
Abstract
Restriction-Modification systems (RMS) are one of the main mechanisms of defence against foreign DNA invasion and can have an important role in the regulation of gene expression. The obligate human pathogen Neisseria gonorrhoeae carries one of the highest loads of RMS in its genome; between 13 to 15 of the three main types. Previous work has described their organization in the reference genome FA1090 and has inferred the associated methylated motifs. Here, we studied the structure of RMS and target methylated motifs in 25 gonococcal strains sequenced with Single Molecule Real-Time (SMRT) technology, which provides data on DNA modification. The results showed a variable picture of active RMS in different strains, with phase variation switching the activity of Type III RMS, and both the activity and specificity of a Type I RMS. Interestingly, the Dam methylase was found in place of the NgoAXI endonuclease in two of the strains, despite being previously thought to be absent in the gonococcus. We also identified the real methylation target of NgoAXII as 5′-GCAGA-3′, different from that previously described. Results from this work give further insights into the diversity and dynamics of RMS and methylation patterns in N. gonorrhoeae.
Similar content being viewed by others


Introduction
Neisseria gonorrhoeae is a sexually-transmitted pathogen that causes gonorrhoea. Antimicrobial resistance in this pathogen is of great public health concern and there have recently been an increasing number of publications aimed at analysing its transmission and the evolution of genetic determinants of antimicrobial resistance using high throughput short-read sequencing1. However, Single Molecule Real-Time (SMRT) PacBio sequencing provides deeper information about microbial genomes, including data on DNA modification, mainly methylation in the form of 6mA, 5mC or 4mC. Several studies have focused on the reference strain N. gonorrhoeae FA1090 to characterize the population of Restriction-Modification systems (RMS) in the gonococcus2,3,4,5 and its methylation landscape6. However, RMS and methylation status are known to vary significantly between strains of the same species, so this provides only limited knowledge about RMS and methylation in the gonococcus as a whole.
N. gonorrhoeae is among the bacterial species with the highest numbers of RMS despite being naturally competent for DNA uptake7. Indeed, highly transformable bacteria have been found to contain a higher number of RMS compared with those which are less competent, acting as a defence system against the invasion of foreign DNA8,9,10. RMS are formed by a restriction endonuclease (REase) and a DNA methyltransferase (MTase) that recognize a specific pattern in the genome and act as multi-subunit complexes (Type I and III) or as independent enzymes (Type II)10. If the MTase is active, it will methylate the associated motifs and thus protect them from cleavage by the cognate REase. Bacteria can contain up to four types of RMS, although only Type I, II and III have been found in Neisseria7,10. Briefly, Type I RMS act as a multi-enzyme complex consisting of two MTase subunits (HsdM), two REase subunits (HsdR) and one specificity unit (HsdS) that recognizes specific asymmetric sequences, methylating both strands10,11. Type II RMS are generally formed by individual MTase and REase enzymes that recognize the same short palindromic motifs and perform a two-strand methylation or cleavage, respectively10,11. Finally, Type III RMS act as a complex of two Mod (MTase) and two Res (REase) subunits12 that recognize specific short asymmetrical motifs10,11. The specificity of Type I RMS is encoded by an independent gene (hsdS) that must be active for the other subunits to be functional, while Type III RMS harbour a DNA recognition domain (DRD) within the MTase10. RMS have also been associated with other roles apart from defending the genome against foreign DNA invasion, such as being involved in the epigenetic control of gene expression8,9,10. They have been described as selfish elements, tending to propagate on mobile genetic elements and promoting their own survival13. They are often associated with mobility genes, such as integrases or transposases and are sometimes flanked by repeats14. Indeed, they have been shown to have an important role in genetic flux among bacteria, as cleavage by REases provides fragments of double-stranded DNA that could be incorporated into the host genome8,15.
The action of some Type I and III RMS is known to be regulated by phase variation in several bacteria7,10. The variation in the number of repeat copies in homopolymers or short tandem repeats, mainly due to slipped-strand mispairing during DNA replication, can cause the switch between a functional, non-functional or a different version of the gene9. It has been hypothesized that phase variation can be used by RMS to regulate genome flux8 and it has been shown to be associated with a random switching in the expression of multiple genes (‘phasevarion’, for phase-variable regulon10,16) in Haemophilus influenzae17,18, N. meningitidis19, N. gonorrhoeae2, Helicobacter pylori20, and Moraxella catarrhalis21. Apart from the Type III RMS, the Type II ‘orphan’ Dam MTase has also been shown to be involved in gene regulation in several Gram-negative bacteria9 and is even required for virulence in some pathogenic organisms such as Escherichia coli, Salmonella, Yersinia or Vibrio species22. The Dam MTase participates in the methyl-directed DNA mismatch repair system and this constrains the flexibility of the bacteria to undergo phase variation, which involves mispairing of bases during replication slippage23. It has been found in some N. lactamica and N. meningitidis strains24, however, there have been no reports of N. gonorrhoeae carrying this enzyme until now. Instead, the gonococcus and other Dam-defective Neisseria contain a dam replacing gene (drg), characterized as an endonuclease24, that has been reported to be important for adhesion and biofilm formation23.
In this study, we characterised the population of RMS and their associated methylation specificities in 25 N. gonorrhoeae strains using SMRT PacBio data. Results from this study give a more comprehensive insight into the link between genomics and epigenomics in the gonococcus.
Results
Detection of RMS and associated DNA methylation
From 13 to 15 complete RMS were found in each of the 25 N. gonorrhoeae strains included in the study (Supplementary Table 1): 2 of Type I, 11 of Type II and 2 of Type III, all of which are present in the REBASE database (Fig. 1). A detailed scan of the genomes for genes with a Pfam annotation associated with a REase or a MTase did not detect any new RMS. Most of the systems were found surrounded by core genes coding for essential functions, such as tRNA aminoacylation during protein translation, ATP binding or magnesium ion binding, but also DNA transposition and isomerase activity (Supplementary Table 2). This could potentially be related to the fact that Type I REases have an important requirement of ATP for cleavage, and Type II REases require magnesium ion as a cofactor11. The amino acid translation of the repetitive region in the two Type III RMS is rich in prolines and, interestingly, the ppiA gene upstream of the methylase in NgoAX is an isomerase that catalyzes the isomerization of peptide bonds in prolyl residues, assisting protein folding25.

