
Abstract
Antibiotics were derived originally from wild organisms and therefore understanding how these compounds evolve among different lineages might help with the design of new antimicrobial drugs. We report the draft genome sequence of Alexander Fleming’s original fungal isolate behind the discovery of penicillin, now classified as Penicillium rubens Biourge (1923) (IMI 15378). We compare the structure of the genome and genes involved in penicillin synthesis with those in two ‘high producing’ industrial strains of P. rubens and the closely related species P. nalgiovense. The main effector genes for producing penicillin G (pcbAB, pcbC and penDE) show amino acid divergence between the Fleming strain and both industrial strains, whereas a suite of regulatory genes are conserved. Homologs of penicillin N effector genes cefD1 and cefD2 were also found and the latter displayed amino acid divergence between the Fleming strain and industrial strains. The draft assemblies contain several partial duplications of penicillin-pathway genes in all three P. rubens strains, to differing degrees, which we hypothesise might be involved in regulation of the pathway. The two industrial strains are identical in sequence across all effector and regulatory genes but differ in duplication of the pcbAB–pcbC–penDE complex and partial duplication of fragments of regulatory genes. We conclude that evolution in the wild encompassed both sequence changes of the effector genes and gene duplication, whereas human-mediated changes through mutagenesis and artificial selection led to duplication of the penicillin pathway genes.
Similar content being viewed by others


Introduction
Clinical use of antibiotics has revolutionised treatment of bacterial infection. The characterization of penicillin1 followed from Alexander Fleming’s discovery of lysis and inhibition of the growth of Staphylococcus on petri dishes colonized by the fungus Penicillium rubens2 (named at the time as P. notatum, and until recently as P. chrysogenum3,4). There followed a golden age in the use of antibiotics to combat bacterial disease5. It was soon realized, however, that this might be short-lived, as more and more pathogenic bacteria evolved resistance to these compounds6,7,8. The result is an arms race between clinical use of antibiotics and the evolution of resistance in target bacteria, which requires new classes of antibiotics and new methods of delivery if medicine is to retain the upper hand.
One potentially useful way to improve antibiotic use and deployment is to draw inspiration from the evolution of antibiotic production and resistance in nature. Micro-organisms produce antibiotics to suppress the growth of their antagonists and reduce competition for space and resources9,10. Accordingly, organisms are under selection to evolve resistance to the antibiotics of others, yet producers are under reciprocal selection to increase the efficacy of their own antibiotics against their antagonists. With such an arms race11, it is expected that as antibiotic resistance evolves, antibiotics themselves will evolve too12. Genome sequencing can reveal variation in antibiotic production pathways among organisms with different antibiotic profiles. For instance, the ability to produce new compounds might evolve by modification of the synthesis genes or metabolic pathways, or there might be changes to existing compounds to counter the degradation of the antibiotic by resistant bacteria. These adaptations could encompass changes in the amino acid sequence of effector genes, changes in regulators of the antibiotic-production pathway, or duplication of effector genes. Gene duplication might lead to increased expression of the enzyme if the copies are conserved, or to production of multiple variants of the antibiotic if paralogous copies diverge in function13. In recent decades, considerable effort has gone into the study of the evolution of antibiotic resistance as a solution to the crisis7,14, but the evolution of antibiotics themselves remains largely neglected. Understanding how antibiotics coevolve in natural arms races with resistant bacteria might help to design methods for countering resistance evolution in the parallel arms race in clinical settings.
In order to gain insights into the evolution of genes underlying the production of the classic antibiotic, penicillin, we here present a draft genome sequence for Fleming’s original isolate, Penicillium rubens (IMI 15378). Cryopreserved living samples of this isolate are kept in numerous global collections, and we revived the fungus from the CABI (IMI) living culture collection for DNA extraction and whole-genome sequencing. We compare the overall genome structure and variation in a set of genes involved in the penicillin-production pathway with closely related Penicillium isolates with sequenced genomes. In particular, we compare Fleming’s isolate to two industrial strains of P. rubens derived from a second isolation from the wild in the USA. These strains were originally misnamed as P. chrysogenum (and still are in some public sequence databases) but were subsequently shown by multigene analysis to belong to the P. rubens clade3,4. The original wild US isolate (NRRL1951), isolated from a mouldy cantaloupe, was subjected to multiple rounds of X-ray, chemical (chlormethine) and ultraviolet mutagenesis and artificial selection15 in order to generate isolates with high production rates of penicillin for industry16, including P2niaD18 and Wisconsin 54-125517. The two lines split after an initial shared phase of both UV and X-ray mutagenesis and selection (via a common ancestor strain of Wis Q-176, see Fig. 1 in16,18,19). Wisconsin 54-1255 was derived from further rounds of UV and nitrogen mustard mutagenesis and selection, while P2niaD18 was derived from a separate round of undocumented improvements by Panlabs Inc, followed by deletion of the nitrate reductase gene niaD16,18,19. Any differences from the Fleming isolate that are shared by these two strains resulted either from evolution in the wild progenitors, or from the first steps of artificial selection and mutagenesis in the lab. In contrast, differences between the two industrial strains are solely the result of mutagenesis and artificial selection for high production: for example, comparison of P2niaD18 to the original Wisconsin 54-1255 genome17 revealed evidence for structural rearrangements and tandem duplication of penicillin-producing genes caused by the mutagenesis20. To provide a broader context for evolution in the wild, we also compare the Fleming genome to another penicillin-producing species from the Penicillium section Chrysogena, P. nalgiovense21,22, Our focus is primarily on short-term evolution among the closely related strains rather than the longer-term acquisition of penicillin production and so we do not repeat previous comparisons among more distantly related organisms23,24.
