independent transgenic lines expressing CcFT3 gene under the control of the AtSUC2 promotor flowered within 16 months after transformation (Fig. 3A–C). Transgenic nucellar seedlings from fruit obtained from the AtSUC2- CcFT3 lines flowered within a year after germination (Fig. 3D). The population was screened using FT and nptII specific primers and both FT and nptII gene fragments were successfully amplified from each of the transgenic lines (Supplementary Fig. S3). To distinguish between the native citrus FT3 and our introduced construct, primers were designed to span a 700 bp fragment of the AtSUC2 promotor and the FT3 gene (Supplementary Table S1). The morphological characteristics of these AtSUC2-CcFT3 transgenic trees were similar to the non-transgenic control. However, thorns in the transgenic lines were visibly smaller and were comparable to those usually seen in mature trees. There were no morphological abnormalities observed in the flowers in any of the transgenic lines (Fig. 3A) and a normal surface morphology of the pollen grains under SEM was observed (Fig. 4A,B). The number of viable pollen grains were similar between the transgenic line and the non-transgenic control (Fig. 4C–F). Controlled pollination between our transgenic lines and the monoembryonic citrus ‘Temple’ resulted in the production of viable hybrid seedlings with a trifoliate leaf phenotype (results not shown).

Pollen grains of a transgenic line showing normal structure and high viability. (A,B) SEM images of pollen grains from the AtSUC2-CcFT3 MP3 transgenic line indicate normal morphology. (B) Brown–red pollen grains stained with 50% Gram’s iodine indicate that the pollen grains are viable compared to the (C) deionized water control. The 1% TTC stain showed that the relative number of viable pollen grains between the (E) MP3 transgenic line and the (F) non-transgenic line was the same, although differential staining was observed. Scale bars represent 10 µm.
Among the seven PCR positive plants (Supplementary Fig. S3), four lines were randomly selected for further examination. The copy number of transgenes integrated in the plant genome of the transgenic lines was first evaluated by Southern blot hybridization. In all the transgenic plants, the presence of positive insertion of CcFT3 transgene was observed compared to its absence in the control plant. Each of the transgenic lines contained two copies of the CcFT3 transgene (Fig. 5A).

Molecular analysis of Citrus transgenic lines expressing CcFT3 under control of the AtSUC2 promoter. (A) Southern blot of selected CcFT3 lines indicating the transgene copy number. The lanes were loaded as following: (1) WT or non-transgenic control, (2) AtSUC2-CcFT3 MP4, (3) AtSUC2-CcFT3 MP3, (4) AtSUC2-CcFT3 MP2 and (5) AtSUC2-CcFT3 MP1. (B) CcFT3 transcript quantification from the selected citrus transgenic lines. CsACTIN was used as reference gene. Data represent the mean (± standard deviation, SD) of three technical replicates and different letters represent a significant difference at P < 0.05 using Student's t-Test (C–F) Western blot showing CcFT3 protein quantification (C) in the mother plants (MP) and (D–F) two individual self-pollinated seedling progenies (SDL) from each MP line. The membranes were probed with CcFT3 specific antibody and Ponceau-S was used as loading control.
Transgenic lines were subsequently evaluated for CcFT3 transcript levels. CcFT3 could not be detected in the juvenile non-transgenic Carrizo citrange plants but was highly upregulated in the selected transgenic lines (Fig. 5B). In addition, the protein levels were correlated with PCR results (Fig. 5C). All the selected transgenic lines showed a high expression of the CcFT3 transgene. The transgene stability was subsequently evaluated in their self-pollinated nucellar seedlings. Stable expression of the transgene in the nucellar seedlings was also detected in all the four lines (Fig. 5D). We also tested the ability of the CcFT3 transgene to induce early flowering in a heterologous species. When the CcFT3 transgene was inserted into Arabidopsis plants, either driven by the 35S or the AtSUC2 promotor, early flowering was observed (Supplementary Fig. S4).
Early flowering transgenic lines upregulate the floral transition process
The effect of the transgene mediating an early flower phenotype in the AtSUC2-CcFT3 transgenic lines by effecting gene expression in the flowering induction pathway was investigated. RT-qPCR was performed using RNA extracted from phloem-rich petioles. We did not observe a difference in the expression of CONSTANS (CO) between transgenic and non-transgenic plants (Fig. 6A). In contrast, SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1) was induced and TERMINAL FLOWER1 (TFL1) was repressed (Fig. 6B,C). The expression of the meristem-identity genes, APETALA1 (AP1) and LEAFY (LFY) were examined. AP1 was highly expressed in the transgenic lines, while LFY was repressed compared to non-transgenic plants (Fig. 6D,E). Similarly, genetic expression analysis from whole leaf showed the induction of SOC1 and AP1 but a reduced expression of LFY (Fig. 6F–H).

Evaluation of flowering pathway related genes. (A–E) Relative expression of flowering related genes for each of the 16-month old transgenic lines compared to the non-transgenic Carrizo citrange shown by RT-qPCR (A) CONSTANS (CO), (B) SUPPRESSOR OF OVEREXPRESSION OF CONSTANS 1 (SOC1), (C) TERMINAL FLOWER1 (TFL1), (D) APETALA1 (AP1) and (E) LEAFY (LFY). RNA was extracted from leaf petioles rich in phloem tissues. (F–H) Total RNA was extracted from leaf tissues to show the expression flowering pathway genes by RT-qPCR (A) SOC1, (B) AP1 and (C) LFY in the AtSUC2-CcFT3 transgenic lines compared to the Carrizo citrange non-transgenic line. CsACTIN was used as the reference gene. Data represent the mean (± standard deviation, SD) of three technical replicates. Different letters represent a significant difference at P < 0.05 using Student's t-Test.
To verify the transgene localization, CcFT3-EGFP fusions were generated. The fusion gene was cloned downstream of either the 35S or the AtSUC2 promoters. Both constructs transiently expressed CcFT3-EGFP in N. benthamiana plants and exhibited cytoplasmic and nuclear localization (Fig. 7). When the fusion gene was driven by the 35S, a stronger EGFP signal was observed indicating higher protein expression when compared to the EGFP signal observed from the same fusion gene under the control of the weaker phloem specific promoter, AtSUC2.

Confocal micrographs showing transient expression of the CcFT3-EGFP fusion protein. Confocal micrographs of GFP, transmission white light (Bright field) and an overlay. Cytoplasmic and nuclear subcellular localization of CcFT3 fused to EGFP under expression of both 35S and AtSUC2 promoters was observed. EGFP was used as positive control.
Transgenic rootstocks induced earlier flowering in non-transgenic juvenile scions
To understand the ability of the transgenic rootstocks to efficiently induce flowering in non-transgenic scions, the MP3 transgenic line was propagated using two node cuttings. Most of the cuttings were capable of rooting, producing morphologically normal plants. Flower bud initiation was observed within 21 days after budding when buds from a 1-year old juvenile ‘Valencia’ seedling were grafted onto pencil-diameter transgenic Carrizo rootstock lines (Fig. 8A). The flowers developed normally (Fig. 8B) and fully open flowers were observed within 10 days from bud initiation (Fig. 8C). When allowed to self-pollinate, fruit formation was observed (Fig. 8D). FT transcript levels increased progressively in the developing scion and was two-fold higher compared to shoots emerging from the non-transgenic rootstocks at the fully open flower stage (Fig. 8E).

