
Abstract
The Kogelberg Biosphere Reserve in the Cape Floral Kingdom in South Africa is known for its unique plant biodiversity. The potential presence of unique microbial and viral biodiversity associated with this unique plant biodiversity led us to explore the fynbos soil using metaviromic techniques. In this study, metaviromes of a soil community from the Kogelberg Biosphere Reserve has been characterised in detail for the first time. Metaviromic DNA was recovered from soil and sequenced by Next Generation Sequencing. The MetaVir, MG-RAST and VIROME bioinformatics pipelines were used to analyse taxonomic composition, phylogenetic and functional assessments of the sequences. Taxonomic composition revealed members of the order Caudovirales, in particular the family Siphoviridae, as prevalent in the soil samples and other compared viromes. Functional analysis and other metaviromes showed a relatively high frequency of phage-related and structural proteins. Phylogenetic analysis of PolB, PolB2, terL and T7gp17 genes indicated that many viral sequences are closely related to the order Caudovirales, while the remainder were distinct from known isolates. The use of single virome which only includes double stranded DNA viruses limits this study. Novel phage sequences were detected, presenting an opportunity for future studies aimed at targeting novel genetic resources for applied biotechnology.
Similar content being viewed by others


Introduction
The Cape Floristic Region situated in the Western Cape province of South Africa is one of five Mediterranean-type ecosystems in the world1 and is recognized as one of the world’s biodiversity hotspots2. Fynbos (fine bush) is the main vegetation type of this region with the Proteaceae, Ericaceae and Restionaceae families dominating Kogelberg Biosphere Reserve Fynbos vegetation. Within this region, the fynbos comprises approximately 9000 plant species of which 70% are endemic to the region1,3. Fynbos vegetation types survive on highly heterogeneous, acidic, sandy, well-leached and infertile soils. The fynbos plants also survive invasions by foreign plants4 and seasonal drought conditions5.
Microorganisms make up a great proportion of the living population in the biosphere. They provide important ecosystem services in edaphic habitats6 and form complex symbiotic relationships with plants7. Plant-associated microorganism studies have shown high microbial diversity in fynbos soils2, where they play a role in sustaining plant communities8. A study focusing on the linkage between fynbos soil microbial diversity and plant diversity showed the presence of novel taxa and of bacteria specifically associated with the rhizospheric zone9. Studies on ammonium-oxidizing bacteria demonstrated that plant-species specific and monophyletic ammonium oxidizing bacterial clades were present in fynbos soils10, where abundance might be driven by the acidic and oligotrophic nature of these soils11. There is evidence that above-ground floral communities are implicated in shaping microbial communities12,13, and that some microbial clades show a high level of plant–host specificity10. This is consistent with the general concept of the mutualistic relationships between the plants and the microbial communities in fynbos soils14.
Soil-borne viruses, including phages, are of great importance in edaphic habitats due to their ability to transfer genes from host to host and as a potential cause of microbial mortality (leading to changes in turnover and concentration of nutrients and gases), processes that can profoundly influence the ecology of soil biological communities15. Virus diversity associated with fynbos plants from Kogelberg Biosphere Reserve fynbos soil has never been thoroughly investigated16. The difficulty of culturing viruses, which are absolutely dependent on a cell host to provide the apparatus for replication and production of progeny virions, presents a barrier to fully accessing viral biodiversity. This is a particular issue in poorly studied habitats, such as fynbos soil, where the true microbial (host) diversity is largely unknown and most microbial phylotypes have never been cultured17. The biodiversity and ecology of viruses in many soils therefore remain poorly investigated and poorly understood18.
Metaviromic surveys of terrestrial environments such as hot desert soil18, rice paddy soil19,20, Antarctic cold desert soil21,22 and hot desert hypolithic niche communities23 have been reported in recent years and have significantly advanced the field of soil viral ecology20,24. These studies have also facilitated the discovery of novel virus genomes20,22,23 and novel viral enzymes25.
However, surveys of viral diversity using NGS sequencing techniques in conjunction with metaviromic databases have focused principally on aquatic environments26,27,28. Studies on taxonomic composition using public metaviromic databases for viral diversity estimations have shown that a majority of environmental virus sequences are unknown19: ~70% of sequences have no homologs in public databases and are therefore typically labelled “viral dark matter”29,30. Bacteriophages constitute the largest known group of viruses found in both aquatic24,31 and soil environments32,33.
Here we report the first investigation of virus diversity in a unique soil type (fynbos soil) using metaviromic approaches. The metavirome of Kogelberg Biosphere Reserve fynbos soil was characterised in terms of diversity and functional composition and adds a new level of understanding to the exceptional biodiversity of this habitat.
Results and Discussion
Viral Morphology
Analysis of the morphology of viruses identified in Kogelberg Biosphere Reserve fynbos soil was carried out by transmission electron microscopy (TEM). TEM analysis of the virus preparations showed that the majority of the isolated virus particles were morphologically similar to known virus taxonomic groups34. The isolated virus particles from the fynbos soil were tailed, spherical or filamentous (Supplementary Fig. S1). Various particles with head-tail morphology, typically belonging to the families Myoviridae, Siphoviridae or Podoviridae, were observed.