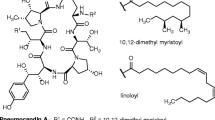
As well as comparing genome structure, we searched for genes involved in the penicillin pathway and compared copy number and sequence divergence among strains. Penicillin is a beta-lactam and encompasses several natural variations of the compound. P. rubens produces penicillin G via a 15 kb gene cluster of 3 genes (pcbAB, pcbC and penDE) present in the genomes of various filamentous fungi25,26,27 (Fig. 1). The first two genes pcbAB and pcbC encode the enzymes delta-(l-alpha-aminoadipyl)-l-cysteinyl-d-valine synthetase (ACVS) and isopenicillin N synthase respectively23,28, which catalyse the formation of the first bioactive molecule in the pathway, isopenicillin N26. All beta-lactams share this two-step pathway (including cephalosporins and cephamycins23,26,28). Evidence indicates that the genes pcbAB and pcbC were horizontally acquired from bacteria by the beta-lactam producing fungi24. The third gene penDE encodes the enzyme isopenicillin N acyltransferase, which catalyses the final step of the biosynthetic pathway that synthesizes penicillin G27,29. This gene is hypothesized to have evolved within the beta-lactam producing fungi rather than being horizontally acquired from bacteria24. In the beta-lactam producing fungi Acremonium chrysogenum, the genes cefD1 and cefD2 encode the isopenicillin N epimerase system and provide an alternative biosynthetic pathway to produce penicillin N from isopenicillin N, and cephalosporin C from penicillin N30,31,32. Several genes have been identified that play a role in regulating the pathway leading to penicillin G production, and evolved within beta-lactam producing fungi, described in more detail below24,33. We hypothesized that selection on antibiotic production should most likely result in changes in the coding sequence or copy number of effector genes at later stages of the pathway (i.e. penDE, which is unique to the production of penicillin G as opposed to other beta-lactams), or of the regulatory genes, rather than the upstream effector genes that generate pre-cursors used by multiple antibiotics.

(A) Synthesis pathway for penicillin G and penicillin N. Bold italics indicate genes producing enzymes that catalyse each step (next to arrows). (B) Regulatory genes considered in this study that either upregulate (+) or downregulate (−) expression of the effector enzymes above.
Materials and methods
Culturing, DNA extraction and sequencing
The fungus Penicillium rubens Biourge (1923)34 IMI 15378 (= ATCC 8537; NRRL 824; CBS 205.57) was obtained from the CABI IMI culture collection. As part of a separate experiment (not reported here), replicates of fungus were grown for 11 weeks at 20 °C on petri dishes with LB Lennox agar media with addition of 20 g/L of sucrose. Fungus from each of 6 treatments was then cultured in LB Lennox Broth with 20 g/L of sucrose at room temperature for a week prior to the DNA extraction. For each of the six treatments, around 100 mg of washed mycelium was ground under liquid nitrogen and DNA was extracted using the DNeasy Plant Mini Kit (Qiagen). DNA libraries were prepared with an Illumina TruSeq PCR-free kit at the Department of Biochemistry, University of Cambridge, and sequenced with Illumina MiSeq v2 technology with 2 × 150 paired-end sequencing and 350 bp insert size. Separate library preparations and sequencing were performed for the 6 separate extractions, but subsequent results indicated no changes had accrued among the treatments, so reads were pooled for the assembly and analyses presented here.
Genome assembly and analysis
Raw reads were filtered for low-quality bases and adapter sequences using BBTools ‘bbduk’ v38.22 (available at https://sourceforge.net/projects/bbmap/) with the parameters ‘ktrim = r k = 23 mink = 11 hdist = 2 maq = 10 minlen = 100 tpe tbo’. An initial assembly was constructed using SPAdes v3.13.035 with default parameters and potential contamination was assessed using BlobTools v1.036. Genome completeness was assessed using Benchmarking Universal Single-Copy Ortholog (BUSCO) gene sets v3.0.2 for Eukaryota (n = 303) and Fungi (n = 290)37. Assembled genomes for the two industrial strains, P2niaD18 and Wisconsin 54-1255, and P. nalgiovense (IBT 13039) were downloaded from NCBI GenBank (Table 1). The P2niaD18 genome was scaffolded into whole chromosomes in the source paper by comparing alignments to the Wisconsin 54-1255 genome. Genomes were aligned using nucmer in the mummer package 4.0beta, and structural changes visualized with dotplots using the DNANexus Dot browser (available at https://dnanexus.github.io/dot/). All raw sequence data have been deposited in the relevant International Nucleotide Sequence Database Collaboration (INSDC) databases under the Study ID PRJEB35151 (see Table S1 for run accessions).
Penicillin pathway genes
We searched for the pcbAB, pcbC and penDE genes in each genome using BLAST, and for a paralog of penDE that was first discovered in the Wisconsin 54-1255 genome17,38 and functionally characterized39. Query sequences are listed in Table S2. We also searched for cefD1 and cefD2 genes, which catalyse an alternative pathway for converting isopenicillin N to penicillin N rather than penicillin G, and were previously discovered in the Wisconsin 54-1255 genome17 and shown to be expressed. In addition, we searched for a suite of genes identified as playing a regulatory role in penicillin production: anBH1, the three subunits of the transcription factor ancF (hapB, hapC, and hapE), pacC, and veA. Functions of these genes are summarised in Fig. 1. In brief, PacC is a wide domain pH and carbon source dependent regulator, which upregulates the pcbAB and pcbC genes in P. chrysogenum in an alkaline environment and/or when the fungus is grown on a depleted carbon source40,41. VeA is a wide domain light dependent regulator in P. chrysogenum, A. chrysogenum and Aspergillus nidulans. It is involved in upregulation of pcbAB and downregulation of pcbC26,42,43. The transcription factor ancF consists of 3 subunits hapB, hapC and hapE and is responsible for the downregulation of the gene pcbAB and upregulation of the genes pcbC and penDE in P. chrysogenum44,45,46. The anBH1 gene produces the basic-region helix loop helix protein (bHLH) that binds to the promoter region upstream of penDE and downregulates the transcription of penDE26,47.