Precocious flowering one-year old non-transgenic ‘Valencia’ scion grafted onto AtSUC2-CcFT3 transgenic rootstock (A) Flower bud emergence within 21 days following budding. Insert shows enlarged image of emerging flower buds (B) fully expanded flower buds, (C) fully open flowers, (D) developing sweet orange fruit and (E) relative FT gene expression in the scion leaves at different stages of flowering. Data represent the mean (± standard deviation, SD) of three technical replicates. Different letters represent a significant difference at P < 0.05 using Student's t-Test.
Expression of CcFT3 gene in the phloem tissues alters the plant’s transcriptome
To reveal the differences in transcript profiles between the transgenic and non-transgenic plants, a transcriptome analysis was performed. The total RNA of three technical replicates of leaf samples from non-transgenic and the AtSUC2-CcFT3 MP3 transgenic plants was subject to next-generation sequencing (RNAseq). The Illumina NovaSeq platform produced on average 22,070,506 and 22,492,946 raw read counts for the Carrizo citrange non-transgenic plants and AtSUC2-CcFT3 (FT3) transgenic lines, respectively (Supplementary Table S2).
After cleaning, an average of 20,980,971 (95.06%) and 21,465,005 (95.43%) read counts remained. The clean reads were mapped to the C. sinensis genome utilizing STAR (version v2.6.0C, https://github.com/alexdobin/STAR) and the C. sinensis 154 v1.1 annotation and genome from Phytozome (version v12.1, https://phytozome.jgi.doe.gov/pz/portal.html). On average there were 17,272,422 and 17,478,222 unique alignments per sample, which accounted for a percent of 82.32% and 81.43%, of the genome respectively (Supplementary Table S2). A principal component analysis (PCA) in two dimensions was performed to demonstrate the variance between the samples of the transgenic and non-transgenic groups (Supplementary Fig. S5). A total of 1492 differentially expressed genes (DEGs) remained from the filtering of DESeq2 software analysis with a |log2FoldChange|≥ 1 and an adjusted P-value ≤ 0.05, of which 938 were upregulated and 554 were downregulated in the MP3 transgenic line when compared to the non-transgenic line (Fig. 9A).

A volcano plot of DEGs and validation of the transcriptome analysis of selected DEGs using RT-qPCR. (A) Volcano plot showing the Log2 fold change difference and the adjusted P-value for 21,635 genes included on all platforms. The red dots represent upregulated genes and the blue dots the downregulated genes in AtSUC2-CcFT3 MP3 transgenic lines (adjusted P < 0.05). (B) The expression levels of DEG candidates in AtSUC2-CcFT3 MP3 transgenic line was calculated using 2-ΔΔCt and those values were compared to non-transgenic control values. Different letters (a, b) represent a significant difference at P ≤ 0.05 using Duncan’s Multiple Range Test and error bars represent SE (n = 3) for RT-qPCR (black) and RNAseq (blue).
To evaluate the accuracy of the RNAseq data and the presence of technical artifacts or errors introduced during the RNAseq library preparations, the expression of several well conserved genes and transcription factors were assayed through RT-qPCR. Six of the selected genes, including orange1.1g042438m (CsAGL14), orange1.1g035470m (CsAGL8), orange1.1g042642m (CsAP1), orange1.1g023148m (CsERF38), orange1.1g037149m (CsAGL6) and orange1.1g009794m (CsWRKY61), were highly upregulated, while orange1.1g041209m (CsHSP70), orange1.1g032690m (CsWRKY23), orange1.1g040046m (CsAP3), orange1.1g019949m (CsGA2OX8), orange1.1g036452m (CsAGL11) and orange1.1g026276m (CsAP2) were downregulated. The RT-qPCR results were consistent with the RNAseq data and the mRNA expression of these genes were significantly either up- or down-regulated by the transgenic expression of CcFT3 (Fig. 9B).
Among the 1492 DEGs, 50 genes which are relevant to flowering time and were either up- or down-regulated were selected to be included in a heatmap. These DEGs were further classified into different groups including flowering induction pathway genes, the APETALA2/ETHYLENE RESPONSE FACTOR (AP2/ERF) family of gene and the jasmonic acid (JA) pathway genes (Fig. 10A and Table 2).

Heat map of RNAseq transcriptome analysis of 50 selected genes clustered by expression patterns and Gene Ontology (GO) enrichment analysis. (A) Heatmap displaying changes in gene expression between the MP3 transgenic line and non-transgenic plants. The DEGs were classified in flowering genes, APETALA2/ETHYLENE RESPONSE FACTOR (AP2/ERF) family, the jasmonic acid (JA) pathway. A color range of dark to light red represents upregulated genes in the transgenic lines with the most upregulated genes being dark red. A color range of dark to light blue represents the downregulated genes in the transgenic line with dark blue being the most downregulated genes (B) Bar graph showing the enrichment of gene ontology terms in differentially expressed genes in the MP3 transgenic line. In the x-axis the enrichment values are represented and GO terms are represented in the y-axis. The blue bars represent biological processes, green bars represent cellular components, and the red bars represent molecular function.
There were 23 genes identified and included in the flowering genes group, as they belong to an integrated network of several genetic pathways converging on flowering. An ortholog of the Arabidopsis AGL14, orange1.1g042438m and two AGL6 like genes, annotated in the sweet orange genome database as orange1.1g037149m and orange1.1g046479m, were upregulated in our MP3 transgenic line. Further, two sweet orange genes (orange1.1g042642m and orange1.1g027470m) identified as the APETALA gene AP1 and AP3, were upregulated. In contrast, a second AP3 orthologue, orange1.1g040046m, was down regulated (Fig. 10A).
The APETALA2/ETHYLENE RESPONSE FACTOR (AP2/ERF) gene family is a group of plant-specific TFs that contain at least one AP2 binding domain. Two sweet orange genes (orange1.1g023148m and orange1.1g029068m) were identified as orthologs of the EARLIER DEHYDRATION RESPONSIVE ELEMENT BINDING PROTEINS (DREB) and two sweet orange orthologs (orange1.1g042174m and orange1.1g044938m) were identified as orthologs of the ETHYLENE RESPONSE FACTOR (ERF) (Fig. 10A).
There were three orange orthologs of the JA pathway genes. Two of them were identified as JASMONATE-ZIM DOMAIN1 (JAZ1), orange1.1g026462m and orange1.1g028982m, and one of them as JAZ10, orange1.1g030695m. Finally, other upregulated genes in the MP3 transgenic associated with the induction of early flowering were orange orthologs of WRKY72 (orange1.1g009794m), TRANSLATION INITIATION FACTOR 4A (EIF4A) (orange1.1g015093m), SYNAPTOSOMAL-ASSOCIATED PROTEIN 25 (SNAP25) (orange1.1g023155m), and SPERMIDINE SYNTHASE (orange1.1g043095m) (Fig. 10A)33,34,35,36.