These results are in a good agreement with previously published findings showing the high dominance of tailed phages in soils from various geographic areas24,33,35. The undetermined spherical or filamentous morphologies in TEM micrographs could be bona fide but uncharacterised viral structures. Spherical particles resembling capsid structures could be members of the Leviviridae, Partitiviridae, Chrysoviridae, Totiviridae or Tectiviridae families, or small plant viruses34. Filamentous particles may possibly correspond to the virus structures of the Inovirus genus, the members of which contain circular ssDNA within flexible filamentous virions. The presence of spherical types and filamentous type of virus particles was also reported for Delaware soils32. The aggressive extraction procedure used in the current study may have resulted in a high incidence of phage tail breakage and the generation of tailless phages36.
Metavirome Assembly
Assembly of the DNA sequence reads yielded 13,595 contigs larger than 500 bp, with an average length of 2,098 bp, accounting for a total of 28,526,478 bp (Table 1). Two different metagenomics pipelines; MetaVir37 and VIROME38, were used for analysis of the contigs, while MG-RAST39 was used for the analysis of the uploaded reads (Table 2). The MetaVir pipeline predicted 51,274 genes, with 5,338 affiliated contigs (i.e., contigs with at least one BLAST hit) and 7880 unaffiliated contigs (Table 2). MetaVir compares reads/contigs to complete viral genomes from the Refseq database and is specifically designed for the analysis of environmental viral communities37. The VIROME pipeline38 predicted 51,242 protein coding regions. Of these, 9555 were assigned as functional proteins, and 31,109 were unassigned (Table 2). Comparisons of functional and taxonomic analysis between Virome and MetaVir indicate that many of the predicted genes were overlapping between the two pipelines with MetaVir on average having a higher predictive potential (Supplementary Table S1). The MG-RAST pipeline predicted 2,555,524 protein coding regions. Of these predicted protein features, 119,220 were assigned a functional annotation using protein databases (M5NR)40 and 2,362,076 had no significant similarities to sequences in the protein databases (ORFans). MG-RAST core analysis and annotation depends heavily on the SEED database which is largely comprised of bacterial and archaeal genomes41. The majority of the annotated sequences in MG-RAST were mapped to bacterial genomes. This high percentage of bacterial sequences in metaviromes may be due to the presence of unknown prophages in bacterial genomes, phages carrying host genes, relatively large size of bacterial genomes compared to viral genomes and larger size of the microbial genome database which is statistically increasing the chance of matching bacterial sequences. The MG-RAST pipeline was used to analyse the reads, not the contigs and shows, therefore a higher number of predicted features, including more partial CDSs42 No rDNA sequences were found with the MG-RAST and VIROME pipelines, confirming the viral origins of the DNA. The fact that more than 80% of the hits in this study, consistent with previous viral metagenomics studies31,43,44, were assigned as hypothetical proteins derived from unknown viruses suggests the presence of a substantial pool of novel viruses.
Viral Diversity Estimation And Taxonomic Composition
The rarefaction curve computed by MG-RAST showed 3952 species clusters at 90% sequence identity for the 3,095,000 reads. The curve did not reach an asymptote (Fig. 1), although extrapolation suggested that approximately 78% of the viral diversity was covered by the metavirome sequence dataset.

Rarefaction curve of the Kogelberg Biosphere Reserve fynbos soil metavirome. Clustering was set at 90% similarity.
MetaVir was used for viral taxonomic composition analysis of the contigs. The taxonomic composition was computed from a BLASTp comparison of the predicted proteins in the contigs with the Viral Refseq protein database (release of 2016-01-19). The results revealed that 37.6% of the contigs represented a significant hit (threshold of 50 on the BLAST bit score). MetaVir identified 18 virus families, in which prokaryotic viruses were the most abundant and dominated by the order Caudovirales, consistent with the TEM observations. The relative abundance ranking of the different families was as follows: tailed bacteriophage families Siphoviridae > Myoviridae > Podoviridae, followed by the algae-infecting family Phycodnaviridae, the archaeal virus family Ampullaviridae and the amoeba-infecting family Mimiviridae (Table 3). Surprisingly, large viruses belonging to the families Phycodnaviridae and Mimiviridae were detected, which should have been removed during the filtration process due to the use of a 0.22-µm filtration step to remove bacterial cells. The identification of Mimiviridae suggests that this filtration process allowed partial mimivirus particles or free-floating DNA to pass through the membrane. Mimiviruses appear to infect only species of Acanthamoeba, which are ubiquitous in nature and have been isolated from diverse environments including freshwater lakes, river waters, salt water lakes, sea waters, soils and the atmosphere35,45,46,47. This suggests the existence of Mimivirus relatives in the KBR soil.
Other viral families and unclassified viruses (dsDNA and ssDNA) were found in low numbers. Putative contamination of Enterobacteria phage phiX174 was also detected in our metavirome sequences. This phage is used for quality control in sample preparation for high-throughput sequencing. Seven sequences from this dataset are similar to the phiX174 genome and were thus disregarded in the taxonomic composition as an artefact of sample processing. Plant viruses were not identified in the dataset, most probably because the majority of plant viruses are RNA viruses which were not sampled in this study.
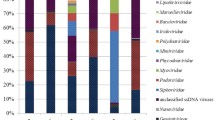
The viral composition of Kogelberg Biosphere Reserve fynbos soil was compared to 12 previously published metaviromes from both similar and dissimilar environments, including fresh water28, soil and hypolithic niche communities22,23, pond water27 and sea water48 (Fig. 2). A comparative metaviromics approach was used to investigate the assumption that certain environments will select for specific viruses49,50.