For each gene, we generated an alignment (including multiple copies where present) using MAFFT 1.3.648 and reconstructed a phylogenetic tree by maximum likelihood using the GTR + invgamma model in PHYML 2.2.349, implemented in Geneious 9.1.8 (Biomatters Ltd, Auckland, New Zealand, https://www.geneious.com). We tested for evidence of positive selection among strains by running codon models in PAML 450 for genes displaying variation: the null model of a single dN/dS ratio across codons, referred to as ω; the neutral model with a fraction p1 of codons that are under purifying selection (dN/dS < 1; ω1) and a fraction p2 evolving neutrally (dN/dS = 1; ω2); and the positive selection model including an additional fraction p3 codons evolving positively (dN/dS > 1; ω3). We used the Akaike Information Criterion to select the best model while penalizing for differences in the number of free parameters (null = 1, neutral = 2, positive = 4). The test is conservative because it requires substantial changes in amino acids to detect positive selection, whereas in reality a single amino acid change could underlie functional divergence. In addition to comparing best models, we plotted the average dN/dS ratio across the genome (ω) as a measure of degree of amino acid conservation among strains, with low values indicating stronger overall purifying selection.
Results
Genome assembly of P. rubens (IMI 15378)
A total of 2.86 Gb trimmed data (82.3% raw; Table S1) were used to generate an initial assembly comprising 274 scaffolds spanning 30.51 Mb. Screens for potential contamination resulted in the removal of 6 scaffolds (15.7 kb) that were not marked as from the genus Penicillium based on sequence similarity to NCBI ‘nt’ and UniRef90 public repositories (Fig S1). Scaffolds less than 500 bp in length were also discarded, resulting in a final draft assembly of 101 scaffolds spanning 30.46 Mb total length (~ 94X coverage), with an N50 scaffold length of 1.62 Mb. Assembly quality, based on the presence of core eukaryotic and fungal genes (BUSCO), indicated a high level of gene completeness (99.0% and 99.3% respectively) and a low level of duplication (0.3% and 1.0% respectively), suggesting a haploid genome assembly in common with the other strains (Table 2). The assembly is marginally smaller than the assemblies of the two industrial strains (32.4 and 32.2 Mb respectively) and has similar GC content (48.9% vs 49% for both industrial strains). The final genome assembly for P. rubens IMI 15378 (nPRUBv1) has been deposited at DDBJ/ENA/GenBank under the accession GCA_902636305.1 (CACPRF010000001–CACPRF010000101).
Structural comparison among Penicillium genomes
The genome of Fleming’s P. rubens (IMI 15378) is broadly colinear with the P2niaD18 genome that was assembled to whole chromosome level, with relatively few cases of translocation or transversions (Fig. 2). More rearrangements are apparent between the Wisconsin 54-1255 strain and the P2niaD18 genome, perhaps indicative of structural mutations caused by mutagenesis during the improvement process as previously reported20. The P. nalgiovense IBT 13039 genome was broadly colinear with P2niaD18, although the greater fragmentation of the assembly makes it harder determine any large-scale rearrangements. All three genomes of P. rubens are highly similar at the sequence level: the Fleming genome is 0.106% divergent from the P2niaD18 genome across aligned regions (31,547 SNPs from 29.7 Mb alignment) whereas the Wisconsin 54-1255 is only 0.0038% divergent (1231 SNPs from 32.2 Mb alignment). P. nalgiovense IBT 13039 is 5.8% divergent (1,484,129 SNPs from 24.8 Mb aligned regions).

Dotplots showing regions of forward alignment (in blue), reversals (green) and repetitive alignments (orange). The 4 nuclear chromosomes of the ‘P. chrysogenum’ P2niaD18 assembly were used as the reference in each case: top panel, Fleming’s strain P. rubens (IMI 15378); middle panel, Wisconsin 54-1255; bottom panel, P. nalgiovense (IBT 13039).
The structure of the penicillin effector genes is conserved across species and always falls into the well characterized cluster of pcbAB, pcbC and penDE genes (Fig. 3). The P2niaD18 genome alone has a single tandem duplication of the whole cluster, rather than multiple complete or partial tandem duplications of the cluster present in other industrial penicillium strains20,51,52. In addition to the main loci, a partial duplicate exhibiting a match to the final 123 bp of pcbAB but with three amino acid substitutions is found in a non-coding region in the two industrial strains, 2704 bp downstream of pcbAB (Fig. 3, Table S3). This fragment, labelled B1 in the previous analysis of Wisconsin 54-1255 by Fierro et al.52 is found in both tandem duplicates in P2niaD18, but absent from the Fleming genome (as confirmed by mapping raw reads of Fleming strain onto the Wisconsin 54-1255 genome, Fig. S2). We speculate that this might play a functional role in the region, for example in regulating expression of pcbAB, but it might simply be a neutral or deleterious side-effect of the mutagenesis during improvement of those strains.

Structure of the penicillin gene cluster in the four strains. Distance between tandem duplicates in P2niaD18 not shown to scale (indicated by dashed line). The asterisk and vertical line in pcbAB indicate a fragment matching to a 36 bp fragment of the cefD1 gene. More detailed view of the region is in figure S2.