Using C. sinensis genome information, gene ontology (GO) enrichment analysis was performed to extract biological meaning of the DEGs obtained from the transcriptome. The C. sinensis genome annotation is composed of 25,380 genes from which 13,309 (52.44%) genes have GO terms. In addition, the total of filtered DEGs found in our RNAseq analysis were 1492 from which 847 (56.77%) have GO terms. In our analysis, the most statistically significant grouped GO terms of up-regulated or down-regulated genes in AtSUC2-CcFT3 transgenic lines were represented in the three main GO categories: ‘biological process’, ‘molecular function’ and ‘cell component’. GO terms with corrected P-values < 0.05 were considered significantly enriched.
The highest 67 GO categories were identified as 10 different biological processes, 7 cellular components and 50 molecular functions. Amongst the 67 GO categories, biological process ‘Terpene Synthase’ was the most enriched. Of the 10 cellular components ‘Tubulin’ and ‘Microtubule Based Process’ were the most enriched. In the following 50 molecular functions, the GO categories most enriched were ‘External Encapsulating Structure’, ‘Carbon–Oxygen Lyase’, ‘Cytoskeletal Protein binding’, ‘Cell wall’ and ‘Enzyme inhibitor’ (Fig. 10B).
To position genes that had the most significant change in expression pattern in response to the transgenic expression of CcFT3 in the flowering induction pathway, a custom MapMan mapping file was created using an Arabidopsis outlined pathway adapted from Kim37 and genes from Phytozome annotation database to compare the DEGs of non-transgenic and transgenic plants. Interestingly, the changes in expression pattern were mostly observed in pathway genes downstream of FT including FT, FD, SOC1, FUL (or AGL8) and AP1. There was a difference of expression in a few genes upstream of the florigen signal including: CO, SVP and ELF4. CO was upregulated in the transgenic lines compared to the non-transgenic line; however, this change was near a one-fold difference. SVP was downregulated in AtSUC2-CcFT3, which is not surprising as SVP is known to play a role in the downregulation of FT38. The repression of SVP may lead to the induction of the flowering pathway.
Discussion
In this study, the expression of the CcFT3 transgene was evaluated to induce early flowering in immature Carrizo citrange rootstock. Stable and uniform precocious flowering that reduces the long juvenile phase will greatly benefit citrus genetic improvement programs. This will decrease the time required to evaluate the fruit quality of potential candidates from a T1, somaclone, or transgenic population. The accumulation of the FT protein is a critical step necessary for the activation of a signaling cascade that promotes flowering in plants. Transgenic plants expressing CcFT3 driven by a constitutive 35S promoter primarily flowered in vitro and did not survive beyond that stage. Genetic expression analysis revealed a several thousand-fold higher expression of CcFT3 under the 35S promoter compared to the other promoter driven lines. While a high level of gene expression is desirable in some cases, this is not an efficient strategy to be used in regulating the flowering genes. The 35S promoter has also showed detrimental effect when used to drive FT expression in trifoliate orange (Poncirus trifoliata L. Raf.) causing an abnormality in the flower phenotype3. In our studies, the few 35S overexpressing lines that were successfully recovered never flowered and were discarded. Since detailed gene expression analysis was not performed on these lines, we speculate that these lines did not have the expression levels necessary to induce flowering or expressed the transgene in non-essential tissues such as the root or random gene silencing39. Heat induction can lead to the efficient expression of transgenes40, however FT could not be induced using this method. Similarly, the NOS promoter which is capable of functioning in the meristematic tissues41 did not induce FT in our studies, apart from the one line that flowered once.
Targeted expression of the Arabidopsis SUC2 promoter in the phloem as described in this study or using a virus-based system for the delivery of the gene efficiently induced early flowering with no alteration of the plant architecture, leaf, flower or fruit morphology4. The Arabidopsis SUC2 promoter has proven to be an excellent phloem specific promoter that has also resulted in the development of phloem specific disease resistant citrus trees28,42. The export of the FT protein from phloem companion cells can trigger flowering, and the Arabidopsis SUC2 promoter can efficiently target transgene expression into those cells30,43. Thus, targeted expression of the FT protein in these cells was sufficient for triggering the early flowering response in our transgenic citrus lines.
The accumulation of the CcFT3 in our transgenic lines resulted in the expression of genes involved in the flowering induction pathway similar to that reported following the endogenous FT gene induction4,44. Among the several genes upregulated in the flowering pathway following accumulation of CcFT3 transgene, three FT endogenous genes were detected. In our transgenic lines, the CcFT3 transgene was unable to induce accumulation of the CONSTANS (CO). In Arabidopsis, CO protein activation occurs upstream of the synthesis of the FT protein45 and SOC1 is a convergence point for several genes in the flowering pathways46. Therefore, the absence of accumulation of CO and the induction of SOC1 observed in our AtSUC2-CcFT3 transgenic lines might lead to the speculation that this genes function similarly to Arabidopsis in citrus, however, more experiments are need to be performed before coming to this conclusion. The repression of the TFL1 gene was observed in the transgenic lines, suggesting a possible opposing relationship between TFL1 and FT gene expression compared to the relationship between SOC1 and FT. Our results suggest that the CcFT3 transgene may be capable of functioning like the endogenous FT gene in citrus.
Sequence analysis of orange1.1g036943m revealed this gene to be a HEADING DATE 3A (Hd3a) gene, an ortholog of FT. In addition, a strong homology between our transgene, orange1.1g036943m and orange1.1g041559m with 90.9% and 87.2% similarity, respectively suggesting that either of these genes can be candidates to CcFT3 homolog. Orange1.1g036943m likely functions similar to AtSUC2-CcFT3 transgene and its induction likely results in an early-flowering phenotype47. Several MADS-box genes, which also participate in the flowering process48, were highly upregulated in our transgenic MP3 line. In Arabidopsis, AGL14 is preferentially expressed in roots49. However, in this study, leaf tissues extractions revealed that the C. sinensis AGL14 homolog orange1.1g042438m was expressed in CcFT3 transgenic line leaves, similar to the findings of Perez-Ruiz et al.50 who reported that the overexpression of AGL14 promoted early flowering in Arabidopsis transgenic lines (35S-AGL14). This demonstrates the ability of the AGL14 to function beyond the root system50. AGL6 is another MADS box gene highly induced in our AtSUC2-CcFT3 line. Since each MADS-box protein has a specific protein–protein interaction pattern, similarities between AGL6 and AP1 suggest that these genes may function as activators for FT and SOC151.
Enhanced expression of the AP1 and LFY meristem-identity genes is the final step in the flowering pathway, resulting in floral induction37. AP1 was highly induced in our transgenic plants, while LFY was weakly expressed. Thus, prolonged AP1 upregulation was sufficient to bypass LFY requirements and alter the competence of the plant to flower. AP1 accumulation in the meristem is necessary for flowering induction52 confirmed by earlier results, which indicated that FT can act as a redundancy gene of LFY to activate AP153. A recent transcriptomic study failed to detect an upregulation of LFY in an early flowering coconut genotype54. However, it is possible that the signaling cascade may differ from the classical Arabidopsis flowering induction. An increase in AP1 expression has been shown to induce early flowering phenotypes in several plant species. Precocious flowering was observed in plants with abnormal overexpression of AP1 including: Arabidopsis, citrus, pear, and roses, among others38,55,56,57,58,59,60.