Comparison of the Kogelberg Biosphere Reserve metavirome taxonomic composition with selected publically available metaviromes. Abundances normalized according to predicted genome size with the GAAS tool. Blue colour represents 0.000 taxon, yellow represents 0.01–19.00, mustard represents 20.00–29.00, light red represents 30.00–49.00, and red represents 50.00–100.00 taxon. More details on the description of metaviromes are described in Supplementary Table S3.
The Caudovirales taxon dominated all metaviromes. In particular, members of the family Siphoviridae were dominant in most metaviromes except for some of the freshwater samples, in which myoviruses were dominant. Within the dsDNA viruses, members of rare taxonomic groupings such as the genera Tectivirus, Asfivirus and Salterprovirus, the families Mimiviridae, Iridoviridae, Marselleviridae, Nudiviridae, Poxviridae and Baculoviridae and the order Herpesvirales were detected in soil samples as well as in hypolith, deep sea, and freshwater metaviromes. Archaeal virus signatures belonging to the family Ampullaviridae have been observed only in the Kogelberg Biosphere Reserve fynbos soil. This family contains viruses with pleomorphic morphologies and a dsDNA genome, and the type species infects the thermoacidophile Acidianus convivator, isolated from Italian hot springs51. Fresh Water Lake, Antarctic soil and coral metaviromes showed a high abundance of ssDNA viruses, results possibly biased by the use of phi29 polymerase amplification (MDA) of the metaviromic DNA during library construction. The amplification of metaviromic DNA using phi29 polymerase amplification (Multiple Displacement Amplification) has been reported to be biased towards ssDNA templates19. It is notable, however, that a high abundance of ssDNA viruses has been observed in beach freshwater samples52, where amplification was not used in the preparation of metagenomic DNA. However, in general, other metaviromes which were not amplified using MDA showed a very low number of ssDNA viruses. In general, soils or soil-associated habitats seem to harbour relatively fewer ssDNA viruses and more tailed phages than aquatic ecosystems.
Consistent with other data22,24,43, it was found that bacteriophage sequences in Kogelberg Biosphere Reserve fynbos soil made up the majority of the virus fraction. Bacteriophages are common in the environment and are the dominant viral type recovered from metaviromics analyses in soil environments18,20,23,30. This finding was not surprising, given the observations from previous studies35,53 which showed high prokaryotic abundances in the Kogelberg soil environment. Nevertheless, signature sequences from large dsDNA eukaryotic virus families such as Mimiviridae 54 were represented in the Kogelberg Biosphere Reserve library despite the use of small pore size filters in sample preparation. Mimivirus signatures have been reported previously in other soil habitats22. Sequences that were found to be most similar to mimivirus ORFs were also obtained from Sargasso sea water samples, suggesting that these viruses, and their hosts, have a rather cosmopolitan distribution46.
Phylogeny Of The Kogelberg Biosphere Reserve Fynbos Soil Metavirome
Specific markers targeting virus families or species were used to analyse the taxonomic affiliations of the annotated ORFs and analyse the diversity within the group (reviewed in ref.55). Phylogenetic trees were drawn from metavirome sequences on the basis of homology to marker gene reference sequences from the PFAM database. Sequences homologous to the marker genes (polB, polB2, T7gp17 and terL (Supplementary Fig. S2, S3, S4 and S5) and reference sequences were used to draw phylogenetic trees.
Using the DNA polymerase family B (polB) marker gene, conserved in all dsDNA viruses, Kogelberg Biosphere Reserve sequences appeared to be distantly related to Rhodothermus phage RM378 (order Caudovirales, family Myoviridae). This phage is the only sequenced representative of the “Far T4” group of myoviruses (i.e., distantly related to Escherichia virus T4) found in a previous diversity analysis of sequences from French lakes28. The Kogelberg polB sequences from this study as well as the gp23 and gp20 marker gene sequences from the French lake study contribute to the expansion of the “Far T4”-like phages dataset.
A DNA polymerase family B (polB2) marker gene, which is conserved in members of Adenoviridae, Salterprovirus, and Ampullaviridae and Podoviridae family viral groups, was analysed. The analysis showed a separate clade of sequences from the Kogelberg Biospheres reserve soil samples. Other polB2 sequences from our dataset were found to be distantly related to members of the Adenoviridae family (isolated from a wide range of animal sources), the Podoviridae family (such as Mycoplasma phage P1, Clostridium phage phi24R, Bacillus phages B103, phi39, Ga1), the Ampullaviridae family (such as Acidianus-bottle-shaped virus) and the Tectiviridae family (such as Bacillus phages G1L16C, Bam35C and AP50).
Analysis of the metavirome sequence database using the marker gene T7gp17 showed the presence of members of the Podoviridae family, subfamily Autographivirinae and genus Phikmvvirus and T7virus. Members of the genus phikmvvirus such as Pseudomonas phage LKA1, and unclassified phiKMV phages such as Ralstonia phage RSB1, were found to be closely related to the Kogelberg Biosphere Reserve sequences. Currently unclassified members of the genus T7virus, such as Klebsiella phage K11 and Yersinia phage φYeO3–12, were also found to be closely related to sequences in the Kogelberg Biosphere Reserve metavirome. The phages in the subfamily Autographivirinae are known to infect a wide range of environmentally important bacteria56.
