
Abstract
Objective was to analyse bacterial composition and abundance of Clostridioides difficile in gut microbiome of patients with C. difficile infection (CDI) in association with clinical characteristics. Whole metagenome sequencing of gut microbiome of 26 CDI patients was performed, and the relative abundance of C. difficile and its toxin genes was measured. Clinical characteristics of the patients were obtained through medical records. A strong correlation between the abundance of C. difficile and tcdB genes in CDI patients was found. The relative abundance of C. difficile in the gut microbiome ranged from undetectable to 2.8% (median 0.089). Patients with fever exhibited low abundance of C. difficile in their gut, and patients with fewer C. difficile organisms required long-term anti-CDI treatment. Abundance of Bifidobacterium and Bacteroides negatively correlated with that of C. difficile at the genus level. CDI patients were clustered using the bacterial composition of the gut: one with high population of Enterococcus (cluster 1, n = 12) and another of Bacteroides or Lactobacillus (cluster 2, n = 14). Cluster1 showed significantly lower bacterial diversity and clinical cure at the end of treatment. Additionally, patients with CDI exhibited increased ARGs; notably, blaTEM, blaSHV and blaCTX-M were enriched. C. difficile existed in variable proportion of the gut microbiome in CDI patients. CDI patients with Enterococcus-rich microbiome in the gut had lower bacterial diversity and poorer clinical cure.
Similar content being viewed by others


Introduction
Clostridioides difficile infection (CDI) is one of the major hospital acquired infections1. Old age (> 65 years), use of antibiotics for non-CDI infections, and exposure to a hospital environment are some of the recognized risk factors for hospital acquired CDI1,2.
Antibiotics have a significant and long-lasting effect on the intestinal microbiota, and reduce colonization resistance against pathogens, including C. difficile. 16S rRNA gene sequence analysis of the gut microbiota of patients with CDI showed a highly variable bacterial composition of Bacteroidetes and Firmicutes, which is at odds with their predominance in the normal flora. Patients with recurrent CDI exhibited a lower species richness than patients with an initial episode of CDI and control subjects3,4,5,6. By altering the community structure of the gut microbiome, antibiotics alter the intestinal metabolome3; metabolic changes in bile acids and short-chain fatty acids are considered to play an important role in the development of CDI3,6,7.
Previous studies have indicated the importance of healthy gut microbiota and intact immune system in the pathogenicity of CDI7,8,9. Generally, CDI occurs in elderly patients with other comorbidities, and having variable states of immune function; these patients generally receive diverse combinations of antibiotics, which interfere with the gut microbiome1,2,9. The bacterial burden and toxin titres in stool do not show any correlation with clinical severity in mice and humans, but inflammatory markers do exhibit an association with clinical severity9,10. However, there are few studies analysing the relative abundance of C. difficile with respect to the bacterial diversity in the gut microbiome of patients with CDI11, and the influence of abundance of the organisms on the clinical presentation has not been investigated yet.
In this study, the population of C. difficile was measured using whole metagenome sequences of gut microbiome in patients with CDI in order to directly analyse the relationship with many clinical and microbiological variables. Firstly, metagenomes were used for analysing the changes in microbial composition in CDI patients. Secondly, we investigated the relationship of each genus or family in gut microbiome with C. difficile by comparing their relative abundance in each patient. Thirdly, despite the low number of CDI patients, the clinical variables such as severity or treatment results and relative abundance of C. difficile in gut microbiome were compared. Fourthly, the patients of CDI were clustered with respect to their metagenome profiles in the gut, and clinical and microbiological characteristics of these two clusters were evaluated. Finally, the distribution of antimicrobial resistance genes (ARGs) in gut of CDI patients was analysed in comparison with healthy individuals.