APETALA3 along with its interacting partner PISTILLATA (PI) form a AP3/PI heterodimer complex which determines petal and stamen morphology and function in Arabidopsis61. While AP1 both directly and indirectly controls the early expression of AP3 and PI, the AP3/PI heterodimer directly acts in concert with other factors to restrict the expression of AP1 during early stages of floral development, thereby acting as a transcriptional/translational feedback loop (TTFL) in the apical meristem or in floral tissues62,63,64.
Genes from AP2/ERF family were also upregulated in our transgenic line. Since timing of flowering is generally induced upon environmental and stress cues, such as cold, drought, salinity, and pathogen infection65,66,67, it is likely that the induction of AP2/ERF is involved in the activation of downstream signals to maximize reproductive success.
Although JA delays flowering through repression of FT expression68, some of the important components of JA signaling pathway (JAZ1 and JAZ10) were also upregulated in our RNAseq data. JAZ1 is known to interact with TARGET OF EAT 1&2 (TOE1, TOE2). These proteins induce late flowering phenotypes upon the transcriptional repression of FT. When the JAZ1 protein forms a complex with TOE1 and TOE2, these proteins become unavailable to repress FT relieving the repression effect of TOE1 on FT transcription68. This indicates that JAZ1 may act as an allosteric inhibitor of TOE1. Similarly, JAZ10 plays an important role in the flowering regulatory process, but there may be potential redundancies between this gene and others in the JA-signaling cascade69.
Since RNAseq data reveals a broad picture of expression results, RT-qPCR indicating a specific fold change upon the expression of the transgene was performed to increase data reliability. In this experiment, the same RNA samples that was sequenced were also used to make cDNA for use in RT-qPCR. The upregulated citrus genes evaluated in this analysis were mapped to their Arabidopsis ortholog AGL14, AGL8, AP1, ERF38, AGL8 or WRKY72 using the phytozome and Arabidopsis databases. At least four of these genes, AGL14, AGL8, AP1 and AGL6 are directly associated with the flowering induction pathway70 and their overexpression in Arabidopsis results in an early flowering phenotype50. In addition, the overexpression of WRKY72 in rice has also led to induction of precocious flowering71. ERF38 has a more indirect effect in flowering phenotype since it is associated with abiotic stress that eventually result in early flowering72. The downregulated genes assayed in this study were mapped as AP2, AP3, AGL11, GA2OX8, HSP70, and WRKY23. WRKY23, AP2 and AP3 have been associated with late flowering phenotypes73. The HSP family of proteins are widely known to play role in abiotic stresses and some of the HSP have a potential to induce the late flowering phenotype in higher plants74. The gibberellin (GA) pathway is one of the main pathways in Arabidopsis involved in flowering induction. In Arabidopsis, GAs could promote flowering by activating the expression of LFY75. Accordingly, the downregulation of GA2OX8 in our CcFT3 transgenic lines could act like the AtGA2OX8 of Arabidopsis that 2β-hydroxylates the C20-GA precursors resulting in a decrease of active GAs levels76, thereby inducing a GA-deficient phenotype such as delayed flowering in different species.
FT induction was stable in our transgenic lines and could be transmitted to the seedling progeny, which also flowered within a year of germination. Trees grew normally, produced normal flowers with viable pollen, and these trees had stable FT expression throughout the five years of this study. Additionally, expression of the CcFT3 transgene resulted in early flowering Arabidopsis plants and the FT protein could be induced through the graft union resulting in precocious flowering on one-year old budded ‘Valencia’ sweet orange shoots. Thus, these FT lines have great potential in shortening the breeding cycle. Stable transmission of the early flowering phenotype from the rootstock to the scion can alleviate many of the long juvenility issues that hampers the rapid development and evaluation of citrus cultivars.
Conclusion
Our results revealed that the phloem-localized expression of the CcFT3 gene sustained precocious flowering in Carrizo citrange rootstocks. Flowers were produced in transgenic lines as early as 16 months from transformation in the rootstocks. Transcriptomic data indicated that the expression of the CcFT3 gene triggered several critical genes in the flowering process. Altogether, our results provide definitive evidence that the CcFT3 transgene when expressed in phloem cells can induce early flowering in citrus.
Material and methods
Plasmid vector construction and transgenic plant production
The Citrus unshiu FT1, FT2 and FT3 sequences available in the NCBI database (AB027456, AB301934 and AB301935) were utilized to find the homologous sequences from the C. clementina genome in the Phytozome database (https://phytozome.jgi.doe.gov). The CcFT1 and CcFT3 DNA sequences (NCBI accession nos: MT707614 and MT602515) were amplified by PCR from C. clementina ‘Nules’ cDNA and modified to remove internal restriction sites to facilitate cloning using the Vector NTI (Life Technologies, NY, USA) bioinformatics software. A BamHI site was inserted immediately upstream of the translation start site and a SacI site was added following the stop codon. The sequences from all the clones were verified using Sanger sequencing and cloned into the complementary sites of the pCAM-CLON plant transformation vector. An Arabidopsis thaliana HSP (AtHSP18.2) promoter29, an AtSUC2 promoter28, or a NOS promoter77 were used to replace the 35S promoter in the pCAM-CLON vector. In total, eight FT based constructs were evaluated in this study (Supplementary Figure S2). The gene sequence of CcFT3 was fused in frame to the egfp gene (NCBI accession no: MT602516) to create a CcFT3-EGFP fusion gene. The fusion gene was artificially synthesized (Twist Bioscience, San Francisco, CA) and similarly cloned into the pCAM-CLON plant transformation vector.
Epicotyl explants of in vitro grown nucellar seedlings of Carrizo citrange (Citrus sinensis Osb. X Poncirus trifoliata L. Raf.) were used in all Agrobacterium genetic transformation experiments as outlined by Dutt and Grosser78. Putative transgenic lines that rooted in a kanamycin supplemented citrus rooting medium (RMM) was transferred to a peat-based commercial potting medium (Metromix 930, Sun Gro Horticulture, WA, USA) and acclimated under a 75% shade cloth in a greenhouse at 32° ± 4 °C. Trees transformed with the AtHSP18.2 promoter were incubated in a growth chamber at 37 °C for 3 h following transfer to the greenhouse. This cycle was repeated at least 6 times in weekly intervals.
Molecular analysis of transformants
PCR was carried out on all putative CcFT3 lines with AtSUC2-FT3-F and AtSUC2-FT3-R primer set to amplify a 700 bp fragment, partly from the 3′ end of the AtSUC2 promoter and the 5′ of the CcFT transgene and NPTII-F and NPTII-R primers to amplify a 500 bp nptII fragment (Supplementary Table S1). Putative transgenic plants were verified by PCR using gene specific primers and an Extract-N-Amp Plant PCR Kit (Sigma-Aldrich, St. Louis, MO). The presence of transgenes was confirmed by visualizing amplicon bands at the expected size on a 1% agarose gel containing GelRed (Biotium, Hayward, CA) under ultraviolet light. All images were recorded and digitized using an Axygen Gel Documentation System (Corning Incorporated, Tewksbury, MA, USA).