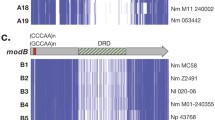
Genome organization of Restriction-Modification systems in N. gonorrhoeae. The nomenclature by Roberts et al.11 is shown between brackets for Type I and Type III RMS. The repeated unit of the phase-variable tandem repeat is shown for the mod genes as well as the alleles (modA and modB) of the DNA recognition domains (DRD). VSR: Very short patch repair endonuclease.
Analysis of DNA modification using PacBio sequencing for the 25 gonococcal strains produced a final list of 12 methylated motifs (Table 1, raw motif summaries in Supplementary Table 3). Manual curation of the raw list of motifs was performed by visualizing the distribution of interpulse duration (IPD) ratios for every base in each motif, which was essential to filter out artefacts produced by noise in the data. For example, some motifs were found to overlap with others methylated in a higher frequency or were inferred because of high IPD values in guanines, which can be caused by a O-6-methylguanine modification, associated with DNA damage by alkylating agents26.
When looking at raw IPD ratios, it is difficult to set a threshold to distinguish between methylated and unmethylated bases. To confirm they can be differentiated, we investigated the distributions of IPD ratios for methylated and unmethylated bases. As expected, 6mA modifications extracted from our final motif list showed the highest IPD ratio values, with a mean value of 5.63 (Supplementary Fig. 1). Cytosine methylations (4mC and 5mC) are difficult to detect using native DNA, especially 5mC, because of the lower effect on delaying the polymerase during PacBio sequencing. Even so, they showed an average IPD ratio of 3.24 and 2.81, respectively, higher than the mean ratios for any unmethylated base (Supplementary Fig. 1). These results confirm that there is enough differentiation between methylated and unmethylated bases to draw further conclusions about methylated regions in the genome.
Type I RMS: NgoAV is multispecific
Two Type I RMS were present in the N. gonorrhoeae strains (Figures 1 and 2). Type I NgoAV RMS showed at least three different regions of genetic variability within the specificity unit (hsdS). An insertion of a T at position 186 of the gene creates a frameshift that causes a premature stop codon in position 62 of the protein, inactivating hsdS, and thus, the whole RMS (Fig. 3). From 0 to 3 repeats of the amino acid sequence LEAT27 were observed in different strains. Some strains also contained a downstream frameshift caused by the deletion of two Gs in a homopolymer, alterations of which have previously been associated with a change of NgoAV specificity4. This second frameshift does not cause the inactivation of the gene, but in combination with the number of LEAT repeats, was found to be associated with a change in the recognition pattern (Fig. 3 and Supplementary Table 4).

Active and inactive Restriction-Modification systems (RMS) in the 25 N. gonorrhoeae strains under study. The Dam methylase is marked in a different colour (see legend). An extra column (‘?’) in each RMS is plotted showing if methylation is expected (orange) for each strain. The tree on the left is a maximum likelihood reconstruction of the core genome SNPs of the 25 strains.

Protein alignment of the HsdS specificity unit of the Type I NgoAV Restriction-Modification system (RMS). The three main regions of variation within the unit are shown as a premature stop codon in position 62 of the protein that causes inactivation of the RMS, a variable number of LEAT repeats and a downstream stop codon in position 201 (caused by a change in the length of a poly-G homopolymer) which, in combination, are related to a different recognition motif. The strains with a full-length protein are labelled in green (left), and the methylated motifs detected using PacBio are shown in different colours on the right. ‘−‘ represents gaps and ‘*’ stop codons.
Only one methylation target is currently shown for NgoAV in REBASE (5′-GCAN{8}TGC-3′). However, we also observed two other methylation targets associated with this RMS. Two strains with 1 LEAT repeat and no frameshift showed two-strand 6mA methylation in the 5′-GACN{6}TGC-3′ motif (Supplementary Table 4 and Supplementary Fig. 2). However, four strains with 2 LEAT repeats instead showed the same starting and ending triplet nucleotides in the recognition pattern but the spacer was 1 bp longer, 5′-GACN{7}TGC-3′ (Supplementary Table 4 and Supplementary Fig. 3). Finally, two strains showed 5′-GCAN{8}TGC-3′ methylation, slightly different in the 5′ end and a further 1 bp longer (Supplementary Table 4 and Supplementary Fig. 4). In this case, they contained 2 LEAT repeats and the frameshift downstream that causes a premature end of the protein, which does not inactivate it but changes its specificity (Fig. 3). The two motifs not annotated in REBASE to NgoAV were assigned to this RMS because they are double-stranded methylated asymmetric motifs and the methylation signal was coherent with the pattern of variability within the NgoAV hsdS. The only exception was strain NCTC12700, which contained complete sequences of the hsdM and hsdS genes, which should translate to full-length proteins. HsdS contains 1 LEAT repeat and no middle frameshift as with WHO M or NCTC10931, but no methylation of the corresponding motif (5′-GACN{6}TGC-3′) is found from the PacBio data (Supplementary Table 4). This could be caused by any of these proteins not being translated due to genetic regulation or posttranslational modifications impeding its action.
The second Type I RMS, NgoAXVII, did not show particular variable regions in the specificity unit or methylase among the strains, as happens with NgoAV hsdS, but only the disruption of one and/or the other was found to cause the inactivation of the whole RMS. All strains with full-length HsdM and HsdS subunits in this RMS showed two-strand 6mA methylation in the 5′-GAGN{5}TAC-3′ motif (Supplementary Fig. 5 and Supplementary Table 5). An additional motif was also detected in WHO O that we hypothesized is an off-target methylation of this enzyme because of the similarity of the motifs: 5′-BGAGN{4}GTTAC-3′, very similar to the main motif methylated by NgoAXVII but one base longer (Table 1). The specificity unit of this enzyme was found to be very conserved, with only two non-synonymous mutations that had no association with the potential off-target methylations.