Results
Demographics and clinical characteristics
During the study period, a total of 26 CDI patients were enrolled. Table 1 presents the demographic and clinical characteristics of the patients. Median age of the patients was 66.5 years, and gender distribution was similar. The patients exhibited a mean score of 3 on the Charlson Comorbidity Index, and diabetes mellitus and malignancy were the common comorbidities. Of the 26 patients, 62% had a history of hospitalization within the previous 2 months and 92% had received antibiotics within 2 months before this episode. As for the clinical parameters at the time of diagnosis, 27% of the patients presented leukocytosis and 7.7% of the patients exhibited elevation of serum creatinine levels (1.5-times baseline). Severity index of CDI was assessed by the two aforementioned methods. Severe CDI was identified in 8 of the 26 patients (31%) based on leukocytosis and acute kidney injury12, and in 12 of the 26 patients (46%), the severity was identified based on the four factors described previously13. As for treatment of CDI, 24 out of the 26 patients were treated with metronidazole (20 [76.9%]) or vancomycin (4 [15.4%]), and recurrence of infection was observed in 7 patients (7/24 [29.1%]); the global cure rate was 54.2% (13/24). Though 3 patients (12.5%) died, none of the patients died because of CDI.
Changes of microbial taxa correlated to CDI in gut microbiome
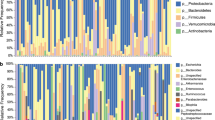
Figure 1A shows bacterial composition in the gut microbiome of 26 patients with CDI in comparison with that of 61 healthy individuals. At the genus level, 15 out of 25 detected genera (average proportion > 1%) showed significant decrease, and among them Bifidobacterium, Ruminococcus, Eubacterium, and Faecalibacterium showed a significant decrease in abundance in the gut of patients with CDI (p < 0.001 for all), whereas the abundance of Enterococcus, Lactobacillus, Escherichia, and Klebsiella increased (p < 0.001, p = 0.031, p = 0.002, and p < 0.001, respectively) (Supplementary Table S1). At the family level, 9 out of 16 detected families (average proportion > 1%) showed significant decrease; Ruminococcaceae, Bifidobacteriaceae, Lachnospiraceae, and Eubacteriaceae showed a decrease in abundance (p < 0.001 for all), whereas Enterococcaceae, Lactobacillaceae, and Enterobacteriaceae increased in abundance in the gut microbiome of patients with CDI (p < 0.001, p = 0.012 and p < 0.001, respectively) (Supplementary Table S2). Principle component analysis (PCA) on bacterial composition at genus level showed a clear separation between healthy individuals and patients with CDI (Fig. 1B), and the diversity of the gut microbiome was significantly lower in these patients (Fig. 1C,D).

Bacterial composition of the gut microbiome in patients with Clostridioides difficile infection (CDI) and healthy individuals. (A) Bacterial composition at the genus level in the gut microbiome of patients with CDI and healthy people. (B) Principal component analysis of bacterial composition at the genus-level in patients with CDI and healthy individuals. (C) Shannon index of gut microbiome in patients with CDI and healthy people. (D) Species diversity of gut microbiome in patients with CDI and healthy people.
Relative abundance of toxigenic C. difficile in the gut microbiome of CDI patients
In order to measure the relative abundance of toxigenic C. difficile in the gut microbiome, we measured the relative abundance of tcdB genes in the metagenome sequences of gut microbiome. The median relative abundance of toxigenic C. difficile in the gut microbiome was 0.089%, ranging from 0 to 2.82% (Fig. 2A). The relative abundance of tcdB genes measured by read mapping on tcdB and RPKM (read per kilobase million reads) showed a strong correlation with the abundance of C. difficile measured by MetaPhlAn clade-specific marker genes (r2 = 0.98) (Fig. 2B). For the reference, the relative abundance of C. difficile in non-CDI population is 0%.

Relative abundance of Clostridioides difficile in the gut microbiome of 26 patients with CDI. (A) Distribution of tcdB abundance in the gut microbiota of 26 CDI patients; it ranges from 0 to 2.82%. (B) Correlation between tcdB abundance and abundance of C. difficile based on clade-specific marker genes.
Factors associated with the relative abundance of toxigenic C. difficile in CDI patients
We observed that the relative abundance of toxigenic C. difficile in the gut microbiome shows an agreement with Ct value in real-time PCR of tcdB (rho = − 0.605, p = 0.002) (Table 2). We analysed the clinical characteristics associated with the abundance of toxigenic C. difficile in the gut microbiome (Table 2). Age, underlying diseases, and the use of antibiotics, proton pump inhibitor, or probiotics within 2 months from the episode of CDI were not associated with the abundance of toxigenic C. difficile. The abundance of toxigenic C. difficile had no effect on the occurrence of leukocytosis, hypoalbuminemia, or acute kidney injury. However, low numbers of toxigenic C. difficile in intestinal metagenomes were associated with fever (rho = − 0.41, p = 0.038), and longer CDI therapy (rho = − 0.405, p = 0.05). Treatment outcomes and the recurrence of CDI were not associated with the abundance of toxigenic C. difficile in the gut.