Other putative beta-lactam effector genes were found in the genomes of all the four strains compared. All four strains contained the paralog of penDE first identified in the Wisconsin 54-1255 genome17. The cefD1 region contains additional 36 to 57 bp long fragments that blast to genome regions outside the main coding region. BLAST confirmed that these are not repeated domains or regions found elsewhere in the genome but represent single partial duplications similar to those observed in pcbAB. Two of these duplicate fragments were found only in the three P. rubens strains and two only in P. nalgiovense. The cefD2 region was not duplicated but recovered as two long sections and one short section in all three genomes, indicating the absence of match across the full-length region found in the P. arizonense gene used for the query.
The genes involved in regulation of penicillin production were scattered across the genome of each strain (Table S3). The hapB gene in the ancF transcription factor complex displays a partial duplicated fragment of 95 bp in the two industrial strains, which is lacking in the other two genomes. A clear hapE match was missing for P. nalgiovense. All other regulatory genes are present in single copy in all four genomes.
Sequence divergence of penicillin effector and regulatory genes
The two industrial strains, P2niaD18 and Wisconsin 54-1255, were identical at the sequence level for all the focal genes and therefore for subsequent analyses only sequences from Wisconsin 54-1255 were used to represent the American isolate of P. rubens. In contrast, penicillin-pathway genes have diverged in amino acid sequence between the Fleming strain and the US strains. All three effector genes encoding enzymes in the penicillin G pathway have diverged, but pcbAB and penDE showed the highest rates of amino acid divergence relative to silent changes whereas pcbC was strongly conserved (Fig. 4, Table S4). The level of divergence in pcbAB is unexpected since this gene functions to produce the initial precursor in the pathway, which is shared in the production of other beta-lactams. The homologs of cefD1 and cefD2 genes were found in all genomes of P. rubens strains. This was unexpected as these genes are involved in the synthesis of the cephalosporin intermediate penicillin N in A. chrysogenum and are not known to have a functional role in P. rubens17,53,54. The penicillin N effector gene cefD2 also showed a high level of amino acid divergence whereas cefD1 was more strongly conserved than other effector genes. The best sequence model for the effector genes plus hapB was a model with most codons being under constraint (dN/dS < < 1) but with a significant proportion of codons being unconstrained (dN/dS = 1). There was no sequence divergence between American and British P. rubens isolates in the penDE paralog, pacC, ancF (hapB, C and E) or veA. To further investigate possible regulatory changes, we looked for sequence variation within transcription factor binding sites within the intergenic region between pcbAB and pcbC, which is a bidirectional promotor region for these genes. Among 28 binding sites previously identified in P. chrysogenum55, all were found in the Fleming genome, and just one site was lost in both industrial strains (GATA to GGTA mutation, Table S5). Thus, the divergence of known binding sites is low, similar to that seen for regulatory proteins.

The average ratio of non-synonymous to synonymous substitutions (dN/dS) for alignments of penicillin pathway genes across the sampled genomes: standard error bars on the estimate from the PAML analysis are shown. The first four genes were sampled for three strains: Wisconsin 54-1255, Fleming (IMI 15378), and P. nalgiovense (IBT 13039). The remaining genes were compared just between the Fleming (IMI 15378) genome and P. nalgiovense (IBT 13039) because of the lack of any variation across the three P. rubens strains.
Discussion
Nearly a century since Alexander Fleming discovered the action of penicillin in bacterial cultures contaminated by P. rubens, we report the first draft genome sequence of his original strain. Very soon after the original discovery and isolation of penicillin, a second wild isolate of P. rubens from the USA was employed for future industrial manufacture owing to its greater rate of penicillin production15. Consequently, two of the strains derived from this isolate have been the focus of previous whole genome sequencing within the P. rubens clade16,17,20. We compared these genomes with each other and a third, more distantly related genome of P. nalgiovense56.
Comparison of the two USA strains provides insights into the industrial mutagenesis and artificial selection process16, which was originally performed by selecting phenotypically useful mutants without knowledge of the underlying genomic basis15. There were no amino acid differences at any genes encoding the enzymes in the penicillin pathway and regulatory genes. Instead, there was evidence for structural rearrangement across the genome, including tandem duplication of the pcbAB–pcbC–penDE cluster in P2niaD18, which has previously been studied in these and other industrial strains51,52. This fits with the type of mutagenesis and artificial selection used for this process. Experimental work showed that tandem duplication of the pcbAB–pcbC–penDE cluster does not directly increase penicillin production over short time periods—a strain of P2niaD18 that was modified to lose one copy did not produce significantly less penicillin over a 96-h assay period57. Substantial copy number multiplication of the region among industrial strains still seems to implicate gene duplication in penicillin production, but perhaps only under specific growth conditions or over longer periods18,51. Another plausible source of variation would be changes in regulatory regions, but experimental evidence indicates that such variation is unlikely to contribute to increased penicillin production18.
Comparison between the UK and US genomes sheds light on both evolved differences between the wild progenitors of the strains, and potential initial changes in the domestication steps prior to the divergence of P2niaD18 and Wisconsin 54-1255. One structural difference shared by the US genomes was the partial duplication of the final portion of the pcbAB gene. Read mapping confirmed that this region is missing from the Fleming genome and not just absent due to assembly artefacts (Fig. S2). Partial duplication and inversion have been documented previously at the ends of the amplified region containing the penicillin synthesis genes for Wisconsin 54-125552. Furthermore, partial duplication has been found to play a role in generating novel diversity previously, e.g., in the case of pathogen resistance in barley58, and could play a role in gene regulation. Without further sequencing, we cannot be certain whether this change occurred in the wild progenitor of the US strains or during initial stages of domestication. Because of the nature of these changes in relation to the predicted effects of mutagenesis, however, and the fact that further such differences arose between P2niaD18 and Wisconsin 54-1255, it seems plausible that shared structural differences of the two industrial strains from the Fleming genome occurred during their initial shared history of mutagenesis prior to their separation. No sequence divergence was observed between the two US strains in any of the genes involved in penicillin G production and regulation of the pathway: mutagenesis and selection for improved function resulted in major structural changes but no substitutions at these loci.