Cytosine methylation by Type II RMS
N. gonorrhoeae contains up to 11 Type II RMS (Figures 1 and 2), 7 of them annotated as associated with cytosine methylation (mC) in REBASE. In spite of apparently showing functional cytosine methylases, only three motifs containing 5mC or 4mC methylation were detected by the PacBio pipeline (Table 1 and Supplementary Table 3). However, as we show above, both types of cytosine modification can be distinguished from an unmethylated base (Supplementary Fig. 1). In order to perform a deeper analysis of the motifs associated with mC in Type II RMS, we specifically targeted those reported in REBASE and evaluated their per-base IPD ratios (Supplementary Table 6). A Mann-Whitney test was performed between the distribution of IPD values for the predicted mCs and a random set of unmethylated cytosines for each strain to assess whether there was a statistically significant difference. Significant cytosine modification was found for the motifs associated with the 7 RMS expressing a functional methylase (Supplementary Fig. 6 and Supplementary Table 6). Four strains showed a gene annotated as “virulence-associated” instead of the NgoAIII RMS and three of those did not show significant signals of methylation (Supplementary Table 6). The motif associated with NgoAIII in REBASE is 5′-GGCGCC-3′ (5mC) and this was detected as a 4mC signal (reported as the degenerate motif 5′-GGCSCCND-3′) in WHO V (Supplementary Table 3), although we showed the signal was in fact present in all the strains with a functional RMS (Supplementary Fig. 6). The WHO G PacBio data produced a non-significant test for mC in the forward strand of 5′-GCGGC-3′, associated with NgoAVII, which could be due to hemi-methylation or poor signal in the data.
RMS NgoAXIII was found as a VSR (Very Short patch Repair) protein followed by a MTase and there is no report of its target motif in REBASE. A previous study experimentally confirmed that this MTase is inactive in FA109028. A series of BLAST searches revealed that only N. polysaccharea M18661 (Genbank accession number CP031325.1) contained a homologous region to NgoAXIII. Instead, genomes of N. lactamica (i.e. Y92-1009, Genbank accession number CP019894.1) showed a longer version of the MTase (362 amino acids compared to 112 in N. gonorrhoeae FA1090 and 133 in the rest). This shows that N. gonorrhoeae does not have a complete NgoAXIII system, but only remnants of a MTase. Interestingly, several N. meningitidis genomes contain the VSR protein followed by a different RMS instead (NmeDI)28.
The 5′-AAANCGGTTNNC-3′ motif was detected by the PacBio SMRT pipeline containing single-strand m4C methylation in the underlined base. The study of the distribution of per-base IPD ratios in the WHO strains revealed that the complementary sequence was probably also methylated at 3′-TTTNGCCAANNG-5′ (Supplementary Fig. 7). A Mann-Whitney test revealed the distribution of IPD values for these two bases to be significantly different than that of an unmethylated cytosine for most of the strains (Supplementary Fig. 8). FA1090 and, especially, the NCTC strains, consistently showed very noisy results, but the test was still statistically significant for them (Supplementary Fig. 8). We hypothesize that the methylation of this motif could result from a secondary target of a Type II RMS although this is unlikely as the fraction of methylated 5′-AAANCGGTTNNC-3′ is very low (Table 1).
6mA methylation by Type II RMS
Three of the Type II RMS contained 6mA methylases. The NgoAVIII system has a BcgI-like structure29 as it is formed by an enzyme with restriction and 6mA methylation properties (RM) and a specificity unit (S). A BLASTn search of the genomic region spanning both units against the NCBI database revealed that this RMS has not been described in any other organism. A transposase immediately upstream of the RM unit indicates that this system may be mobile and acquired from an unknown source. The target motif in REBASE for this RMS is that of BcgI, 5′-GACN{5}TGA-3′, although we do not detect this motif as methylated or any other motif with 6mA methylation that could be associated with NgoAVIII (Supplementary Table 3), despite the system appearing intact. Interestingly, BcgI-like MTases are known to prefer hemi-methylated DNA, although their recognition sequences are very similar to those of Type I MTases, which methylate both strands30,31. Thus, NgoAVIII may have a maintenance role for the motifs targeted by Type I MTases, methylating the growing strand immediately after chromosomal replication, a point when those motifs will be hemi-methylated.
The Dam methylase was found in place of the restriction enzyme in NgoAXI in two strains (NCTC10931 and NCTC12700), which showed double-strand 6mA 5′-GATC-3′ methylation (Supplementary Fig. 9). This MTase has been reported to be substituted by the Dam-replacing endonuclease (Drg) in many Neisseria species, but has not been found in N. gonorrhoeae until now23,24. Finally, the NgoAXVI RMS is formed by two methylases with 5mC and 6mA methylation specificities, respectively, and a restriction enzyme. A clear 6mA signal is found in the associated 5′-GGTGA-3′ motif in all the strains (Supplementary Fig. 10). A more detailed comparative analysis of the distribution of IPD values of the three cytosines in the reverse complement of this motif against unmethylated Cs for each strain identified the middle cytosine as the most plausible candidate for 5mC methylation (3′-CCACT-5′ Supplementary Fig. 11).
Type III RMS: the target of NgoAXII is 5′-GCAGA-3′
Two Type III RMS were present in the N. gonorrhoeae strains (Figures 1 and 2). These contain a phase-variable methylase (Mod) that controls the activation or inactivation of the system and includes a DNA-recognition domain (DRD) that controls its specificity. NgoAX3 contained a variable number of 5′-CCCAA-3′ repeats that switch the system ON/OFF and the modB1 DRD allele (Supplementary Table 7). In contrast, NgoAXII contained a variable number of 5′-AGCC-3′ repeats and the modA13 DRD allele (Supplementary Table 8). Both modA and modB DRD alleles are those typically found in N. gonorrhoeae5,32.
Active NgoAX RMS showed 6mA 5′-CCACC-3′ methylation in 6 strains (Supplementary Fig. 12 and Supplementary Table 7), and active NgoAXII showed 6mA 5′-GCAGA-3′ methylation in 3 strains (Supplementary Fig. 13 and Supplementary Table 8). A previous report suggested the methylated motif for the modA13 allele of this enzyme was 5′-AGAAA-3′ in strain FA1090 by evaluating the digestion pattern of ApoI and other enzymes2. This is the recognition motif associated with NgoAXII in REBASE, although it has not been detected in PacBio studies6. The sequence of the DRD region was identical among the three strains and FA1090, providing further evidence that there are no mutations associated that could explain a different specificity among FA1090 and the three strains in this study. However, our analysis of the distribution of the per-base IPD ratios for both motifs in the three strains with an active modA (Fig. 4) revealed that less than 0.1% of the AGAAA motifs contained an IPD ratio above 3, while most of the GCAGA (>79%) showed high IPDs in the underlined base (Supplementary Table 9). We only observed between 4 and 8 motifs in the genomes that included both motifs overlapping with each other and that expanded to include a complete ApoI recognition sequence (5′-RAATTY-3′), 5′-GCAGAAATTY-3′. Therefore, the digestion in these cases could have been impeded by the methylation of the second A instead (in bold). A low fraction of motifs with high IPDs in the first A of 5′-GCAGA-3′ was found in the strains with an active Type I NgoAV, and this may be due to overlapping signals between both RMS (Supplementary Figures 2–4 and 13).