A Southern blot was performed to confirm the stable integration of the gene fragment and to determine the copy number in selected CcFT3 transgenic plants. Genomic DNA was extracted from young leaves using a cetyl trimethylammonium bromide (CTAB) protocol79, and was enzymatically digested using EcoRI, to cut within the T-DNA, but outside of the probe region. After overnight electrophoresis in 0.8% agarose gel at 25 V, DNA fragments were denatured, depurinated, and blotted onto Hybond N + membrane (Roche, Indianapolis, IN) according to manufacturer’s protocol. The digoxigenin (DIG) labelled FT3 probe (Roche, Indianapolis, IN) was UV cross-linked, and hybridized to the membrane and processed following the manufacturer’s instructions. The FT3 probe was synthesized by PCR using a DIG labelling kit (Roche). The amplification of amplicons by PCR was performed using the primers listed in the Supplementary Table S1 under the following cycling conditions: 2 min at 95 °C, 30 cycles of 30 s at 95 °C, 30 s at 66 °C, 40 s at 72 °C and a final extension of 10 min at 72 °C, yielding a 525 bp product. Finally, the blot was exposed to a Kodak X-ray film for 30 min prior to image analysis.
RNA extraction, cDNA synthesis and quantitative PCR
Citrus RNA was isolated either from 100 mg of leaf blades or leaf petioles using a Direct-zol RNA Miniprep Plus Kit (Zymo Research, Irvine, CA) according to the manufacturer’s protocol. The cDNA was synthesized using RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s instructions. Transcripts were quantified by real-time quantitative PCR (RT-qPCR). The RT-qPCR reaction mix consisted of SYBR Green PCR Master Mix (Applied Biosystems, Foster City, CA) and primers as outlined in Supplementary Table S1 for a 20 μL reaction. The reaction conditions consisted of initial denaturation at 95 °C for 30 s, followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. A no template/water control and non-transgenic cDNA were used as RT-qPCR negative controls. The relative mRNA levels were compared to those of the endogenous C. sinensis ACTIN gene80 and calculated using the 2-ΔΔCT method81.
Protein extraction and immunoblot analysis
Protein analysis was carried out by western blotting. Approximately 100 mg of young citrus leaves were flash frozen in liquid nitrogen and crushed using chilled mortars and pestles. The tissue was homogenized with 500 μL of CelLytic P Cell Lysis Reagent containing protease inhibitors (Sigma-Aldrich Corp., St. Louis, MO). The protein extract was transferred to a microcentrifuge tube and centrifuged 17,949×g for 10 min at 4 °C. The supernatant was transferred to another microcentrifuge tube, and this process was repeated three times. The protein concentration was measured using a Quick Start Bradford protein assay (Bio-rad Laboratories, Inc, Hercules, CA). Crude protein was separated on a 12% SDS/PAGE gel cast from TGX FastCast Acrylamide Kit (Bio-Rad) and transferred to a PVDF membrane. The PVDF membrane was incubated for 5 min in Ponceau-S solution (40% methanol (v/v), 15% acetic acid (v/v), 0.25% Ponceau-S), and subsequently de-stained with deionized water before the membrane was photographed. The Ponceau-S solution was incubated in tap water until complete removal of the dye, before blocking the membranes in 5% dry milk prepared in 1 × TBST for 30 min. The membrane was probed with a CcFT3 polyclonal antibody (ABclonal, Woburn, MA) and incubated overnight with agitation at 4 °C. The membranes were washed three times using 1 × TBST and then probed with a secondary antibody (goat anti-rabbit IgG Antibody, house radish peroxidase (HRP)-conjugate). Immunoblots were developed using an ECL detection kit and imaged using X-ray film autography (Thermo-Fisher Scientific, Waltham, MA).
Citrus propagation and precocious flowering phenotype assessment
The AtSUC2-CcFT3 number 3 transgenic line (MP3) was selected for further analyses. Single node cuttings were produced from two- and half-year-old trees at the time of propagation, in a mist bed. Non-transgenic Carrizo citrange cuttings of a similar age were used as controls. Budwood from a 1-year old ‘Valencia’ sweet orange seedling were grafted onto six-month-old clonally propagated rootstocks. Precocious flowering phenotype evaluation was performed weekly. Leaf samples for RNA isolation were collected periodically. The budded plants were photographed for documentation.
Microscopy
Pollen was collected from anthers of the MP3 transgenic line and non-transgenic controls and placed on a slide. Pollen grains were incubated in 50% Gram’s iodine solution diluted with deionized water on a slide for 5 min, or a 1% solution of 2,3,5-Triphenyltetrazolium chloride (TTC) on a microscope slide for two hours, before imaging. Controls were also examined as described above by incubating pollen grains in deionized water.
For SEM images of pollen grains, anthers were collected from fully expanded, unopened flowers and fixed in a 4% paraformaldehyde solution in 1X PBS. Anthers were rinsed three times for 10 min in 1X PBS and dehydrated in an ethanol series (30%, 50%, 70%, 80%, 90%, 95%, 100%, 100%, 100%). Samples were critical point dried using a Ladd 28000 critical point dryer (Ladd Research Industries, Williston VT, USA). Anthers were cut open and pollen was retrieved and placed on a double-sided 12 mm Carbon sticker (Electron Microscopy Sciences, Hatfield PA, USA). The samples were sputter coated with palladium/gold using a Ladd 30800 sputter Coater (Ladd Research Industries). Pollen grains were observed, and images were recorded using a Hitachi S4000 scanning electron microscope (Hitachi, Tokyo, Japan).
Transcriptome profiling of AtSUC2-CcFT3 transgenics
RNA extracted from three technical replicates of MP3 transgenic lines and control non-transgenic plants were sequenced using the Illumina NovaSeq 6000 platform attuned for a 2 × 150 read length configuration. To obtain clean reads, adaptors were removed by AdapterRemoval (version v2.2.2, https://github.com/MikkelSchubert/adapterremoval)82. The reads were filtered based on quality and length using Trimmomatic (version v0.39, http://www.usadellab.org/cms/?page=trimmomatic)83 by removing the adaptors and reads with less than 100 bases and an average quality of 16 or less. The following parameter were applied: A minimum length of 100 bases, a trailing and leading of 16, and a sliding window of 16:25. A final inspection of the reads was carried out by using FastQC (version v0.11.8, https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) in order to ensure proper cleaning was performed84. The raw data has been deposited into the National Center for Biotechnology Information’s Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra) with a SRA accession number: PRJNA668159.
Mapping of the reads, transcript count and differential expression analysis
The cleaned reads were mapped to the C. sinensis genome using STAR85. The C. sinensis genome and annotation used in the mapping were obtained from Phytozome86. The STAR output bam files were sorted and indexed by SAMtools (version v1.7, https://github.com/samtools)87. Read counts of each gene from the sorted and indexed bam files were counted by FeatureCounts v1.6.088. The outputted read counts for each gene and sample were placed into a comma-separated values format master file. The construction of a metadata file was also performed, which contained the samples names and trial type.