In contrast, penicillin-pathway enzymes have diverged in amino acid sequence between the Fleming strain and the US strains, especially pcbAB, penDE and cefD2. While it is possible in principle that these changes were caused by mutagenesis during domestication of the US strains, we think that this is unlikely: subsequent rounds of the same process led to no sequence divergence between the US strains, and the numbers of substitutions involved would seem more commensurate with longer periods of time elapsing. Instead, these differences are likely to have accrued during evolutionary divergence of the UK and US strains of P. rubens in the wild.
Although the level of divergence did not meet the statistical criteria for detecting significant evidence of positive selection, a low level of constraint on protein sequence of these genes could still indicate a history of divergent selection at a subset of codons. Alternatively, it could indicate that the function of these proteins is less dependent on amino acid identity at several sites than is the case for the other genes. In A. nidulans, the aatA gene (an ortholog of penDE) encodes the enzyme isopenicillin N acyltransferase26,59. It has been found that disruption of this gene does not disrupt penicillin production in A. nidulans. A paralog of aatA, aatB compensates for this as it encodes a homolog of isopenicillin N acyltransferase59. It should be noted that the isopenicillin N acyltransferase encoded by aatA is only 55.2% similar to its homolog encoded by aatB and the two genes themselves are only 58% similar59.
Additionally, the liquid chromatography–mass spectrometry (LC–MS) data for penicillin compounds synthesized by either of the genes indicate unexplained significant peaks in proximity to the peaks representing standard penicillin V or penicillin G compounds synthesized by these genes. These unexplained peaks could represent penicillin analogues synthesized by aatB and aatA. It would be worthwhile to investigate further how the differences in penicillin effector genes translate into altered function of the enzymes encoded, such as variation in the substrate specificity or efficiency of the enzymes60. Such variation in specificity of the enzymes could result in synthesis of penicillin G analogues. Furthermore, presence of a penDE paralog, and cefD1 and cefD2 homologs in all the genomes compared in this study suggest the possibility that these genes encode homologs of isopenicillin N acyltransferase and isopenicillin N epimerase respectively17,39,53,61. These enzymes could potentially synthesize analogues of penicillin G and penicillin N. Other beta-lactam gene variants such as homologs of the gene encoding 7-alpha-cephemmethoxylase subunit, cmcJ, have also been identified in the genome of P. chysogenum17. Studies suggest that many of these gene variants are expressed but further work is needed to elucidate the functional importance of these genes, which is currently unclear17,39,54.
The biosynthesis of penicillin G in P. chrysogenum and P. rubens consists of a simple three gene pathway, but in certain bacteria such as S. clavuligerus, as many as twelve genes can be involved in the synthesis of beta-lactams such as cephamycin C26,62. Much of what is known regarding the evolution of diversity of natural antibiotics stems from the concept of rearrangement of genes in an existing biosynthetic gene cluster, or by addition of novel genes to existing clusters via processes such as horizontal gene transfer12,63. Our analyses indicate that individual genes of beta-lactam biosynthetic pathways can themselves vary between species. Evidence indicates that many penicillin producing species such as P. chrysogenum are genetically diverse, and allelic variation within wild P. chrysogenum populations can impact penicillin production within these populations64,65. Thus, it is plausible that sequence variation in the genomes that we describe could account for the production of novel penicillin analogues. Subtle variation in chemical structure of antibiotics has been identified for other antibiotics such as antimycins produced by Streptomyces63,66,67. Future work to sample variation more widely in P. rubens and measure the impacts of variation on chemical structure of penicillin compounds is needed to distinguish these alternatives.
In conclusion, our results provide preliminary evidence that genes involved in the production of penicillin display relatively high rates of amino acid divergence between populations, as predicted if antibiotics evolve in an arms race with antagonistic microbes. Moreover, the results indicate that natural changes involving point mutation and amino acid substitutions were not fully explored by the classical industrial mutagenesis approach, which instead produced larger structural rearrangements. Thus, the mutagenesis approach employed previously may have missed some solutions for optimizing penicillin design compared to natural selection in the wild, especially in the context of robustness to evolving antibiotic resistance. Future approaches could use solutions explored by nature as a template for the development of novel antibiotic varieties.
References
Chain, E. et al. Penicillin as a chemotherapeutic agent. The Lancet 236, 226–228. https://doi.org/10.1016/S0140-6736(01)08728-1 (1940).
Fleming, A. On the antibacterial action of cultures of a Penicillium, with special reference to their use in the isolation of B. influenzae. Br. J. Exp. Pathol. 10, 226–236 (1929).
Houbraken, J., Frisvad, J. C. & Samson, R. A. Fleming’s penicillin producing strain is not Penicillium chrysogenum but P. rubens. IMA Fungus 2, 87–95. https://doi.org/10.5598/imafungus.2011.02.01.12 (2011).
Houbraken, J. et al. New penicillin-producing Penicillium species and an overview of section Chrysogena. Persoonia 29, 78–100. https://doi.org/10.3767/003158512X660571 (2012).
Gaynes, R. The discovery of Penicillin—new insights after more than 75 years of clinical use. Emerg. Infect. Dis. 23, 849–853. https://doi.org/10.3201/eid2305.161556 (2017).
Fair, R. J. & Tor, Y. Antibiotics and bacterial resistance in the 21st century. Perspect. Med. Chem. 6, 25–64. https://doi.org/10.4137/PMC.S14459 (2014).
Ventola, C. L. The antibiotic resistance crisis: part 2: management strategies and new agents. P&T Peer-Rev. J. Formul. Manag. 40, 344–352 (2015).