Comparison of the per-base distribution of IPD ratios for the 5′-GCAGA-3′ motif detected in three strains as associated with NgoAXII, the distributions for the previously inferred 5′-AGAAA-3′ and the motif overlapping between the previous two and the recognition pattern of the ApoI restriction enzyme, 5′-RAATTY-3′2 in the three strains with a functional NgoAXII system. Two boxplots are shown per base, corresponding to the IPD ratios of the forward (red) and reverse (blue) strands.
Strangely, results from the motif analysis in FA1090 did not show signals of 5′-GCAGA-3′ methylation, although the mod gene was apparently complete in the reference genome (NCBI:AE004969). An assembly of the PacBio reads from which the methylation data was obtained for this strain6 revealed that mod in the NgoAXII RM system in the subclone of FA1090 used for sequencing in the cited work contained 35 AGCC repeats instead of 37 in the reference genome, causing a protein truncation and thus a lack of 5′-GCAGA-3′ methylation. This difference in the number of repeats was caused by phase variation probably during subculturing, and inactivation of the mod gene has been associated with a change in gene expression17.
Orphan methylases in N. gonorrhoeae include the Dam methylase
Disrupted methylases can cause the inactivation of the whole RMS due to mechanisms that avoid self-degradation of the chromosomal DNA10 in this situation. However, methylases can be functional with a disrupted or absent endonuclease. These methylases have been associated with a regulatory rather than defensive role and have been described as ‘orphan’. In the strains under study we observed multiple examples of this in the Type I NgoAXVII (Supplementary Table 5) and Type II NgoAII RMS (Supplementary Table 6). The Dam methylase in NgoAXI acts as an ‘orphan’ methylase, as it completely replaces the endonuclease of the system, and the downstream MTase is disrupted. A BLASTp search at the NCBI non-redundant protein database revealed that the gonococcal Dam sequence has 97–98% amino acid identity to N. polysaccharea, 97% to N. flavescens, 96–97% to N. lactamica, and 92–96% to N. meningitidis. A broader screening of the Dam protein in the Neisseria genus showed a reasonable amount of diversity at the amino acid level, with recombination swapping sequence variants among Neisseria species (Supplementary Fig. 14).
A scan of the Pfam domains of the proteome of the 25 N. gonorrhoeae strains did not reveal further DNA methylases without an accompanying restriction enzyme in the main chromosomes. However, the strains carrying the conjugative plasmid (WHO G, L, M, N, O, W, and NCTC10931) showed an orphan MTase in this element that has a 100% amino acid and nucleotide identity to the Type II M.Ngo8107ORF11P and M.Ngo5289ORFAP enzymes in REBASE (Supplementary Fig. 15). No further matches were found to any other bacteria in a BLASTn search against a non-redundant nucleotide database.
Discussion
Restriction-Modification systems have been shown to perform several other roles apart from being a defence system against foreign DNA8. For example, some of them, especially those from Type III, are involved in the control of gene expression through phase variable repeats in the Mod unit16. Previous papers have characterised different RMS mostly in the N. gonorrhoeae FA1090 reference strain2,3,4,5,33 and also data on its methylation status has been described6. In this manuscript, we provide a detailed view of the structure and variability of DNA MTases and specificity units in RMS in 25 strains of N. gonorrhoeae and link them to the detected methylated motifs.
The analysed gonococcal genomes showed from 13 to 15 complete RMS, all of them described in the reference strain FA1090 in REBASE34. Twelve motifs were detected as methylated in the 25 strains, most of them containing 6mA methylation (Table 1). In the case of the Type I NgoAV, earlier work confirmed the functionality of the naturally truncated hsdS in FA1090, which was predicted to recognize the 5′-GCAN{8}TGC-3′ motif35. Later, it was proven that the cause of this truncation was a phase-variable G homopolymer and that the restitution of a complete form of this unit changed the specificity of the MTase to 5′-GCAN{7}STGC-3′4. Our study reveals further insight into the diversity of specificities of the NgoAV RMS as two other different motifs (5′-GACN{6}TGC-3′ and 5′-GACN{7}TGC-3′) were detected in strains with a functional RMS. The change in specificity was found to be related to a combination of two factors: the number of LEAT repeats in the protein sequence of hsdS and the presence of the downstream frameshift that causes the previously-described truncation4 (Fig. 3 and Supplementary Table 4). The presence of amino acid repeats in the centre region of HsdS and its impact on specificity was first described in Escherichia coli (TAEL repeats)36 and has also been described in previous works in N. gonorrhoeae (LEAT or EATL repeats)27,35, but not its role in sequence specificity in combination with the downstream frameshift. Off-target methylated motifs were associated with the other Type I RMS NgoAXVII, although no associated variability was found in hsdS in this system.
Seven out of 11 Type II RMS in N. gonorrhoeae are associated with cytosine methylation. However, this type of DNA modification is difficult to detect by current PacBio sequencing, and high coverage in combination with Tet-conversion is preferable37. Only three motifs associated with 5mC or 4mC methylation were detected in a subset of the strains (Table 1). Nonetheless, a statistical comparison between the IPD values of target cytosines in the motifs annotated in REBASE for these RMS to those from unmethylated cytosines revealed significantly higher values for those in the strains carrying active 5mC MTases (Supplementary Table 5). 5mC can be converted into T by deamination, generating mismatches that can produce mutations. Bacteria can modulate this effect by methylating 4mC instead8. In fact, double-stranded methylation of 5′-CCGCGG-3′ by NgoAIII was detected by the PacBio system as 4mC, apart from the potential off-target 5′-AAANCGGTTNNC-3′ motif (Table 1). Interestingly, secondary methylation by these types of enzymes has been observed in H. influenzae38.