Tailed phages of the order Caudovirales were the most commonly observed DNA viruses in the Kogelberg Biosphere Reserve sequences, consistent with other environmental samples23,33,57. A phylogenetic tree built from a Caudovirales-specific terminase large subunit marker gene (terL) was used to visualise the diversity of the Kogelberg Biosphere Reserve fynbos soil Caudovirales (Fig. 3). The Kogelberg Biosphere Reserve sequences clustered with all three families of tailed phages, indicating high phage richness in our sample set. These results were consistent with the taxonomic affiliations of contigs in the virus families shown in Table 3.

terL phylogenetic tree. Viral sequence origin of Caudovirales indicated with different colours on the contigs names. Kogelberg Biosphere Reserve fynbos soil - Red, Siphoviridae - green, Myoviridae - purple, Podoviridae - blue, unclassified viruses - grey.
Analysis of A Near-Complete Phage Genome
MetaVir assemblies predicted 352 genes from the 6 contigs larger than 40 kb, as well as 758 genes predicted from 19 contigs of between 20 kb and 40 kb. The 6 largest contigs were predicted to be linear, double stranded genomes. The sizes of the genomes were predicted to be 47 kb long with 63 genes for the largest contig (Fig. 4), followed by 44 kb with 58 genes, 42 kb with 61 genes, 42 kb with 53genes, 40 kb with 68 genes and 40 kb with 49 genes. The genes in these contigs were predicted to show similarity to members of the order Caudovirales.

Gene annotation of contig 414. Arrowed blocks are open reading frames (ORFs), showing their orientation. Numbers within the contiguous genome are nucleotide positions, starting within gene number 1 and onwards in a clockwise orientation.
The largest contig represents a near-complete phage genome in the family Podoviridae. Members of this family typically contain double stranded and linear genomes of around 40–45 kb in length with approximately 55 genes58. Four of the genes in this assembled genome (genes 15, 16, 34 and 41) showed similarity to members of both Podoviridae and Siphoviridae families. The translated products of two of these genes (15 and 16) were identified as putative terminase large subunit (gene 15) and terminase small subunit (gene 16) genes, with 88% and 89% amino acid identity to Puniceispirillum phage HMO-2011 and Pseudomonas phage vB_PaeP_Tr60_Ab31, respectively. Both Puniceispirillum phage HMO-2011 and Pseudomonas phage vB_PaeP_Tr60_Ab31 belong to the family Podoviridae. The terL phylogenetic tree (Supplementary Fig. S4) showed a distant relatedness to members of the Podoviridae clade. Both terminase large and small subunits, together termed the terminase complex, are involved in the cleavage and packaging of concatemeric phage dsDNA59. The large terminase subunit is involved in DNA cleavage and translocation into the procapsid while the small terminase subunit is involved in packaging initiation and stimulation of the ATPase activity of the large terminase. These DNA packaging mechanisms are used by most members of the Caudovirales.
The translated product of gene 34 was identified as a putative ERF superfamily protein and showed 55% amino acid identity to a homologue encoded by the unclassified Clostridium phage phiCP34O (order Caudovirales, family Siphoviridae). The ERF superfamily proteins are involved in the recombination of phage genomes60. The translated product of gene 41 was identified as a putative gp77 and showed 95% amino acid similarity to a homologue encoded by Mycobacterium phage Che9d (order Caudovirales, family Siphoviridae, genus Che8likevirus). gp77 proteins are known to function as shut-off genes during early stages of phage replication61.
Fifty nine of the translated products of genes in the assembled phage genome showed identity to hypothetical proteins. Of these hypothetical proteins, 56 showed no sequence similarity to known virus families in BLASTp comparison to the RefseqVirus protein database. Three of the genes were predicted to encode glucosaminidase (a hydrolytic enzyme), Phage integrase (a site-specific recombinase that mediates controlled DNA integration and excision) and PDDEXK_1 (nuclease superfamily). Members of this PDDEXK_1 family belong to the PD-(D/E) XK nuclease superfamily. The PD-(D/E)XK nuclease superfamily contains type II restriction endonucleases and many other enzymes involved in DNA recombination and repair62.
The protein sequences identified in this analysis indicated the presence of a putative ERF superfamily protein, Phage integrase and PDDEXK_1 family; all proteins implicated in DNA recombination. The ERF superfamily protein encoded by gene 34, whose sequences are expressed during recombination of temperate phages, catalyses annealing of single-stranded DNA chains and pairing of ssDNA with homologous dsDNA, which may function in RecA-dependent and RecA-independent DNA recombination pathways63.
A few large contigs contained some predicted ORFs with similarities to phage sequences and coding for specific conserved phage proteins, including terminases, structural proteins (mainly related to Caudovirales tail structures) and phage DNA polymerases (Supplementary Table S2).
Cluster analysis
Contig datasets from nine metaviromes from various aquatic and soil habitats were selected for dinucleotide frequency comparisons64.
A comparison of the dinucleotide frequencies of the 9 metaviromes shows a clear bimodal clustering (Fig. 5). Group 1, composed of soil-associated habitat and deep sea sediment metaviromes, is further subdivided into soil, hypolith and sediments clades. Group 2 was restricted to freshwater habitats. The Arctic and Atlantic deep sea sediment and freshwater lake28 metaviromes clustered in single independent nodes. Such clustering reflects significant genetic similarity between these metaviromes, despite the geographical distances between sample locations.