We observed the relationship between the abundance of microbial families or genera and the abundance of toxigenic C. difficile in the gut microbiome (Supplementary Table S3). At the genus level, the abundance of Bifidobacterium and Bacteroides showed a negative correlation with the abundance of toxigenic C. difficile (rho = − 0.417, p = 0.034; rho = − 0.403, p = 0.041, respectively). As these genera are the main constituents of the families, Bifidobacteriaceae and Bacteroidaceae, their populations also showed a negative correlation with the abundance of toxigenic C. difficile (rho = − 0.411, p = 0.037; rho = − 0.403, p = 0.041, respectively).
Antibiotic resistance genes in gut microbiome of CDI patients
A total of 53 ARG families from 20 classes were screened in the gut microbiome. Compared to the microbiome of healthy individuals, the gut microbiome of the patients with CDI exhibited a fourfold higher abundance of ARGs. The resistome in the healthy population ranged from 49.7 to 292.5 GPM with a median value of 89.7, while in CDI patients, it ranged from 141.5 to 1095.8 with a median value of 356.7 GPM. Figure 3A presents the abundance of 15 ARG classes, which were significantly different between the healthy individuals and the CDI patients (p value < 0.01 in t-test). Notably, resistance genes against β-lactam, aminoglycoside, polymyxin, LMS, and glycopeptide were markedly enhanced in patients with CDI (5.1, 4.3, 18.5, 3.1, and 7.7-fold, respectively). Figure 3B–D present the differential prevalence of β-lactam, aminoglycoside, and tetracycline resistance genes in healthy individuals and patients with CDI patients. In particular, class A β-lactamase genes such as blaTEM, blaSHV, and blaCTX-M genes were markedly enhanced; class C plasmid-mediated AmpC genes such as blaCMY and blaDHA were also enhanced in patients with CDI. Notably, KPC and NDM carbapenemase genes were observed in one and two patients, respectively. As for aminoglycoside genes, ANT(3′), AAC(3), and AAC(6′)-APH(2″) were markedly enhanced in the gut microbiome of CDI patients (Fig. 3C). However, tetracycline resistance genes showed only a slight increase (1.5-fold), and the distribution of individual tetracycline resistance genes was significantly different; tet32, tet44, tetM, and tetQ were fewer, but tetO, tetS, and tet34 appeared more in the gut microbiota of patients with CDI than in the gut microbiota of healthy individuals (Fig. 3D).

Differential distribution of antibiotic resistance genes in patients with Clostridioides difficile infection and healthy people. (A) Distribution of AR class in healthy individuals and patients with CDI (p value < 0.01 in t-test). (B) Differential distribution of beta-lactam resistance genes in patients with CDI and healthy people. (C) Differential distribution of aminoglycoside resistance genes in patients with CDI and healthy people. (D) Differential distribution of tetracycline resistance genes in patients with CDI and healthy individuals.
Distinct bacterial community in two different groups of CDI patients
When the patients with CDI were clustered with respect to the bacterial composition, two different groups were observed with different major constituents in their bacterial communities (Fig. 4): a cluster of samples with high abundance of Enterococcus (cluster 1, n = 12), and a cluster with high abundance of Bacteroides or Lactobacillus (cluster 2, n = 14). The bacterial diversity was significantly low in cluster 1, compared to cluster 2 (p < 0.001), which was evident in the genus distribution shown in Fig. 4. The proportion of C. difficile was not different between the 2 clusters (p = 0.129). Interestingly, the abundance of ARGs was differentially distributed between the two groups. In particular, aminoglycoside, diaminopyrimidine, and LMS resistance genes were overrepresented in the patients with high abundance of Enterococcus (p value < 0.05). However, the recurrence status, severity scores, and the total abundance of ARGs did not show any significant difference between these two groups. Clinical characteristics were compared between the 2 clusters (Table 3); more patients in Bacteroides group took proton pump inhibitor (p = 0.018) or fluoroquinolone marginally (p = 0.065) within previous 2 months. Disease severity was not different between the 2 groups but clinical cure was achieved in more patients of Bacteroides group (p = 0.031) and all fatal cases came from the Enterococcus group.