To obtain the differentially expressed genes (DEGs), DESeq2 v3.10 was used89. The metadata file and counts master file were inputted into DESeq2. The counts for the comparison were normalized and then estimated dispersion followed. The output list of DEGs were then filtered to remove any DEGs without a |log2FoldChange|≥ 1 and a P adjusted value (FDR) ≤ 0.05.
Go enrichment and pathway analysis
The list of statistically significant and high log2FoldChange DEGs were analyzed using agriGO (version v2, http://systemsbiology.cau.edu.cn/agriGOv2/)90 in order to obtain a significant grouped Gene Ontology (GO) terms. A significance level of p-value (P < 0.05) was selected as the cutoff in agriGO v2. The statistically significant GO terms were then run with REVIGO (http://revigo.irb.hr/) to remove redundant GO terms91. MapMan (version v3.5.1, https://mapman.gabipd.org/mapman) was then used to analyze the DEGs and for a more in-depth analysis on individual pathways and other factors92.
The filtered DEGs from DESeq2 had their PacID attached and were then imported into MapMan. A custom MapMan mapping file and pathway were generated using genes from the photoperiod, vernalization, autonomous and gibberellin pathways acquired from the Arabidopsis and Phytozome annotation databases. The DEGs with the PacID attached were compared to the custom MapMan file generated to show the changes between non-transgenic and transgenic plants.
Confocal microscopy
Protein expression and localization assays were performed using CcFT3-EGFP fusion driven by 35S and AtSUC2 promoters. The clones were transiently expressed in Nicotiana benthamiana plants according the method described by Sparkes et al.93 with minor modifications. A single colony of Agrobacterium strain EHA105 containing each construct was grown in Luria–Bertani broth containing rifampicin (25 mg/L) and kanamycin (100 mg/L) incubated at 28 °C overnight. The culture was centrifuged and re-suspended in infiltration buffer (10 mM 2-(N-morpholino) ethanesulfonic acid (MES), pH 5.85; 10 mM MgCl2) containing 200 µm acetosyringone. The bacterial suspension was incubated at room temperature for 3–4 h and the leaves were infiltrated with bacteria using a 1 mL needleless syringe. Leaves were imaged 3 days post-infiltration. Leaf discs were punched using a hole puncher, mounted on a slide and imaged using a Leica SP8 laser-scanning confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL, USA).
References
Nishikawa, F. Regulation of floral induction in citrus. J. Jpn. Soc. Horticult. Sci. 82, 283–292 (2013).
Peña, L. et al. Constitutive expression of Arabidopsis LEAFY or APETALA1 genes in citrus reduces their generation time. Nat. Biotechnol. 19, 263 (2001).
Endo, T. et al. Ectopic expression of an FT homolog from Citrus confers an early flowering phenotype on trifoliate orange (Poncirus trifoliata L. Raf.). Transgen. Res. 14, 703–712 (2005).
Velázquez, K. et al. Precocious flowering of juvenile citrus induced by a viral vector based on Citrus leaf blotch virus: A new tool for genetics and breeding. Plant Biotechnol. J. 14, 1976–1985 (2016).
Furr, J. R., Cooper, W. C. & Reece, P. C. An investigation of flower formation in adult and juvenile citrus trees. Am. J. Bot. 34, 1–8 (1947).
Spiegel-Roy, P. & Goldschmidt, E. E. The Biology of Citrus (Cambridge University Press, Cambridge, 1996).
Hong, Y. & Jackson, S. Floral induction and flower formation—the role and potential applications of miRNAs. Plant Biotechnol. J. 13, 282–292 (2015).
Poethig, R. S. Phase change and the regulation of shoot morphogenesis in plants. Science 250, 923–930 (1990).
Wilkie, J. D., Sedgley, M. & Olesen, T. Regulation of floral initiation in horticultural trees. J. Exp. Bot. 59, 3215–3228 (2008).
Pineiro, M. & Coupland, G. The control of flowering time and floral identity in Arabidopsis. Plant Physiol. 117, 1–8 (1998).
Boss, P. K., Bastow, R. M., Mylne, J. S. & Dean, C. Multiple pathways in the decision to flower: Enabling, promoting, and resetting. Plant Cell 16, S18–S31 (2004).
Wigge, P. A. et al. Integration of spatial and temporal information during floral induction in Arabidopsis. Science 309, 1056–1059 (2005).
Abe, M. et al. FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 309, 1052–1056 (2005).
Blázquez, M. A., Green, R., Nilsson, O., Sussman, M. R. & Weigel, D. Gibberellins promote flowering of Arabidopsis by activating the LEAFY promoter. Plant Cell 10, 791–800 (1998).
Eriksson, S., Böhlenius, H., Moritz, T. & Nilsson, O. GA4 is the active gibberellin in the regulation of LEAFY transcription and Arabidopsis floral initiation. Plant Cell 18, 2172–2181 (2006).
Corbesier, L. et al. FT protein movement contributes to long-distance signaling in floral induction of Arabidopsis. Science 316, 1030–1033 (2007).
Kazan, K. & Lyons, R. The link between flowering time and stress tolerance. J. Exp. Bot. 67, 47–60 (2016).
Hecht, V. et al. The pea GIGAS gene is a FLOWERING LOCUS T homolog necessary for graft-transmissible specification of flowering but not for responsiveness to photoperiod. Plant Cell 23, 147–161 (2011).
Imamura, T., Nakatsuka, T., Higuchi, A., Nishihara, M. & Takahashi, H. The gentian orthologs of the FT/TFL1 gene family control floral initiation in Gentiana. Plant Cell Physiol. 52, 1031–1041 (2011).
Jaeger, K. E. & Wigge, P. A. FT protein acts as a long-range signal in Arabidopsis. Curr. Biol. 17, 1050–1054 (2007).
Kong, F. et al. Two coordinately regulated homologs of FLOWERING LOCUS T are involved in the control of photoperiodic flowering in soybean. Plant Physiol. 154, 1220–1231 (2010).
Lin, M. K. et al. FLOWERING LOCUS T protein may act as the long-distance florigenic signal in the cucurbits. Plant Cell. 19, 1488–1506 (2007).
Tamaki, S., Matsuo, S., Wong, H. L., Yokoi, S. & Shimamoto, K. Hd3a protein is a mobile flowering signal in rice. Science 316, 1033–1036 (2007).
Lifschitz, E. et al. The tomato FT ortholog triggers systemic signals that regulate growth and flowering and substitute for diverse environmental stimuli. Proc. Natl. Acad. Sci. 103, 6398–6403 (2006).
Nishikawa, F. et al. Increased CiFT abundance in the stem correlates with floral induction by low temperature in Satsuma mandarin (Citrus unshiu Marc.). J. Exp. Bot. 58, 3915–3927 (2007).
Samach, A. Congratulations, you have been carefully chosen to represent an important developmental regulator!. Ann. Bot. 111, 329–333 (2013).
Pajon, M., Febres, V. J. & Moore, G. A. Expression patterns of flowering genes in leaves of ‘Pineapple’sweet orange [Citrus sinensis (L.) Osbeck] and pummelo (Citrus grandis Osbeck). BMC Plant Biol. 17, 146 (2017).