Hierarchical clustering of nine metaviromes (assembled into contigs) based on dinucleotide frequencies. The types of biome are differentiated by colour with Kogelberg Biosphere Reserve - red, freshwater - dark green, hyperarid desert - light blue, hypersaline - yellow, hypolith - dark blue, seawater - light green and unknown biomes - gold. The x-axis denotes eigenvalues distances. The tree was constructed using MetaVir server pipeline according to the method in64. More details on sample names are described in supplementary Table S3.
Both hypolithic metaviromes (i.e., cold Antarctic and hot Namib Desert hypolithic biomass samples) clustered as a single node, despite their widely differing habitat-associated environmental characteristics (dominated by an est. 50 °C mean annual temperature difference) and substantial spatial separation (approx. 55 degrees of latitude), suggesting that aridity and not temperature may be the dominant driver of host and viral diversity22,65. Interestingly, soil related metaviromes (from Kogelberg Biosphere Reserve fynbos soil, Peruvian rainforest soil and Antarctic Dry Valley desert soil) clustered together and were clearly distinct from soils which were geographically much closer.
The Kogelberg Biosphere Reserve soil metavirome clustered at a single sub-node with the Peruvian rainforest soil metavirome. Both of these habitats experience high annual rainfall and warm temperatures and are characterised by heavily leached and low nutrient status soils, suggesting that soil composition and/or nutrient status may be the strong driver of the host and viral diversity66,67. These observations suggest a niche-dependent pattern, where spatially distinct niche environments cluster together and separate from their geographically closer soil counterparts65.
Previous study reported that cluster analysis of hypolith and open soil metaviromes from Antarctic and Namib Desert soil has shown that both hypolith metaviromes clustered at a single node and also that both open soil metaviromes displayed an identical pattern65. Similarly to our study, related habitat types harboured more closely related viral communities, despite the great geographic distances or differing environmental conditions. The common factor in these hyperarid environments is water scarcity, which may be a key driver of community speciation and recruitment in these environments. We conclude that these adaptations and the nature of soil habitat compared to the ‘refuge’ habitat of quartz stones for hypolithic communities, may be the driving force between both communities not to cluster together.
Functional Properties of The Kogelberg Biosphere Reserve Fynbos Soil Metavirome
The functional implication of the reads was explored using MG-RAST. The Kogelberg Biosphere Reserve metavirome sequences exhibited a high proportion of uncharacterized ORFs, with 2,362,076 sequences showing no significant similarities to proteins in the databases (ORFans). Twelve functional categories were annotated by MG-RAST, each subdivided into distinct subsystems (Fig. 6). The database searches against SEED in the MG-RAST subsystem resulted in 9360 hits. The highest percentage hits (20.3%) in the functional annotation belonged to the “Phage, prophages, transposable elements and plasmids” subsystem category, with r1t-like streptococcal phages, phage packaging machinery and phage replication annotations most commonly identified.

Functional assignment of predicted ORFs. Functional annotation was performed at 60% similarity cut-off as predicted by MG-RAST.
The other functional subsystem categories showed “Clustering-based subsystems (e.g., biosynthesis of galactoglycans and related lipopolysaccharides; catabolism of an unclassified compound etc., and other clusters identified as unclassified). The “Protein metabolism” and “DNA metabolism” functional categories were also dominant annotations. Many proteins in these functional categories, such as terminases, HNH homing endonucleases, DNA helicases, DNA polymerases and DNA primases, could potentially be of phage origin. These functional groups have also been found to be highly represented in previous metaviromic datasets23,68,69.
Analysis of the metavirome reads using the KEGG Orthology (KO) database showed metabolism protein families (carbohydrate metabolism, amino acid metabolism and nucleotide metabolism) to be the most commonly identified. Members of the genetic information procession protein family, including replication and repair, transcription and translation proteins, were also commonly identified. Deeper analysis of a subset of annotated contigs identified genes encoding numerous virus structures (e.g., phage capsid, terminase, tail fibre protein etc.) and DNA manipulating enzymes (e.g., endonuclease, DNA methylase, primase-polymerase, DNA primase/helicase, DNA polymerase I, integrase, ssDNA annealing protein, exonuclease, transferase, site-specific DNA methylase, ligase, recombinase etc.).
From this analysis, we demonstrate that phage-related genes and metabolic genes are highly represented. The virome displayed a strong enrichment in phage-like genes (e.g. phages, prophages, transposable elements, plasmids) and lacked typical cellular categories rarely observed in sequenced phages (e.g. ‘cofactors, vitamins, prosthetic groups, pigments’). Cellular categories commonly identified in known phages were retrieved (e.g. ‘nucleosides and nucleotides’, ‘DNA metabolism’). The highly abundance of virome-associated metabolic genes shows that the phages may have the potential to interfere with the metabolism of their hosts. Our virome analysis, consistent with other virome studies, demonstrate the unexpected picture of global ‘viral’ metabolism, suggesting that viruses might actively dictate the metabolism of infected cells on a global scale69.