Clusters of patients with Clostridioides difficile infection with respect to the bacterial composition.
Discussion
In this study, we measured the relative abundance of C. difficile in the gut microbiome of CDI patients using metagenome sequences of gut microbiome. The gene tcdB was used as a target gene to estimate the relative abundance of toxigenic C. difficile in the gut microbiome. In general, quantitation of C. difficile culture presented as CFU (colony forming unit)/g faeces, or quantitative PCR of stool is used to evaluate the burden of C. difficile9,10. Our metagenomic approach applied in this study has an advantage over such existing methods in the sense that it can calculate the proportion of C. difficile population in the gut microbiome. Interestingly, the relative abundance of toxigenic C. difficile in the gut microbiome ranged from undetectable to 2.82% of the organisms in the gut, and in 12 of the 26 patients (46%) the C. difficile population amounted to > 0.1% of the gut microbiome. The mammalian gut is colonized by trillions of microorganisms14, and although the number of microorganisms in the gut might be reduced due to antibiotic use that predisposed the patients to CDI, the C. difficile population in the gut of patients with CDI might approach to billions.
Several studies have shown that the burden of C. difficile was not associated with the severity of CDI, and that the clinical severity of CDI was associated with the inflammatory response in the gut and the virulence of infecting organisms in human and mice9,10,15. The relative abundance of the organisms did not correlate with clinical severity in our study as well. However, unexpectedly, the C. difficile burden showed a negative correlation with the occurrence of fever and the treatment duration. This finding indicates that patients with sufficient immunity might have developed an inflammation to cause fever so as to control the pathogen levels in their gut, and the treatment duration was prolonged due to the inflammation in their gut.
We analysed the taxonomic composition of the intestinal microbiome using whole metagenome sequencing. The change in the bacterial composition of the CDI patients was similar to that reported in previous studies, which used amplification of 16S rRNA gene for identifying microbial taxa3,4,5,16,17,18. Among many genera that are known to be enhanced in healthy people compared with CDI patients, it is interesting that only the genera of Bifidobacterium and Bacteroides showed a significant negative association with toxigenic C. difficile when analysed using the proportion in the gut microbiome.
Age, gender, and underlying diseases are known to influence the microbiome structures of gut11,14, and hospitalization and usage of antibiotics have a huge impact11. In this study, the age and underlying diseases were not matched between the patients with CDI and healthy people. In addition, hospitalization and antibiotic usage were not matched because of the inclusion criteria of healthy people, which might contribute to the difference in the structure of gut microbiome and distribution of ARGs between CDI patients and healthy people.
We found that the bacterial composition is an important discriminator to cluster the patients of CDI into two groups: one group with high abundance of Enterococcus, and the other with high abundance of Bacteroides or Lactobacillus. Compared with the previous report based on 16S rRNA amplicon sequencing data5, bacterial composition patterns were generally consistent, but we further characterized the two groups clinically and microbiologically. The bacterial diversity was significantly lower in Enterococcus cluster, which suggests that gut microbiome structure was more destroyed. The total days of antibiotic usage were marginally higher in Enterococcus cluster. Since we counted the antibiotic usage only within 2 months from the enrolment, we suspect that probably more antibiotics might have been used in the cluster. Interestingly, more clinical cure was achieved in Bacteroides cluster, and all fatal cases came from the Enterococcus cluster despite no significant difference in demographics, comorbidities, and clinical severity of the diseases. These findings suggest a poor prognosis of Enterococcus cluster with more destruction of gut microbiome structure.
In terms of ARGs in gut microbiome, a four-fold increase in the number of ARGs was detected in CDI patients relative to that in healthy people. Recent admission history and antibiotic usage in CDI patients would contribute to the enrichment of ARGs in gut microbiome of CDI patients. Above all, class A β-lactamase genes, which include clinically important extended-spectrum β-lactamase genes, were markedly increased with the enhancement of Enterobacteriaceae in patients with CDI. Furthermore, plasmid-mediated carbapenemase genes were detected in three patients. A marked increase in ARGs along with high carriage number of C. difficile organisms in the gut of CDI patients reinforced the necessity of contact precaution of CDI patients.