Dutt, M., Ananthakrishnan, G., Jaromin, M., Brlansky, R. & Grosser, J. Evaluation of four phloem-specific promoters in vegetative tissues of transgenic citrus plants. Tree Physiol. 32, 83–93 (2012).
Takahashi, T., Naito, S. & Komeda, Y. The Arabidopsis HSP18. 2 promoter/GUS gene fusion in transgenic Arabidopsis plants: A powerful tool for the isolation of regulatory mutants of the heat-shock response. Plant J. 2, 751–761 (1992).
Truernit, E. & Sauer, N. The promoter of the Arabidopsis thaliana SUC2 sucrose-H+ symporter gene directs expression of β-glucuronidase to the phloem: Evidence for phloem loading and unloading by SUC2. Planta 196, 564–570 (1995).
An, G., Costa, M. A., Mitra, A., Ha, S. B. & Márton, L. Organ-specific and developmental regulation of the nopaline synthase promoter in transgenic tobacco plants. Plant Physiol. 88, 547–552 (1988).
Kay, R., Chan, A., Daly, M. & McPherson, J. Duplication of CaMV 35S promoter sequences creates a strong enhancer for plant genes. Science 236, 1299–1302 (1987).
Bush, M. S., Crowe, N., Zheng, T. & Doonan, J. H. The RNA helicase, eIF 4A–1, is required for ovule development and cell size homeostasis in Arabidopsis. Plant J. 84, 989–1004 (2015).
Imamura, T., Fujita, K., Tasaki, K., Higuchi, A. & Takahashi, H. Characterization of spermidine synthase and spermine synthase–The polyamine-synthetic enzymes that induce early flowering in Gentiana triflora. Biochem. Biophys. Res. Commun. 463, 781–786 (2015).
Zhu, Z. X., Ye, H. B. & Xuan, Y. H. Overexpression of a SNARE protein AtBS14b alters BR response in Arabidopsis. Bot. Stud. 55, 55 (2014).
Yu, X. & Michaels, S. D. The Arabidopsis Paf1c complex component CDC73 participates in the modification of FLOWERING LOCUS C chromatin. Plant Physiol. 153, 1074–1084 (2010).
Kim, D. H. Current understanding of flowering pathways in plants: focusing on the vernalization pathway in Arabidopsis and several vegetable crop plants. Horticult. Environ. Biotechnol. 2, 1–19 (2020).
Han, Y. et al. RcAP1, a homolog of APETALA1, is associated with flower bud differentiation and floral organ morphogenesis in Rosa chinensis. Int. J. Mol. Sci. 20, 3557 (2019).
Anand, A., Trick, H. N., Gill, B. S. & Muthukrishnan, S. Stable transgene expression and random gene silencing in wheat. Plant Biotechnol. J. 1, 241–251 (2003).
Barnett, T., Altschuler, M., McDaniel, C. N. & Mascarenhas, J. P. Heat shock induced proteins in plant cells. Dev. Genet. 1, 331–340 (1979).
An, G., Costa, M. A. & Ha, S.-B. Nopaline synthase promoter is wound inducible and auxin inducible. Plant Cell 2, 225–233 (1990).
Dutt, M., Barthe, G., Irey, M. & Grosser, J. Transgenic citrus expressing an Arabidopsis NPR1 gene exhibit enhanced resistance against Huanglongbing (HLB; Citrus Greening). PLoS ONE 10, e0137134 (2015).
Mathieu, J., Warthmann, N., Küttner, F. & Schmid, M. Export of FT protein from phloem companion cells is sufficient for floral induction in Arabidopsis. Curr. Biol. 17, 1055–1060 (2007).
Shalom, L. et al. Alternate bearing in citrus: changes in the expression of flowering control genes and in global gene expression in on-versus off-crop trees. PLoS ONE 7, e46930 (2012).
Simpson, G. G. Evolution of flowering in response to day length: Flipping the CONSTANS switch. BioEssays 25, 829–832 (2003).
Marín, I. C. et al. Nitrate regulates floral induction in Arabidopsis, acting independently of light, gibberellin and autonomous pathways. Planta 233, 539–552 (2011).
Kojima, S. et al. Hd3a, a rice ortholog of the Arabidopsis FT gene, promotes transition to flowering downstream of Hd1 under short-day conditions. Plant Cell Physiol. 43, 1096–1105 (2002).
Liu, Z. & Mara, C. Seminars in Cell & Developmental Biology 80–86 (Elsevier, Amsterdam, 2020).
Garay-Arroyo, A. et al. The MADS transcription factor XAL2/AGL14 modulates auxin transport during Arabidopsis root development by regulating PIN expression. EMBO J. 32, 2884–2895 (2013).
Pérez-Ruiz, R. V. et al. XAANTAL2 (AGL14) is an important component of the complex gene regulatory network that underlies Arabidopsis shoot apical meristem transitions. Mol. Plant 8, 796–813 (2015).
Alvarez-Buylla, E. R. et al. An ancestral MADS-box gene duplication occurred before the divergence of plants and animals. Proc. Natl. Acad. Sci. 97, 5328–5333 (2000).
Teper-Bamnolker, P. & Samach, A. The flowering integrator FT regulates SEPALLATA3 and FRUITFULL accumulation in Arabidopsis leaves. Plant Cell 17, 2661–2675 (2005).
Ruiz-García, L. et al. Different roles of flowering-time genes in the activation of floral initiation genes in Arabidopsis. Plant Cell 9, 1921–1934 (1997).
Xia, W. et al. Alternative splicing of flowering time gene FT is associated with halving of time to flowering in coconut. Sci. Rep. 10, 11640 (2020).
Mandel, M. A., Gustafson-Brown, C., Savidge, B. & Yanofsky, M. F. Molecular characterization of the Arabidopsis floral homeotic gene APETALA1. Nature 360, 273–277 (1992).
Fernando, D. D. & Zhang, S. Constitutive expression of the SAP1 gene from willow (Salix discolor) causes early flowering in Arabidopsis thaliana. Dev. Genes. Evol. 216, 19 (2006).
Liu, Y. et al. Isolation and characterization of an APETALA1-like gene from pear (Pyrus pyrifolia). Plant Mol. Biol. Report. 31, 1031–1039 (2013).
Sun, L. M., Zhang, J. Z., Mei, L. & Hu, C. G. Molecular cloning, promoter analysis and functional characterization of APETALA 1-like gene from precocious trifoliate orange (Poncirus trifoliata L. Raf.). Sci. Horticult. 178, 95–105 (2014).
Sawettalake, N., Bunnag, S., Wang, Y., Shen, L. & Yu, H. DOAP1 promotes flowering in the orchid Dendrobium Chao Praya Smile. Front. Plant Sci. 8, 400 (2017).
Tang, M., Tao, Y. B. & Xu, Z. F. Ectopic expression of Jatropha curcas APETALA1 (JcAP1) caused early flowering in Arabidopsis, but not in Jatropha. PeerJ 4, e1969 (2016).