N. gonorrhoeae harbours two VSR endonucleases to correct T:G mismatches, V.NgoAXIII and V.NgoAXIV33. In this study, we observed a truncated NgoAXIII MTase in all strains, as previously described for FA109028. However, the VSR may still be functional, as it has been found to recognize T:G mismatches in every nucleotide context33. V.NgoAXIV has also been described to recognize mismatches in sequences other than 5′-CCGG-3′33. No methylated motif has been found for the Bcg-like RMS NgoAVIII, however, previous reports describe this type of MTase as having a strict preference for hemi-methylated sites30,31 in Type I-like motifs. Thus, we propose that NgoAVIII may have a maintenance role, methylating the replicated strand in sites targeted by Type I RMS during DNA replication.
The Dam methylase is present in some strains of N. lactamica and N. meningitidis24. However, it has not been described in N. gonorrhoeae until now. Instead, Neisseria species or strains lacking this enzyme are known to carry an endonuclease encoded by drg (dam replacing gene) recognizing the 5′-GATC-3′ motif24,39. Here, we observed two N. gonorrhoeae strains (NCTC10931 and NCTC12700) carrying the dam MTase gene instead of the drg gene in the NgoAXI RMS locus, followed by a truncated MTase. Thus, Dam is acting as an orphan enzyme, a form that has also been shown to regulate gene expression40. Interestingly, previous publications have suggested that strains carrying the drg gene have an advantage over those carrying dam because they have more flexibility for phase-variation, as the Dam MTase participates in DNA mismatch repair during replication23,39. Also, the same publications experimentally showed that strains carrying drg form more stable biofilms and have better adhesion to human cells during infection.
Several works have studied the ‘phasevarion’ in N. gonorrhoeae and N. meningitidis, referred to the set of genes with an altered expression due to phase variation in a MTase2,10,17. Type I NgoAV has been considered to regulate a phasevarion as the phase variation of the G homopolymer is involved in a change in specificity4,16. In our work, we complement this result, as we show that the change in specificity is produced as a combination of the phase-variable poly-G and the number of LEAT repeats27. However, the best examples of phasevarion are the phase-variable Type III RMS10, in which for example modD1 has been associated with hypervirulent N. meningitidis clonal complexes19 and modA13 with enhanced biofilm formation and intracellular survival16. In a recent publication, the NgoAX Mod was experimentally inactivated in N. gonorrhoeae and a deregulation of the expression of 121 genes was observed, along with an effect on the adherence to and invasion of host epithelial cells, which are essential for infection5. In contrast, inactivation of the NgoAXII Mod enzyme showed a deregulation of 54 genes under iron-limiting conditions2. The N. gonorrhoeae population has been reported to carry a very conserved modB1 allele compared to modA13, which has led to the suggestion that NgoAX Mod is the main regulator of the epigenome, while NgoAXII regulates in specific growth conditions5. Here, we observe that indeed all the strains under study carry the modA13 and modB1 alleles, with all possible combinations of activity apparent: one of the two Mod active (methylates the target motif), both active or both inactive (Supplementary Table 7 and Supplementary Table 8). In this work we show that the real methylation target of NgoAXII is 5′-GCAGA-3′, different from that annotated in REBASE for this RMS, highlighting the necessity of using experimental data such as PacBio kinetic information, to update this and other databases.
Results from this study shed further light onto the path to understanding RMS and the role of methylation in bacteria, and particularly in N. gonorrhoeae. However, we want to emphasize that the activation or inactivation of many of the RMS are produced in a reversible manner through phase variation. This means that different culture conditions or growth phases of the same strain can have a different picture of functional RMS. We have indeed observed this in the present work with the inactivation of the NgoAXII Mod unit in the subclone of FA1090 used to produce the PacBio data used in this study6. It is also important to mention the limitations of high throughput sequencing approaches, which can struggle to report the real length of a repeat, especially if there is a subpopulation with different repeat lengths and a consensus sequence is given. Further studies on larger collections of N. gonorrhoeae, different culture conditions and growth phases will allow a better understanding of when RMS are activated and the proportions of active RMS in the gonococcal population.
In summary, genetic diversity created through phase variation or hypervariable domains controls the activation or inactivation of RMS in N. gonorrhoeae, along with a variation of specificity in the Type I NgoAV RMS. A change in the number of tandem repeats and homopolymers through phase variation can happen in a reversible manner to control the ON/OFF switching of an RMS according to the selection acting on the bacteria, i.e. defence against a particular stress, immune evasion, etc. This work gives further insight into the RMS in N. gonorrhoeae and the consequent methylation landscape, which can change to modulate gene expression. We also show the importance of detailed PacBio SMRT analysis to enhance and complete the methylation information contained in public databases.
Methods
Genomes included in the study
We analysed the genomes of a total of 25 N. gonorrhoeae strains sequenced using PacBio. Of those, 14 complete genomes were obtained from the 2016 WHO reference panel41 and 10 additional genomes were selected from the Public Health England NCTC 3000 collection (http://www.sanger.ac.uk/resources/downloads/bacteria/nctc/) avoiding overlap with the WHO panel, to further improve the representativeness of the N. gonorrhoeae genome diversity (Supplementary Table 1). Additionally, PacBio raw data and predicted motifs were retrieved for the reference genome N. gonorrhoeae FA1090 from Blow et al.6. Sequencing was run using native DNA in all cases. Assembly and annotation were performed as reported in the publications cited above, using an automatic and improved pipeline at the Wellcome Sanger Institute41. FA1090 was reassembled using the PacBio data with Canu v1.642 to compare the number of tandem repeats in the Type III MTases with those from the original assembly available in the public databases (GenBank accession AE004969).
Analysis of DNA methylation
Genome-wide base modifications and predicted modified motifs were called using the RS_Modification_and_Motif_Analysis protocol from the PacBio SMRT Analysis software v2.3.0. Coordinates of the predicted motifs were localized in all the genomes and plasmids using the EMBOSS application fuzznuc43. Per-base IPD ratios for the predicted motifs were extracted from the raw data and visualized in the 25 strains using R44.