Ventola, C. L. The antibiotic resistance crisis: part 1: causes and threats. P&T Peer-Rev. J. Formul. Manag. 40, 277–283 (2015).
Abrudan, M. I. et al. Socially mediated induction and suppression of antibiosis during bacterial coexistence. Proc. Natl. Acad. Sci. 112, 11054–11059. https://doi.org/10.1073/pnas.1504076112 (2015).
Granato, E. T., Meiller-Legrand, T. A. & Foster, K. R. The evolution and ecology of bacterial warfare. Curr. Biol. 29, R521–R537. https://doi.org/10.1016/j.cub.2019.04.024 (2019).
Brockhurst, M. et al. Running with the Red Queen: the role of biotic conflicts in evolution. Proc. R. Soc. Lond. B https://doi.org/10.1098/rspb.2014.1382 (2014).
Pathak, A., Kett, S. & Marvasi, M. Resisting antimicrobial resistance: lessons from fungus farming ants. Trends Ecol. Evol. 34, 974–976. https://doi.org/10.1016/j.tree.2019.08.007 (2019).
Jia, N., Ding, M.-Z., Luo, H., Gao, F. & Yuan, Y.-J. Complete genome sequencing and antibiotics biosynthesis pathways analysis of Streptomyces lydicus 103. Sci. Rep. 7, 44786–44786. https://doi.org/10.1038/srep44786 (2017).
Blair, J. M. A., Webber, M. A., Baylay, A. J., Ogbolu, D. O. & Piddock, L. J. V. Molecular mechanisms of antibiotic resistance. Nat. Rev. Microbiol. 13, 42. https://doi.org/10.1038/nrmicro3380 (2014).
Backus, M. P. & Stauffer, J. F. The production and selection of a family of strains in Penicillium chrysogenum. Mycologia 47, 429–463. https://doi.org/10.1080/00275514.1955.12024468 (1955).
Salo, O. V. et al. Genomic mutational analysis of the impact of the classical strain improvement program on β-lactam producing Penicillium chrysogenum. BMC Genomics 16, 937–937. https://doi.org/10.1186/s12864-015-2154-4 (2015).
van den Berg, M. A. et al. Genome sequencing and analysis of the filamentous fungus Penicillium chrysogenum. Nat. Biotechnol. 26, 1161 (2008).
Newbert, R. W., Barton, B., Greaves, P., Harper, J. & Turner, G. Analysis of a commercially improved Penicillium chrysogenum strain series: involvement of recombinogenic regions in amplification and deletion of the penicillin biosynthesis gene cluster. J. Ind. Microbiol. Biotechnol. 19, 18–27. https://doi.org/10.1038/sj.jim.2900411 (1997).
Barreiro, C., Martín, J. F. & García-Estrada, C. Proteomics shows new faces for the old Penicillin producer Penicillium chrysogenum. J. Biomed. Biotechnol. https://doi.org/10.1155/2012/105109 (2012).
Specht, T., Dahlmann, T. A., Zadra, I., Kürnsteiner, H. & Kück, U. Complete sequencing and chromosome-Scale Genome Assembly of the Industrial Progenitor Strain P2niaD18 from the Penicillin Producer Penicillium chrysogenum. Genome Announc. 2, e00577-e1514. https://doi.org/10.1128/genomeA.00577-14 (2014).
Laich, F., Fierro, F., Cardoza, R. E. & Martin, J. F. Organization of the gene cluster for biosynthesis of penicillin in Penicillium nalgiovense and antibiotic production in cured dry sausages. Appl. Environ. Microbiol. 65, 1236–1240 (1999).
Andersen, S. J. & Frisvad, J. C. Penicillin production by Penicillium nalgiovense. Lett. Appl. Microbiol. 19, 486–488. https://doi.org/10.1111/j.1472-765X.1994.tb00988.x (1994).
Aharonowitz, Y., Cohen, G. & Martin, J. F. Penicillin and cephalosporin biosynthetic genes: structure, organization, regulation, and evolution. Annu. Rev. Microbiol. 46, 461–495. https://doi.org/10.1146/annurev.mi.46.100192.002333 (1992).
Brakhage, A. A., Al-Abdallah, Q., Tüncher, A. & Spröte, P. Evolution of β-lactam biosynthesis genes and recruitment of trans-acting factors. Phytochemistry 66, 1200–1210. https://doi.org/10.1016/j.phytochem.2005.02.030 (2005).
Díez, B. et al. The cluster of penicillin biosynthetic genes. Identification and characterization of the pcbAB gene encoding the alpha-aminoadipyl-cysteinyl-valine synthetase and linkage to the pcbC and penDE genes. J. Biol. Chem. 265, 16358–16365 (1990).
Brakhage, A. A. et al. Aspects on evolution of fungal β-lactam biosynthesis gene clusters and recruitment of trans-acting factors. Phytochemistry 70, 1801–1811. https://doi.org/10.1016/j.phytochem.2009.09.011 (2009).
Weber, S. S., Polli, F., Boer, R., Bovenberg, R. A. L. & Driessen, A. J. M. Increased penicillin production in Penicillium chrysogenum production strains via balanced overexpression of isopenicillin N acyltransferase. Appl. Environ. Microbiol. 78, 7107–7113. https://doi.org/10.1128/AEM.01529-12 (2012).
Cooper, R. D. G. The enzymes involved in biosynthesis of penicillin and cephalosporin; their structure and function. Bioorg. Med. Chem. 1, 1–17. https://doi.org/10.1016/S0968-0896(00)82098-2 (1993).