Riechmann, J. L., Krizek, B. A. & Meyerowitz, E. M. Dimerization specificity of Arabidopsis MADS domain homeotic proteins APETALA1, APETALA3, PISTILLATA, and AGAMOUS. Proc. Natl. Acad. Sci. 93, 4793–4798 (1996).
Lamb, R. S., Hill, T. A., Tan, Q. K. G. & Irish, V. F. Regulation of APETALA3 floral homeotic gene expression by meristem identity genes. Development 129, 2079–2086 (2002).
Ng, M. & Yanofsky, M. F. Activation of the Arabidopsis B class homeotic genes by APETALA1. Plant Cell 13, 739–753 (2001).
Sundström, J. F., Nakayama, N., Glimelius, K. & Irish, V. F. Direct regulation of the floral homeotic APETALA1 gene by APETALA3 and PISTILLATA in Arabidopsis. Plant J. 46, 593–600 (2006).
Li, W. et al. The U-Box/ARM E3 ligase PUB13 regulates cell death, defense, and flowering time in Arabidopsis. Plant Physiol. 159, 239–250 (2012).
Riboni, M., Robustelli Test, A., Galbiati, M., Tonelli, C. & Conti, L. Environmental stress and flowering time: The photoperiodic connection. Plant Signal. Behav. 9, e29036 (2014).
Yaish, M. W., Colasanti, J. & Rothstein, S. J. The role of epigenetic processes in controlling flowering time in plants exposed to stress. J. Exp. Bot. 62, 3727–3735 (2011).
Zhai, Q. et al. Transcriptional mechanism of jasmonate receptor COI1-mediated delay of flowering time in Arabidopsis. Plant Cell 27, 2814–2828 (2015).
Chini, A., Gimenez-Ibanez, S., Goossens, A. & Solano, R. Redundancy and specificity in jasmonate signalling. Curr. Opin. Plant Biol. 33, 147–156 (2016).
Purugganan, M. D. The MADS-box floral homeotic gene lineages predate the origin of seed plants: Phylogenetic and molecular clock estimates. J. Mol. Evol. 45, 392–396 (1997).
Song, Y., Chen, L., Zhang, L. & Yu, D. Overexpression of OsWRKY72 gene interferes in the abscisic acid signal and auxin transport pathway of Arabidopsis. J. Biosci. 35, 459–471 (2010).
Cheng, Z. et al. Over-expression of ERF38 gene enhances salt and osmotic tolerance in transgenic poplar. Front. Plant Sci. 10, 1375 (2019).
Du, C. et al. Reaumuria trigyna transcription factor RtWRKY23 enhances salt stress tolerance and delays flowering in plants. J. Plant Physiol. 239, 38–51 (2019).
Duck, N., McCormick, S. & Winter, J. Heat shock protein hsp70 cognate gene expression in vegetative and reproductive organs of Lycopersicon esculentum. Proc. Natl. Acad. Sci. 86, 3674–3678 (1989).
Blázquez, M. A. & Weigel, D. Integration of floral inductive signals in Arabidopsis. Nature 404, 889–892 (2000).
Schomburg, F. M., Bizzell, C. M., Lee, D. J., Zeevaart, J. A. & Amasino, R. M. Overexpression of a novel class of gibberellin 2-oxidases decreases gibberellin levels and creates dwarf plants. Plant Cell 15, 151–163 (2003).
Kim, Y., Buckley, K., Costa, M. A. & An, G. A 20 nucleotide upstream element is essential for the nopaline synthase (nos) promoter activity. Plant Mol. Biol. 24, 105–117. https://doi.org/10.1007/BF00040578 (1994).
Dutt, M. & Grosser, J. Evaluation of parameters affecting Agrobacterium-mediated transformation of citrus. Plant Cell Tissue Organ Cult. 98, 331–340 (2009).
Porebski, S., Bailey, L. G. & Baum, B. R. Modification of a CTAB DNA extraction protocol for plants containing high polysaccharide and polyphenol components. Plant Mol. Biol. Report. 15, 8–15 (1997).
Mafra, V. et al. Reference genes for accurate transcript normalization in citrus genotypes under different experimental conditions. PLoS ONE 7, 2 (2012).
Livak, K. J. & Schmittgen, T. D. Analysis of relative gene expression data using real-time quantitative PCR and the 2− ΔΔCT method. Methods 25, 402–408 (2001).
Lindgreen, S. AdapterRemoval: Easy cleaning of next-generation sequencing reads. BMC Res. Notes 5, 337 (2012).
Bolger, A. M., Lohse, M. & Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014).
Andrews, S. B. Bioinformatics (Babraham Institute, Cambridge, 2010).
Dobin, A. et al. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013).
Goodstein, D. M. et al. Phytozome: A comparative platform for green plant genomics. Nucleic Acids Res. 40, D1178–D1186 (2012).
Li, H. et al. The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079 (2009).
Liao, Y., Smyth, G. K. & Shi, W. featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550 (2014).
Tian, T. et al. AgriGO v2.0: A GO analysis toolkit for the agricultural community, 2017 update. Nucleic Acids Res. 45, W122–W129 (2017).
Supek, F., Bošnjak, M., Škunca, N. & Šmuc, T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS ONE 6, 2 (2011).
Thimm, O. et al. MAPMAN: A user-driven tool to display genomics data sets onto diagrams of metabolic pathways and other biological processes. Plant J. 37, 914–939 (2004).
Sparkes, I. A., Runions, J., Kearns, A. & Hawes, C. Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat. Protoc. 1, 2019 (2006).
Acknowledgements
This work was partially supported by a grant from the Citrus Research and Development Foundation (CRDF-547).
Author information
Authors and Affiliations
Contributions
J.M.S.—conducted Arabidopsis related work, western blots and evaluated protein localization and wrote the manuscript with M.D. K.C.W.—transcriptome data analysis. W.Q., L.M.M. and P.H.—gene expression analysis. D.S.—evaluation of transgenic pollen. H.W and J. Z.—Southern blot analysis. K.A.J.—transgenic and control rootstock propagation and T1 seed germination. M.D.—developed the FT constructs, generated, screened the transgenic plants, budded the rootstocks. M.D. and J.W.G.—designed the study, obtained funding, and supervised the project. All authors read and edited the document.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Soares, J.M., Weber, K.C., Qiu, W. et al. The vascular targeted citrus FLOWERING LOCUS T3 gene promotes non-inductive early flowering in transgenic Carrizo rootstocks and grafted juvenile scions. Sci Rep 10, 21404 (2020). https://doi.org/10.1038/s41598-020-78417-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-78417-9
- Springer Nature Limited
This article is cited by
-
Genome-wide identification and characterization of flowering genes in Citrus sinensis (L.) Osbeck: a comparison among C. Medica L., C. Reticulata Blanco, C. Grandis (L.) Osbeck and C. Clementina
BMC Genomic Data (2024)
-
Evolutionary assessment of SQUAMOSA PROMOTER BINDING PROTEIN-LIKE genes in citrus relatives with a specific focus on flowering
Molecular Horticulture (2023)
-
Gene editing in tree and clonal crops: progress and challenges
In Vitro Cellular & Developmental Biology - Plant (2021)