The functional assignments from the SEED database of Kogelberg Biosphere Reserve fynbos soil was clustered with SEED database functional assignments of the 12 previously published metaviromes from both similar and dissimilar environments (fresh water28, soil and hypolithic niche communities22,23, pond water27 and sea water48 mentioned in Fig. 2. A cluster analysis of the SEED database subsystem classification revealed different functional patterns between the metaviromes and no clear soil clustering (Fig. 7). The sequences from Kogelberg Biosphere Reserve clustered amongst the sequences from three of the fresh water lakes and the Namib hypolith metaviromes. Antarctic samples (Antarctic open soil and Antarctic hypolith) were more distinct and formed a heterogeneous clade with the other fresh water samples. This can be potentially be explained by the larger number of cellular contamination in some of these metaviromes. This finding suggests that different biomes can share similar functional patterns and, conversely, that taxonomically similar viromes can encode different functional genes. It may also indicate that certain phage groups are more prevalent in certain biogeographic regions.

Cluster analysis of functional assignment of predicted ORFs. Viromes were clustered with the hclust algorithm in R according to the abundance of SEED database functional categories present. SEED categories were assigned using Megan6 after blastp-based comparison with the non-redundant protein database of NCBI. More details on the description of metaviromes are described in Supplementary Table 2.
This study is not without limitations. The major limitation to this study is the use of only a single virome that includes only double stranded DNA viruses.
Conclusion
We have successfully used the metaviromics approach to explore the diversity and functional composition of a previously unexplored Kogelberg Biosphere Reserve fynbos soil virome. Our quantitative comparison of taxonomic and functional composition of the Kogelberg soil metavirome with other published viromes is a valuable and novel contribution that will enhance the repertoire of publicly available datasets and advance our understanding of viral ecology. Furthermore, contigs corresponding to novel virus genomes were assembled in the current work; this presents an opportunity for future studies aimed at targeting these novel genetic resources for applied biotechnology.
Experimental Design
Sample Site Location
Samples were collected from the Kogelberg Biosphere Reserve, situated to the east of Cape Town, South Africa in the Boland Mountains (GPS coordinates: 34°19′48′′.0 S, 18°57′21.0′′ E). Open soil samples were collected aseptically during the winter of 2014. Approximately 20 kg of soil was collected at depth of 0–4 cm. Soil samples were stored in sterile containers at −80 °C.
Sample Processing, Dna Extraction
Samples were collected in the open soil. Only 3 samples were collected. The DNA of these samples where pooled together for NGS sequencing. Soil samples were processed as previously described70 with some modifications. 8 kg of soil and 1X SM buffer (8 L) (0.1 M NaCl, 8 mM MgSO4, 50 mM Tris-HCl, pH 7.5) were mixed and shaken vigorously in a sterile container until soil was well suspended and left overnight at 4 °C to settle. The supernatant was centrifuged at 10000 g for 15 min to pellet any remaining soil particles and other debris and passed through a 0.22 µm filter (Millipore, streicup 500 ml). The filtrate was treated with DNase. Viral particles were precipitated with 10% (w/v) polyethylene glycol (PEG) 8000 overnight at 4 °C and centrifuged for 15 min at 11000 g. After removing the supernatant the viral pellet was resuspended in TE buffer, pH 7.670.
The absence of bacterial and eukaryotic DNA was confirmed by PCR with primers pairs E9F (5′-GAG TTT GAT CCT GGC TCA G-3′) and U1510R (5′-GGT TAC CTT GTT ACG ACT T-3′) and ITS1 (5′- TCCGTAGGT GAACCTGCGG-3′) and ITS4 (5′- TCCTCCGCTTATTGATATGC-3′)71.
Transmission Electron Microscopy
Aliquots of viral suspensions isolated from soil were fixed with 2% glutaraldehyde for three hours at 4 °C and 10 µl of the phage suspension was overlaid on a carbon coated grid of 200 Mesh72. The suspension was allowed to dry on the grid, which was then negatively stained with 2% uranyl acetate. Excess stain was removed using filter paper and allowed to air-dry prior to examination using a Philips (FEI) CM100 TEM.
Dna Extraction Sequencing
DNA was extracted from virus particle preparations using a ZR soil microbe DNA MidiPrepTM kit according to manufacturer’s instructions (Zymo Research). Extracted metaviromic DNA (unamplified) was sequenced using an Illumina MiSeq platform (Inqaba Biotechnical Industries). Briefly, following DNA quantification using NanoDrop Fluorospectrometer 3300, 1 ng of isolated metavirome DNA was used to prepare 4 individually indexed NexteraXT libraries. They were then sequenced using the MiSeq v3 (600 cycles) sequencing kit, generating 2 × 300 bp reads. The raw reads were trimmed and demultiplexed, resulting in four fastq files.
Sequence Data Analysis
The quality of the raw read files was checked with CLC Genomics Workbench version 6.0.1 (CLC, Denmark). The reds were then filtered and trimmed, with the removal of low quality (sequence limit of 0.05), ambiguous reads (maximal of 2 and minimum length of 15). This yielded 1,488,462,918 reads with an average length of 212.05 bp. The post-QC reads were assembled using CLC Genomics Workbench as paired files (3 × 2 read files per site). The assembly resulted in 28,511,204 contigs with a minimum length of 1,002 bases at an N50 of 2,047 and a maximum of 47,854 bases.