To the best of our knowledge, this is the first attempt to investigate the association of the abundance of C. difficile with the clinical and microbiological characteristics in gut microbiome. The distribution of ARGs in the gut microbiome of CDI patients was compared with that of healthy individuals for the first time as well. Despite the methodological advantages, there are certain limitations in this study. The number of enrolled patients is relatively small, and healthy controls were not matched with CDI patients in age, underlying diseases, and antibiotic usage.
To summarize, the population of C. difficile in the gut of patients with CDI varied significantly, but did not influence the clinical severity. Regarding the bacterial composition in the gut, the patients of CDI could be discriminated into Enterococcus-rich clusters with low bacterial diversity, and Bacteroides-rich clusters with preserved bacterial diversity, and the patients belonging to the latter cluster led to a better clinical cure.
Methods
Study design and sample collection
The study was conducted at the Hanyang University Seoul Hospital, a 900-bed tertiary care facility and the Hanyang University Guri Hospital, a 600-bed secondary care facility, in South Korea. Through July 2016 to June 2018, individuals diagnosed with CDI were screened. CDI patients who could provide subsample over 50 g of faeces from CDI diagnosed stool sample were enrolled.
For the comparison, existing metagenomic data of 61 healthy Korean individuals were used19. Briefly, healthy person was defined as zero score according to the Charlson comorbidity index20 and no admission history within the past year, and individuals aged between 30 and 59 were enrolled from June to October 2017 at the Hanyang University Health Promotion Center for the health screening services.
The institutional review boards of Hanyang University Hospital and Hanyang University Guri Hospital approved these protocols, and written informed consent was obtained from all the participants. All methods were performed in accordance with relevant guidelines and regulations.
Definitions and collection of data
CDI was diagnosed when the C. difficile isolates from stool culture showed positive toxin genes (tcdA, tcdB, cdtA, or cdtB) by multiplex PCR, positive results in toxin assay A&B with commercial kit (VIDAS C. difficile Toxin A & B; BioMerieux SA, Marcy l’Etoile, France), and/or pseudomembrane on endoscopy21. Real-time PCR for tcdB was performed with Xpert CD assay (Cepheid, USA) according to manufacturer’s instructions; cyclic threshold (Ct) value was used for semi-quantitative analysis of tcdB in the stool. Demographic and clinical characteristics data were collected retrospectively using medical records20. The severity of CDI was assessed by two methods as described12,13. Recurrence was defined as the resurgence of symptoms with diagnosis as CDI after cessation of treatment, at least 10 days after the first episode. Global cure was defined as patients who were cured at the end of treatment and did not have a recurrence22,23.
Faecal DNA preparation, sequencing, and sequence filtering
Faeces was collected into a sterile container and stored at −80 °C deep freezer prior to DNA extraction. Methods for faecal DNA preparation, sequencing, and sequence filtering were followed by Human Metagenome Project- core microbiome sampling protocol A24. Total DNA was extracted using the Fast DNA SPIN Kit for Feces (MP Biomedicals, #116,570,200) and Illumina HiSeqX Platform (Illumina, San Diego, USA) was used to sequence the samples. Low-quality reads were removed using Sickle25. Four Gb of read sequences were retained in each sample for further quantitative analysis.
Metagenomic analysis of tcdB, taxonomic composition and antibiotic resistance genes
Filtered reads were assembled into contigs using MEGAHIT26 with default options. Genes were predicted from contigs (> 500 bps) using FragGeneScan27. MetaPhlAn28,29, which uses clade-specific marker genes to profile bacterial compositions with the whole metagenome sequencing data, was used to find the taxonomic composition of each sample. The relative abundance of C. difficile was also reported based on this profile result. The Partitioning Around Medoids (PAM) algorithm in R package was used to cluster the samples based on bacterial composition. The optimal number of clusters was selected as two after calculating the silhouette score (= 0.36). The relative abundance of tcdB (accession no. NC_009089.1:786021-805508) was measured by mapping reads to the genes using Bowtie to measure toxigenic C. difficile. The relative abundance was reported by RPKM30. To identify ARGs, genes predicted in the metagenomic data set were searched against the ARGs annotated in the CARD database31 using Blastp32 with the threshold of an e-value less than 1 × 10–10, similarity over 70%, and reference coverage over 70%. The resistance genes were classified into 53 ARG subclasses and 20 ARG classes based on the gene ontology31 (Supplementary Table S4). For normalization, RPKM (read per kilobase million read) was used for measuring the abundance of tcdB; GPM (gene per million genes) was used as a measure of the abundance of ARGs in each sample:
The images for figures were generated by using R package.