Tobin, M. B., Fleming, M. D., Skatrud, P. L. & Miller, J. R. Molecular characterization of the acyl-coenzyme A: isopenicillin N acyltransferase gene (penDE) from Penicillium chrysogenum and Aspergillus nidulans and activity of recombinant enzyme in Escherichia coli. J. Bacteriol. 172, 5908–5914. https://doi.org/10.1128/jb.172.10.5908-5914.1990 (1990).
Martín, J. F., Ullán, R. V. & García-Estrada, C. Regulation and compartmentalization of β-lactam biosynthesis. Microb. Biotechnol. 3, 285–299. https://doi.org/10.1111/j.1751-7915.2009.00123.x (2010).
Ullán, R. V. et al. A novel epimerization system in fungal secondary metabolism involved in the conversion of Isopenicillin N into Penicillin N in Acremonium chrysogenum. J. Biol. Chem. 277, 46216–46225 (2002).
Ullán, R. V., Casqueiro, J., Naranjo, L., Vaca, I. & Martín, J. F. Expression of cefD2 and the conversion of isopenicillin N into penicillin N by the two-component epimerase system are rate-limiting steps in cephalosporin biosynthesis. Mol. Genet. Genomics 272, 562–570. https://doi.org/10.1007/s00438-004-1087-4 (2004).
García-Estrada, C., Domínguez-Santos, R., Kosalková, K. & Martín, J.-F. Transcription factors controlling primary and secondary metabolism in filamentous fungi: the β-lactam paradigm. Fermentation 4, 47. https://doi.org/10.3390/fermentation4020047 (2018).
Biourge, P. Les moissisures du groupe Penicillium Link. La Cellule 33, 7–331 (1923).
Bankevich, A. et al. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol. 19, 455–477. https://doi.org/10.1089/cmb.2012.0021 (2012).
Laetsch, D. R. & Blaxter, M. L. BlobTools: interrogation of genome assemblies. F1000Research 6, 1287 (2017).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. https://doi.org/10.1093/bioinformatics/btv351 (2015).
Van Den Berg, M. A. Penicillium chrysogenum: genomics of an antibiotics producer. In Genomics of Soil- and Plant-Associated Fungi Soil Biology, Ch. 10 (eds Horwitz, B. A. et al.) 229–254 (Springer, Berlin, 2013).
García-Estrada, C. et al. Molecular characterization of a fungal gene paralogue of the penicillin penDE gene of Penicillium chrysogenum. BMC Microbiol. 9, 104. https://doi.org/10.1186/1471-2180-9-104 (2009).
Suárez, T. & Peñalva, M. A. Characterization of a Penicillium chrysogenum gene encoding a PacC transcription factor and its binding sites in the divergent pcbAB–pcbC promoter of the penicillin biosynthetic cluster. Mol. Microbiol. 20, 529–540. https://doi.org/10.1046/j.1365-2958.1996.5421065.x (1996).
Bergh, K. T. & Brakhage, A. A. Regulation of the Aspergillus nidulans penicillin biosynthesis gene acvA (pcbAB) by amino acids: implication for involvement of transcription factor PACC. Appl. Environ. Microbiol. 64, 843 (1998).
Kato, N., Brooks, W. & Calvo, A. M. The expression of sterigmatocystin and penicillin genes in Aspergillus nidulans is controlled by veA, a gene required for sexual development. Eukaryot. Cell 2, 1178–1186. https://doi.org/10.1128/EC.2.6.1178-1186.2003 (2003).
Martín, J. F. Key role of LaeA and velvet complex proteins on expression of β-lactam and PR-toxin genes in Penicillium chrysogenum: cross-talk regulation of secondary metabolite pathways. J. Ind. Microbiol. Biotechnol. 44, 525–535. https://doi.org/10.1007/s10295-016-1830-y (2017).
Bergh, K. T., Litzka, O. & Brakhage, A. A. Identification of a major cis-acting DNA element controlling the bidirectionally transcribed penicillin biosynthesis genes acvA (pcbAB) and ipnA (pcbC) of Aspergillus nidulans. J. Bacteriol. 178, 3908–3916. https://doi.org/10.1128/jb.178.13.3908-3916.1996 (1996).
Brakhage, A. A. et al. HAP-Like CCAAT-binding complexes in filamentous fungi: implications for biotechnology. Fungal Genet. Biol. 27, 243–252. https://doi.org/10.1006/fgbi.1999.1136 (1999).
Steidl, S. et al. AnCF, the CCAAT binding complex of Aspergillus nidulans, contains products of the hapB, hapC, and hapE genes and is required for activation by the pathway-specific regulatory gene amdR. Mol. Cell. Biol. 19, 99–106. https://doi.org/10.1128/MCB.19.1.99 (1999).
Caruso, M. L., Litzka, O., Martic, G., Lottspeich, F. & Brakhage, A. A. Novel Basic-region helix–loop–helix transcription factor (AnBH1) of Aspergillus nidulans counteracts the CCAAT-binding complex AnCF in the promoter of a Penicillin biosynthesis gene. J. Mol. Biol. 323, 425–439. https://doi.org/10.1016/S0022-2836(02)00965-8 (2002).
Katoh, K., Misawa, K., Kuma, K.-I. & Miyata, T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 30, 3059–3066. https://doi.org/10.1093/nar/gkf436 (2002).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. https://doi.org/10.1093/sysbio/syq010 (2010).
Yang, Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. https://doi.org/10.1093/molbev/msm088 (2007).
van den Berg, M. A., Westerlaken, I., Leeflang, C., Kerkman, R. & Bovenberg, R. A. L. Functional characterization of the penicillin biosynthetic gene cluster of Penicillium chrysogenum Wisconsin 54-1255. Fungal Genet. Biol. 44, 830–844. https://doi.org/10.1016/j.fgb.2007.03.008 (2007).