The processed reads were assembled de novo using CLC Genomics Workbench version 6.0.1 using the default settings. Reads and contigs were uploaded to the MetaVir37 (http://metavir-meb.univ-bpclermont.fr), VIROME (http://virome.dbi.udel.edu/)38 and MG-RAST (http://metagenomics.anl.gov/)39 servers for virus diversity estimations. The viromes were uploaded in 2015 and analysed in 2017. The taxonomic composition was computed from a BLAST comparison with the Refseq complete viral genomes protein sequence database from NCBI (release of 2016-01) using BLASTp with a threshold of 50 on the BLAST bitscore. The assembled sequences were searched for open reading frames (ORFs) and compared to the RefSeq complete viral database using MetaVir and MG-RAST. Functional and organism assignments were based on annotation and other information obtained from the following databases: GenBank, Integrated Microbial Genomes (IMG), Kyoto Encyclopaedia of Genes and Genomes (KEGG), Pathosystems Resource Integration Center (PATRIC), RefSeq, SEED, Swiss-Prot, tremble, and eggnog; and for the assignment of functional hierarchy, COG (clusters of orthologous groups), KEGG Orthology (KO), and NOG databases were used. The Genome relative Abundance and Average Size (GAAS)73 tools were used for normalization of the total composition, estimation of the mean genome length and for the estimation of relative abundance and size for each taxon. The phylogenetic tree were generated by an open-source JavaScript library called jsPhyloSVG74. The phylogenetic trees were based on the reference sequences and the Kogelberg Biosphere Reserve virome sequences, and computed with 100 bootstraps. Further analysis of the sequences was performed using METAGENassist (a web server that provides a broad range of statistical tools for comparative metagenomics)75. Functional assignments produced by VIROME using 120 identified functional subsystems were used for the statistical analysis with METAGENassist.
Clustering analysis comparison was plotted as a clustering tree and computed with pvclust computed by MetaVir (an R package for assessing the uncertainty in hierarchical clustering)76 (Fig. 6). Hierarchical clustering using dinucleotide comparisons was used to quantify the grouping behaviour of nine published metaviromes and the comparison were plotted and demonstrated as a clustering dendrograms. Only metaviromes containing more than 50,000 sequences and with an average sequence length of over 100 bp were used, as this comparison is based on a normalised virome sub-sample. Metaviromes that did not match these criteria were not listed for nucleotide composition bias comparison. Hence, only 9 metaviromes were suitable for comparison using dinucleotide frequencies in the MetaVir sever. The largest contigs were analysed by MetaVir. The SEED classification clustering of the 12 metaviromes was assessed using BLASTp against the nr database of NCBI (release 2017-05)77. Differences between the virome SEED functional components were transformed into a Bray Curtis dissimilarity matrix using the vegan package in RStudio, clustered using the hclust algorithm (method = average), and represented as a dendrogram78,79.
Data availability
Viral sequences from Kogelberg Biosphere Reserve fynbos soil sample are available on MetaVir under the project KBR under the names “KBR 1 and KBR 2”.
References
Bergh, E. W. & Compton, J. S. South African Journal of Botany A one-year post- fi re record of macronutrient cycling in a mountain sandstone fynbos ecosystem, South Africa. South African J. Bot. 97, 48–58 (2015).
Slabbert, E., Kongor, R. Y., Esler, K. J. & Jacobs, K. Microbial diversity and community structure in Fynbos soil. Mol. Ecol. 19, 1031–1041 (2010).
Van Wyk, A. E. & Smith, G. Regions of Floristic Endemism in Southern Africa. Umdaus 5–17 (2001).
Sprent, J. I. & Parsons, R. Nitrogen fixation in legume and non-legume trees. F. Crop. Res. 65, 183–196 (2000).
Mucina, L. & Wardell-Johnson, G. W. Landscape age and soil fertility, climatic stability, and fire regime predictability: beyond the OCBIL framework. Plant Soil 341 (2011).
Jeanbille, M. et al. Soil Parameters Drive the Structure, Diversity and Metabolic Potentials of the Bacterial Communities Across Temperate Beech Forest Soil Sequences. Microb. Ecol. 71, 482–493 (2016).
Rappé, M. S. & Giovannoni, S. J. The uncultured microbial majority. Annu. Rev. Microbiol. 57, 369–394 (2003).
van der Heijden, M. G. A., Bardgett, R. D. & van Straalen, N. M. The unseen majority: soil microbes as drivers of plant diversity and productivity in terrestrial ecosystems. Ecol. Lett. 11, 296–310 (2008).
Stafford, W. H. L., Baker, G. C., Brown, S. A., Burton, S. G. & Cowan, D. A. Bacterial diversity in the rhizosphere of Proteaceae species. Environ. Microbiol. 7, 1755–1768 (2005).
Ramond, J. B., Lako, J. D. W., Stafford, W. H. L., Tuffin, M. I. & Cowan, D. A. Evidence of novel plant-species specific ammonia oxidizing bacterial clades in acidic South African fynbos soils. J. Basic Microbiol. 55, 1040–1047 (2015).
Prosser, J. I. & Nicol, G. W. Relative contributions of archaea and bacteria to aerobic ammonia oxidation in the environment. Environ. Microbiol. 10, 2931–41 (2008).
Hamilton, E. W. & Frank, D. A. Can plants stimulate soil microbes and their own nutrient supply? Evidence from a grazing tolerant grass. Ecology 82, 2397–2402 (2001).