Statistical analysis
To compare the demographics and clinical characteristics, SPSS version 24.0 for Windows (SPSS Inc., Armonk, NY, USA) was used. Pearson’s chi-square test or Fisher’s exact test were used to analyse categorical variables, and Mann–Whitney U-test was used to analyse continuous variables. Spearman’s rank correlation test was performed to evaluate the relationship between two variables. A p value of < 0.05 by a two-tailed test was considered to be statistically significant.
Ethics approval and consent to participate
The study protocol was approved by the institutional review boards (IRB No. HYUH 2016-05-031 and HYUH 2017-06-001 from Hanyang University Hospital and GURI 2016-05-003 from Hanyang University Guri hospital), and written informed consent was obtained from all the participants.
Consent of publication
All participants in this study provided consent for publication.
Data availability
All raw sequencing data described in this study is available at European Nucleotide Archive (ENA) with the accession Nos. PRJEB35738 and PRJEB33013.
References
Freeman, J. et al. The changing epidemiology of Clostridium difficile infections. Clin. Microbiol. Rev. 23(3), 529–549 (2010).
Dubberke, E. R. et al. Clostridium difficile–associated disease in a setting of endemicity: identification of novel risk factors. Clin. Infect. Dis. 45(12), 1543–1549 (2007).
Theriot, C. M. & Young, V. B. Interactions between the gastrointestinal microbiome and Clostridium difficile. Annu. Rev. Microbiol. 69, 445–461 (2015).
Chang, J. Y. et al. Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 197(3), 435–438 (2008).
Seekatz, A. M., Rao, K., Santhosh, K. & Young, V. B. Dynamics of the fecal microbiome in patients with recurrent and nonrecurrent Clostridium difficile infection. Genome Med. 8(1), 47 (2016).
Baktash, A. et al. Mechanistic insights in the success of fecal microbiota transplants for the treatment of clostridium difficile infections. Front Microbiol. 9, 1242 (2018).
Theriot, C. M. et al. Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat. Commun. 5, 3114 (2014).
Kyne, L., Warny, M., Qamar, A. & Kelly, C. P. Asymptomatic carriage of Clostridium difficile and serum levels of IgG antibody against toxin A. N Engl J Med. 342(6), 390–397 (2000).
Lewis, B. B. et al. Pathogenicity locus, core genome, and accessory gene contributions to clostridium difficile virulence. MBio. 8(4), e00885-17. https://doi.org/10.1128/mBio.00885-17 (2017).
El Feghaly, R. E. et al. Markers of intestinal inflammation, not bacterial burden, correlate with clinical outcomes in Clostridium difficile infection. Clin. Infect. Dis. 56(12), 1713–1721 (2013).
Vincent, C. et al. Bloom and bust: intestinal microbiota dynamics in response to hospital exposures and Clostridium difficile colonization or infection. Microbiome. 4, 12 (2016).
Cohen, S. H. et al. Clinical practice guidelines for Clostridium difficile infection in adults: 2010 update by the society for healthcare epidemiology of America (SHEA) and the infectious diseases society of America (IDSA). Infect. Control Hosp. Epidemiol. 31(5), 431–455 (2010).
Zar, F. A., Bakkanagari, S. R., Moorthi, K. M. & Davis, M. B. A comparison of vancomycin and metronidazole for the treatment of Clostridium difficile-associated diarrhea, stratified by disease severity. Clin. Infect. Dis. 45(3), 302–307 (2007).
Kurilshikov, A., Wijmenga, C., Fu, J. & Zhernakova, A. Host Genetics and Gut Microbiome: Challenges and Perspectives. Trends Immunol. 38(9), 633–647 (2017).