Fierro, F. et al. The penicillin gene cluster is amplified in tandem repeats linked by conserved hexanucleotide sequences. Proc. Natl. Acad. Sci. U. S. A. 92, 6200–6204. https://doi.org/10.1073/pnas.92.13.6200 (1995).
Kiel, J. A. K. W. et al. Matching the proteome to the genome: the microbody of penicillin-producing Penicillium chrysogenum cells. Funct. Integr. Genomics 9, 167–184. https://doi.org/10.1007/s10142-009-0110-6 (2009).
Jami, S., Barreiro, C., García-Estrada, C. & Martin, J. Proteome analysis of the penicillin producer Penicillium chrysogenum: characterization of protein changes during the industrial strain improvement. Mol. Cell. Proteom. 9, 1182–1198. https://doi.org/10.1074/mcp.M900327-MCP200 (2010).
Martín, J. F. Molecular control of expression of penicillin biosynthesis genes in fungi: regulatory proteins interact with a bidirectional promoter region. J. Bacteriol. 182, 2355–2362. https://doi.org/10.1128/jb.182.9.2355-2362.2000 (2000).
Nielsen, J. C. et al. Global analysis of biosynthetic gene clusters reveals vast potential of secondary metabolite production in Penicillium species. Nat. Microbiol. 2, 17044. https://doi.org/10.1038/nmicrobiol.2017.44 (2017).
Ziemons, S., Koutsantas, K., Becker, K., Dahlmann, T. & Kück, U. Penicillin production in industrial strain Penicillium chrysogenum P2niaD18 is not dependent on the copy number of biosynthesis genes. BMC Biotechnol. 17, 16–16. https://doi.org/10.1186/s12896-017-0335-8 (2017).
Rajaraman, J. et al. Evolutionarily conserved partial gene duplication in the Triticeae tribe of grasses confers pathogen resistance. Genome Biol. 19, 116. https://doi.org/10.1186/s13059-018-1472-7 (2018).
Spröte, P. et al. Identification of the novel penicillin biosynthesis gene aatB of Aspergillus nidulans and its putative evolutionary relationship to this fungal secondary metabolism gene cluster. Mol. Microbiol. 70, 445–461. https://doi.org/10.1111/j.1365-2958.2008.06422.x (2008).
Pavela-Vrancic, M., Dieckmann, R. & von Döhren, H. ATPase activity of non-ribosomal peptide synthetases. Biochim. Biophys. Acta (BBA) Proteins Proteom. 1696, 83–91. https://doi.org/10.1016/j.bbapap.2003.09.012 (2004).
Ullán, R. V., Campoy, S., Casqueiro, J., Fernández, F. J. & Martín, J. F. Deacetylcephalosporin C production in Penicillium chrysogenum by expression of the isopenicillin N epimerization, ring expansion, and acetylation genes. Chem. Biol. 14, 329–339. https://doi.org/10.1016/j.chembiol.2007.01.012 (2007).
Alexander, D. C. & Jensen, S. E. Investigation of the Streptomyces clavuligerus Cephamycin C gene cluster and its regulation by the CcaR protein. J. Bacteriol. 180, 4068–4079 (1998).
Seipke, R. F. & Hutchings, M. I. The regulation and biosynthesis of antimycins. Beilstein J. Org. Chem. 9, 2556–2563. https://doi.org/10.3762/bjoc.9.290 (2013).
Wong, V. L., Ellison, C. E., Eisen, M. B., Pachter, L. & Brem, R. B. Structural variation among wild and industrial strains of Penicillium chrysogenum. PLoS ONE 9, e96784. https://doi.org/10.1371/journal.pone.0096784 (2014).
Henk, D. A. et al. Speciation despite globally overlapping distributions in Penicillium chrysogenum: the population genetics of Alexander Fleming’s lucky fungus. Mol. Ecol. 20, 4288–4301. https://doi.org/10.1111/j.1365-294X.2011.05244.x (2011).
Seipke, R. F. et al. A single Streptomyces symbiont makes multiple antifungals to support the fungus farming ant Acromyrmex octospinosus. PLoS ONE 6, e22028. https://doi.org/10.1371/journal.pone.0022028 (2011).
Joynt, R. & Seipke, R. F. A phylogenetic and evolutionary analysis of antimycin biosynthesis. Microbiology 164, 28–39. https://doi.org/10.1099/mic.0.000572 (2018).
Acknowledgements
This work was funded in part by a Natural Environment Research Council Grant NE/S010866/1 and a project bursary from the Masters by Research in Computational Methods in Ecology and Evolution at Imperial College London.
Author information
Authors and Affiliations
Contributions
A.P. and T.G.B. conceived the study. M.R. provided materials. A.P. and C.G.W. grew cultures and extracted DNA. A.P., R.W.N. and T.G.B. analysed the data. A.P. and T.G.B. led writing the manuscript with further inputs from R.W.N., M.R. and C.G.W. We thank 3 anonymous reviewers for extremely helpful comments on earlier drafts.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pathak, A., Nowell, R.W., Wilson, C.G. et al. Comparative genomics of Alexander Fleming’s original Penicillium isolate (IMI 15378) reveals sequence divergence of penicillin synthesis genes. Sci Rep 10, 15705 (2020). https://doi.org/10.1038/s41598-020-72584-5
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-020-72584-5
- Springer Nature Limited
This article is cited by
-
Engineered and total biosynthesis of fungal specialized metabolites
Nature Reviews Chemistry (2024)
-
Genome sequencing and analysis of penicillin V producing Penicillium rubens strain BIONCL P45 isolated from India
International Microbiology (2024)
-
An efficient Agrobacterium-mediated system based on the pyrG auxotrophic marker for recombinant expression in the filamentous fungus Penicillium rubens
Biotechnology Letters (2023)