The distribution of IPD values for each of the four bases outside any of the predicted motifs was used as the distribution of values for the four unmethylated bases. A random subsample of 10,000 unmethylated sites of each base was used. Cytosine methylation from Type II RMS was inferred by evaluating the distribution of IPD ratios in the target base in the associated motif in REBASE34 compared to that of unmethylated cytosines. A Mann-Whitney test was performed to evaluate statistical significance and p-values were corrected using Bonferroni45.
Detection and specificity of RMS
An extended and manually curated list of Pfam domains associated with REases and MTases from a previous work46 was used to detect these genes in the annotations of the genomes under study. HMMER47 hmmscan was run independently on the proteome of each genome (.faa files) against Pfam(A) v3048 to complement the annotation information. R language44 was used to parse the annotation files and the results from hmmscan and extract genes with an inferred Pfam domain in the list of target REases and MTases. Genes predicted to be an REase or an MTase that were less than 10 genes distant were considered to be part of the same RMS. The rest were tagged as ‘orphan’. Results were compared to the RMS annotated for FA1090 in REBASE34. The presence or absence of every RMS in each strain was visualized using phandango49.
The motif annotated in REBASE34 for each MTase was compared to the list of predicted motifs obtained by the PacBio SMRT Analysis software for all the strains. In REBASE, many methylation targets are inferred across RMS that have similar amino acid sequences. However, a small change in the sequence of the DNA recognition domain of a methylase or the specificity unit among species with orthologous RMS can alter the sequence that is methylated. This means that matching the methylated motifs obtained by PacBio with the annotated methylases in REBASE is sometimes not straightforward and new methylation signals can be found that are associated with existing annotated methylases. These cases were further investigated and assigned to a RMS using the genomic data and according to the characteristics of the newly-found methylated motifs (one/two-strand methylation, palindromic/asymmetric motif, length, etc)10. Nucleotide and protein sequences of every unit of the RMS were extracted for each strain and compared using SeaView v4.6.150 to look for sources of variability within the MTases or the specificity units. These and the flanking genes were visualized using Artemis v16.0.051. Disrupted proteins were confirmed by a protein-protein BLAST against a non-redundant protein database52.
Representative protein sequences of the Dam methylase in the Neisseria genus were downloaded from the Identical Protein Groups (IPG) tool of the NCBI database (accessed on 21/01/2019). These sequences were aligned using SeaView v4.6.150 and the resulting alignment trimmed using Gblocks v0.91b53 considering only positions in which at least half of the sequences do not have a gap. PhyML54 was used to build a maximum likelihood tree using the LG model and performing 100 bootstrap replicates. Final tree and metadata were visualised using iToL55.
GO enrichment analysis
A Gene Ontology (GO) enrichment analysis was performed for the genes flanking all the RMS using the topGO R package56. The three ontologies were scanned (‘BP’, biological process; ‘MF’, molecular function; and ‘CC’, cellular component). The classic and weight01 algorithms were used which do not, and do, use hierarchy information for scoring a particular GO term, respectively57. The statistical significance of the enrichment was calculated using a Fisher’s exact test between the observed and expected number of genes in each term for each algorithm. Terms with a p-value < 0.05 in both algorithms were considered as significant in the results. P-values were not corrected for multiple testing to avoid excluding significant GO terms near the cut-off. Besides, the tests are not independent when using the weight01 algorithm as it is conditioned on neighbouring terms56.
Data Availability
Scripts used to perform the analyses and plots in this work are available in the GitHub repository https://github.com/leosanbu/MethylationProject. The 14 N. gonorrhoeae PacBio raw data and complete genomes from the 2016 WHO panel are available under the ENA Bioproject PRJEB14020 (Sample accessions SAMEA2448460-SAMEA2448470 and SAMEA2796326-SAMEA2796328)41. Accession numbers for the PacBio data from the 10 strains downloaded from the NCTC3000 project are available in the following link: https://www.sanger.ac.uk/resources/downloads/bacteria/nctc/ (Sample accessions SAMEA3174297-SAMEA3174299, SAMEA4076737, SAMEA4076741, SAMEA4076765, SAMEA4076768-SAMEA4076770, SAMEA4076773). GFF files for the WHO and NCTC sequence data are available in the GitHub repository https://github.com/leosanbu/MethylationProject. The N. gonorrhoeae FA1090 reference genome was obtained from NCBI accession number AE004969 and the PacBio raw data from the study by Blow et al.6. Supplementary Table 1 contains the detailed information for each strain.
References
Unemo, M., Golparian, D. & Eyre, D. W. Antimicrobial resistance in Neisseria gonorrhoeae and treatment of gonorrhea. Methods Mol Biol 1997, 37–58, https://doi.org/10.1007/978-1-4939-9496-0_3 (2019).
Srikhanta, Y. N. et al. Phasevarions mediate random switching of gene expression in pathogenic Neisseria. PLoS Pathog 5, e1000400, https://doi.org/10.1371/journal.ppat.1000400 (2009).
Adamczyk-Poplawska, M., Lower, M. & Piekarowicz, A. Characterization of the NgoAXP: phase-variable Type III restriction-modification system in Neisseria gonorrhoeae. FEMS Microbiol Lett 300, 25–35, https://doi.org/10.1111/j.1574-6968.2009.01760.x (2009).
Adamczyk-Poplawska, M., Lower, M. & Piekarowicz, A. Deletion of one nucleotide within the homonucleotide tract present in the hsdS gene alters the DNA sequence specificity of Type I restriction-modification system NgoAV. J Bacteriol 193, 6750–6759, https://doi.org/10.1128/JB.05672-11 (2011).
Kwiatek, A., Mrozek, A., Bacal, P., Piekarowicz, A. & Adamczyk-Poplawska, M. Type III methyltransferase M.NgoAX from Neisseria gonorrhoeae FA1090 regulates biofilm formation and interactions with human cells. Front Microbiol 6, 1426, https://doi.org/10.3389/fmicb.2015.01426 (2015).
Blow, M. J. et al. The epigenomic landscape of prokaryotes. PLoS Genet 12, e1005854, https://doi.org/10.1371/journal.pgen.1005854 (2016).
Rotman, E. & Seifert, H. S. The genetics of Neisseria species. Annu Rev Genet 48, 405–431, https://doi.org/10.1146/annurev-genet-120213-092007 (2014).
Vasu, K. & Nagaraja, V. Diverse functions of restriction-modification systems in addition to cellular defense. Microbiol Mol Biol Rev 77, 53–72, https://doi.org/10.1128/MMBR.00044-12 (2013).