See, I. et al. NAP1 strain type predicts outcomes from Clostridium difficile infection. Clin. Infect. Dis. 58(10), 1394–1400 (2014).
Seekatz, A. M. & Young, V. B. Clostridium difficile and the microbiota. J. Clin. Invest. 124(10), 4182–4189 (2014).
Rousseau, C. et al. Clostridium difficile colonization in early infancy is accompanied by changes in intestinal microbiota composition. J. Clin. Microbiol. 49(3), 858–865 (2011).
Schubert, A. M. et al. Microbiome data distinguish patients with Clostridium difficile infection and non-C. difficile-associated diarrhea from healthy controls. MBio. 5(3), e01021-14 (2014).
Pai. H, Kim. J, Rho. M, Seo. M-R, Kim. B. Metagenomic analyses of antibiotic resistance genes in gut microbiome of healthy people in Korea[abstract O1017]. In 29th European Congress for Clinical Microbiology and Infectious Diseases (Amsterdam, Netherlands, 2019).
Quan, H. et al. Updating and validating the Charlson comorbidity index and score for risk adjustment in hospital discharge abstracts using data from 6 countries. Am. J. Epidemiol. 173(6), 676–682 (2011).
Kim, J. et al. Epidemiology of Clostridium difficile infections in a tertiary-care hospital in Korea. Clin. Microbiol. Infect. 19(6), 521–527 (2013).
Louie, T., Miller, M., Donskey, C., Mullane, K. & Goldstein, E. J. Clinical outcomes, safety, and pharmacokinetics of OPT-80 in a phase 2 trial with patients with Clostridium difficile infection. Antimicrob. Agents Chemother. 53(1), 223–228 (2009).
Kim, J., Pai, H., Seo, M. R. & Kang, J. O. Epidemiology and clinical characteristics of Clostridium difficile infection in a Korean tertiary hospital. J. Korean Med. Sci. 26(10), 1258–1264 (2011).
Sites PL. Human Microbiome Project–Core Microbiome Sampling Protocol A. 2010.
Joshi NA FJ. Sickle: A sliding-window, adaptive, quality-based trimming tool for FastQ files (Version 1.33) [Software]. Available at https://github.com/najoshi/sickle (2011).
Li, D., Liu, C. M., Luo, R., Sadakane, K. & Lam, T. W. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31(10), 1674–1676 (2015).
Rho, M., Tang, H. & Ye, Y. FragGeneScan: predicting genes in short and error-prone reads. Nucleic Acids Res. 38(20), e191 (2010).
Truong, D. T. et al. MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat. Methods. 12(10), 902–903 (2015).
Segata, N. et al. Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods. 9(8), 811–814 (2012).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods. 9(4), 357–359 (2012).
McArthur, A. G. et al. The comprehensive antibiotic resistance database. Antimicrob. Agents Chemother. 57(7), 3348–3357 (2013).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215(3), 403–410 (1990).
Funding
This research was supported by a fund (2017ER540702) by Research of Korea Centres for Disease Control and Prevention and by the National Research Foundation of Korea (NRF) grant funded by the Korea government (MSIT) (No. 2015R1C1A1A01055646). The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Author information
Authors and Affiliations
Contributions
H.P. and M.R. designed the overall study. H.P. and M.R. equally contributed as the corresponding author. J.K. and M.B. contributed to the data acquisition and analysis. Y.C. and MS performed the experiments and analyzed data. J.K. and B.K. provided clinical information of patients and analysis of data. J.K., H.P., M.R. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Kim, J., Cho, Y., Seo, MR. et al. Quantitative characterization of Clostridioides difficile population in the gut microbiome of patients with C. difficile infection and their association with clinical factors. Sci Rep 10, 17608 (2020). https://doi.org/10.1038/s41598-020-74090-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-74090-0
- Springer Nature Limited
This article is cited by
-
Comparative analyses of the faecal resistome against β-lactam and quinolone antibiotics in humans and livestock using metagenomic sequencing
Scientific Reports (2023)
-
Gut metabolites predict Clostridioides difficile recurrence
Microbiome (2022)
-
High fecal carriage of blaCTX-M, blaCMY-2, and plasmid-mediated quinolone resistance genes among healthy Korean people in a metagenomic analysis
Scientific Reports (2021)