Casadesus, J. & Low, D. Epigenetic gene regulation in the bacterial world. Microbiol Mol Biol Rev 70, 830–856, https://doi.org/10.1128/MMBR.00016-06 (2006).
Srikhanta, Y. N., Fox, K. L. & Jennings, M. P. The phasevarion: phase variation of Type III DNA methyltransferases controls coordinated switching in multiple genes. Nat Rev Microbiol 8, 196–206, https://doi.org/10.1038/nrmicro2283 (2010).
Roberts, R. J. et al. A nomenclature for restriction enzymes, DNA methyltransferases, homing endonucleases and their genes. Nucleic Acids Res 31, 1805–1812, https://doi.org/10.1093/nar/gkg274 (2003).
Roberts, R. J., Vincze, T., Posfai, J. & Macelis, D. REBASE: restriction enzymes and methyltransferases. Nucleic Acids Res 31, 418–420, https://doi.org/10.1093/nar/gkg069 (2003).
Kobayashi, I. Behavior of restriction-modification systems as selfish mobile elements and their impact on genome evolution. Nucleic Acids Res 29, 3742–3756, https://doi.org/10.1093/nar/29.18.3742 (2001).
Furuta, Y., Abe, K. & Kobayashi, I. Genome comparison and context analysis reveals putative mobile forms of restriction-modification systems and related rearrangements. Nucleic Acids Res 38, 2428–2443, https://doi.org/10.1093/nar/gkp1226 (2010).
Oliveira, P. H., Touchon, M. & Rocha, E. P. Regulation of genetic flux between bacteria by restriction-modification systems. Proc Natl Acad Sci USA 113, 5658–5663, https://doi.org/10.1073/pnas.1603257113 (2016).
Seib, K. L., Jen, F. E., Scott, A. L., Tan, A. & Jennings, M. P. Phase variation of DNA methyltransferases and the regulation of virulence and immune evasion in the pathogenic Neisseria. Pathog Dis 75, ftx080, https://doi.org/10.1093/femspd/ftx080 (2017).
Srikhanta, Y. N., Maguire, T. L., Stacey, K. J., Grimmond, S. M. & Jennings, M. P. The phasevarion: a genetic system controlling coordinated, random switching of expression of multiple genes. Proc Natl Acad Sci USA 102, 5547–5551, https://doi.org/10.1073/pnas.0501169102 (2005).
Fox, K. L. et al. Haemophilus influenzae phasevarions have evolved from Type III DNA restriction systems into epigenetic regulators of gene expression. Nucleic Acids Res 35, 5242–5252, https://doi.org/10.1093/nar/gkm571 (2007).
Seib, K. L. et al. A novel epigenetic regulator associated with the hypervirulent Neisseria meningitidis clonal complex 41/44. FASEB J 25, 3622–3633, https://doi.org/10.1096/fj.11-183590 (2011).
Srikhanta, Y. N. et al. Phasevarion mediated epigenetic gene regulation in Helicobacter pylori. PLoS One 6, e27569, https://doi.org/10.1371/journal.pone.0027569 (2011).
Blakeway, L. V. et al. ModM DNA methyltransferase methylome analysis reveals a potential role for Moraxella catarrhalis phasevarions in otitis media. FASEB J 28, 5197–5207, https://doi.org/10.1096/fj.14-256578 (2014).
Wion, D. & Casadesus, J. N6-methyl-adenine: an epigenetic signal for DNA-protein interactions. Nat Rev Microbiol 4, 183–192, https://doi.org/10.1038/nrmicro1350 (2006).
Kwiatek, A., Bacal, P., Wasiluk, A., Trybunko, A. & Adamczyk-Poplawska, M. The dam replacing gene product enhances Neisseria gonorrhoeae FA1090 viability and biofilm formation. Front Microbiol 5, 712, https://doi.org/10.3389/fmicb.2014.00712 (2014).
Cantalupo, G. et al. Evolution and function of the neisserial dam-replacing gene. FEBS Lett 495, 178–183, https://doi.org/10.1016/S0014-5793(01)02388-2 (2001).
Unal, C. M. & Steinert, M. Microbial peptidyl-prolyl cis/trans isomerases (PPIases): virulence factors and potential alternative drug targets. Microbiol Mol Biol Rev 78, 544–571, https://doi.org/10.1128/MMBR.00015-14 (2014).
Sedgwick, B. Repairing DNA-methylation damage. Nat Rev Mol Cell Biol 5, 148–157, https://doi.org/10.1038/nrm1312 (2004).
Adamczyk-Poplawska, M., Kondrzycka, A., Urbanek, K. & Piekarowicz, A. Tetra-amino-acid tandem repeats are involved in HsdS complementation in Type IC restriction-modification systems. Microbiology 149, 3311–3319, https://doi.org/10.1099/mic.0.26497-0 (2003).
Kwiatek, A., Kobes, M., Olejnik, K. & Piekarowicz, A. DNA methyltransferases from Neisseria meningitidis and Neisseria gonorrhoeae FA1090 associated with mismatch nicking endonucleases. Microbiology 150, 1713–1722, https://doi.org/10.1099/mic.0.27011-0 (2004).
Loenen, W. A., Dryden, D. T., Raleigh, E. A. & Wilson, G. G. Type I restriction enzymes and their relatives. Nucleic Acids Res 42, 20–44, https://doi.org/10.1093/nar/gkt847 (2014).
Kong, H. Analyzing the functional organization of a novel restriction modification system, the BcgI system. J Mol Biol 279, 823–832, https://doi.org/10.1006/jmbi.1998.1821 (1998).
Smith, R. M., Jacklin, A. J., Marshall, J. J., Sobott, F. & Halford, S. E. Organization of the BcgI restriction-modification protein for the transfer of one methyl group to DNA. Nucleic Acids Res 41, 405–417, https://doi.org/10.1093/nar/gks1000 (2013).
Tan, A. et al. Distribution of the Type III DNA methyltransferases modA, modB and modD among Neisseria meningitidis genotypes: implications for gene regulation and virulence. Sci Rep 6, 21015, https://doi.org/10.1038/srep21015 (2016).
Kwiatek, A., Luczkiewicz, M., Bandyra, K., Stein, D. C. & Piekarowicz, A. Neisseria gonorrhoeae FA1090 carries genes encoding two classes of Vsr endonucleases. J Bacteriol 192, 3951–3960, https://doi.org/10.1128/JB.00098-10 (2010).
Roberts, R. J., Vincze, T., Posfai, J. & Macelis, D. REBASE–a database for DNA restriction and modification: enzymes, genes and genomes. Nucleic Acids Res 43, D298–299, https://doi.org/10.1093/nar/gku1046 (2015).
Piekarowicz, A., Klyz, A., Kwiatek, A. & Stein, D. C. Analysis of Type I restriction modification systems in the Neisseriaceae: genetic organization and properties of the gene products. Mol Microbiol 41, 1199–1210, https://doi.org/10.1046/j.1365-2958.2001.02587.x (2001).
Price, C., Lingner, J. & Bickle, T. A. Basis for changes in DNA recognition by the EcoR124 and EcoR124/3 Type I DNA restriction and modification enzymes. J Mol Biol 205, 115–125, https://doi.org/10.1016/0022-2836(89)90369-0 (1989).
Clark, T. A. et al. Enhanced 5-methylcytosine detection in single-molecule, real-time sequencing via Tet1 oxidation. BMC Biol 11, 4, https://doi.org/10.1186/1741-7007-11-4 (2013).
Cohen, H. M., Tawfik, D. S. & Griffiths, A. D. Promiscuous methylation of non-canonical DNA sites by HaeIII methyltransferase. Nucleic Acids Res 30, 3880–3885, https://doi.org/10.1093/nar/gkf507 (2002).
Bucci, C. et al. Hypermutation in pathogenic bacteria: frequent phase variation in meningococci is a phenotypic trait of a specialized mutator biotype. Mol Cell 3, 435–445, https://doi.org/10.1016/s1097-2765(00)80471-2 (1999).
Low, D. A., Weyand, N. J. & Mahan, M. J. Roles of DNA adenine methylation in regulating bacterial gene expression and virulence. Infect Immun 69, 7197–7204, https://doi.org/10.1128/IAI.69.12.7197-7204.2001 (2001).
Unemo, M. et al. The novel 2016 WHO Neisseria gonorrhoeae reference strains for global quality assurance of laboratory investigations: phenotypic, genetic and reference genome characterization. J Antimicrob Chemother 71, 3096–3108, https://doi.org/10.1093/jac/dkw288 (2016).
Koren, S. et al. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27, 722–736, https://doi.org/10.1101/gr.215087.116 (2017).
Rice, P., Longden, I. & Bleasby, A. EMBOSS: the European Molecular Biology Open Software Suite. Trends Genet 16, 276–277, https://doi.org/10.1016/s0168-9525(00)02024-2 (2000).
R Development Core Team, R. l. R: A Language and Environment for Statistical Computing (2008).
Miller, R. G. Simultaneous statistical inference, 2nd edition (ed New York: Springer-Verlag), https://doi.org/10.1007/978-1-4613-8122-8 (1981).
Croucher, N. J. et al. Diversification of bacterial genome content through distinct mechanisms over different timescales. Nat Commun 5, 5471, https://doi.org/10.1038/ncomms6471 (2014).
Eddy, S. R. Accelerated Profile HMM Searches. PLoS Comput Biol 7, e1002195, https://doi.org/10.1371/journal.pcbi.1002195 (2011).
Finn, R. D. et al. The Pfam protein families database: towards a more sustainable future. Nucleic Acids Res 44, D279–285, https://doi.org/10.1093/nar/gkv1344 (2016).
Hadfield, J. et al. Phandango: an interactive viewer for bacterial population genomics. Bioinformatics, 1–2, https://doi.org/10.1093/bioinformatics/btx610 (2017).
Gouy, M., Guindon, S. & Gascuel, O. SeaView version 4: A multiplatform graphical user interface for sequence alignment and phylogenetic tree building. Mol Biol Evol 27, 221–224, https://doi.org/10.1093/molbev/msp259 (2010).
Carver, T., Harris, S. R., Berriman, M., Parkhill, J. & McQuillan, J. A. Artemis: an integrated platform for visualization and analysis of high-throughput sequence-based experimental data. Bioinformatics 28, 464–469, https://doi.org/10.1093/bioinformatics/btr703 (2012).
Johnson, M. et al. NCBI BLAST: a better web interface. Nucleic Acids Res 36, W5–9, https://doi.org/10.1093/nar/gkn201 (2008).
Castresana, J. Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17, 540–552, https://doi.org/10.1093/oxfordjournals.molbev.a026334 (2000).
Guindon, S. et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol 59, 307–321, https://doi.org/10.1093/sysbio/syq010 (2010).
Letunic, I. & Bork, P. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44, W242–W245, https://doi.org/10.1093/nar/gkw290 (2016).
Alexa, A. & Rahnenfuhrer, J. topGO: Enrichment Analysis for Gene Ontology. R package version 2.26.0. (2016).
Alexa, A., Rahnenfuhrer, J. & Lengauer, T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics 22, 1600–1607, https://doi.org/10.1093/bioinformatics/btl140 (2006).
Acknowledgements
This work was supported by Wellcome grant number 098051 (L.S.-B., S.R.H and J.P.) and the Foundation for Medical Research at Örebro University Hospital, Örebro, Sweden (D.G. and M.U.). We thank them for the financial support.
Author information
Authors and Affiliations
Contributions
All co-authors planned and designed the study. L.S.-B. and S.R.H. performed the bioinformatic analyses, with support from D.G., M.U. and J.P., L.S.-B. and S.R.H. drafted the manuscript and all co-authors commented and approved the final version.
Corresponding authors
Ethics declarations
Competing Interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Sánchez-Busó, L., Golparian, D., Parkhill, J. et al. Genetic variation regulates the activation and specificity of Restriction-Modification systems in Neisseria gonorrhoeae. Sci Rep 9, 14685 (2019). https://doi.org/10.1038/s41598-019-51102-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-019-51102-2
- Springer Nature Limited
This article is cited by
-
Genome-wide methylome analysis of two strains belonging to the hypervirulent Neisseria meningitidis serogroup W ST-11 clonal complex
Scientific Reports (2021)
-
Adaptation to the cervical environment is associated with increased antibiotic susceptibility in Neisseria gonorrhoeae
Nature Communications (2020)