Nüsslein, K. & Tiedje, J. M. Soil bacterial community shift correlated with change from forest to pasture vegetation in a tropical soil. Appl. Environ. Microbiol. 65, 3622–6 (1999).
Keluskar, R., Nerurkar, A. & Desai, A. Mutualism between autotrophic ammonia-oxidizing bacteria (AOB) and heterotrophs present in an ammonia-oxidizing colony. Arch. Microbiol. 195, 737–747 (2013).
Kimura, M., Jia, Z. J., Nakayama, N. & Asakawa, S. Ecology of viruses in soils: Past, present and future perspectives. Soil Sci. Plant Nutr. 54, 1–32 (2008).
Cowan, D. A., Rybicki, E. P., Tuffin, M. I., Valverde, A. & Wingfield, M. J. Biodiversity: So much more than legs and leaves. S. Afr. J. Sci. 109, 1–9 (2013).
Schoenfeld, T. et al. Functional viral metagenomics and the next generation of molecular tools. Trends Microbiol. 18, 20–9 (2010).
Zablocki, O., Adriaenssens, E. M. & Cowan, D. Diversity and Ecology of Viruses in Hyperarid Desert Soils. Appl Env. Microbio 82, 770–777 (2016).
Kim, K. H. et al. Amplification of uncultured single-stranded DNA viruses from rice paddy soil. Appl. Environ. Microbiol. 74, 5975–5985 (2008).
Kim, M.-S., Whon, T. W. & Bae, J.-W. Comparative viral metagenomics of environmental samples from Korea. Genomics Inform. 11, 121–8 (2013).
Srinivasiah, S. et al. Direct assessment of viral diversity in soils by random PCR amplification of polymorphic DNA. Appl. Environ. Microbiol. 79, 5450–7 (2013).
Zablocki, O. et al. High-Level Diversity of Tailed Phages, Eukaryote-Associated Viruses, and Virophage-Like Elements in the Metaviromes of Antarctic Soils. Appl. Environ. Microbiol. 80, 6888–6897 (2014).
Adriaenssens, E. M. et al. Metagenomic analysis of the viral community in Namib Desert hypoliths. Environ. Microbiol. 17, 480–495 (2015).
Fancello, L. et al. Viruses in the desert: a metagenomic survey of viral communities in four perennial ponds of the Mauritanian Sahara. ISME J. 7, 359–69 (2013).
Gudbergsdóttir, S. R., Menzel, P., Krogh, A., Young, M. & Peng, X. Novel viral genomes identified from six metagenomes reveal wide distribution of archaeal viruses and high viral diversity in terrestrial hot springs. Environ. Microbiol. n/a-n/a, https://doi.org/10.1111/1462-2920.13079 (2015).
Breitbart, M. Marine viruses: truth or dare. Ann. Rev. Mar. Sci. 4, 425–48 (2012).
Rodriguez-Brito, B. et al. Viral and microbial community dynamics in four aquatic environments. Isme J 4, 739–751 (2010).
Roux, S. et al. Assessing the Diversity and Specificity of Two Freshwater Viral Communities through Metagenomics. PLoS One 7, e33641 (2012).
Roux, S., Hallam, S. J., Woyke, T. & Sullivan, M. B. Viral dark matter and virus-host interactions resolved from publicly available microbial genomes. Elife 4 (2015).
Hatfull, G. F. Dark matter of the biosphere: The amazing world of bacteriophage diversity. J. Virol. 89, 8107–8110 (2015).
Alhamlan, F. S., Ederer, M. M., Brown, C. J., Coats, E. R. & Crawford, R. L. Metagenomics-based analysis of viral communities in dairy lagoon wastewater. J. Microbiol. Methods 92, 183–8 (2013).
Williamson, K. E., Radosevich, M. & Wommack, K. E. Abundance and diversity of viruses in six Delaware soils. Appl. Environ. Microbiol. 71, 3119–3125 (2005).
Reavy, B. et al. Distinct circular single-stranded DNA viruses exist in different soil types. Appl. Environ. Microbiol. 81, 3934–3945 (2015).
Ackermann, H. W. 5500 Phages examined in the electron microscope. Arch. Virol. 152, 227–243 (2007).
Zablocki, O., Adriaenssens, E. & Cowan, D. Diversity and ecology of viruses in hyperarid desert soils. Appl. Environ. Microbiol. 82, AEM.02651–15 (2015).
Ackermann, H.-W. Classification of bacteriophages. The bacteriophages 2, 8–16 (2006).
Roux, S., Tournayre, J., Mahul, A., Debroas, D. & Enault, F. Metavir 2: new tools for viral metagenome comparison and assembled virome analysis. BMC Bioinformatics 15, 76 (2014).
Wommack, K. E. et al. VIROME: a standard operating procedure for analysis of viral metagenome sequences. Stand. Genomic Sci. 6, 427 (2012).
Meyer, F. et al. The metagenomics RAST server - a public resource for the automatic phylogenetic and functional analysis of metagenomes. BMC Bioinformatics 9, 386 (2008).
Wilke, A. et al. The M5nr: a novel non-redundant database containing protein sequences and annotations from multiple sources and associated tools. BMC Bioinformatics 13, 141 (2012).
Overbeek, R., Disz, T. & Stevens, R. The SEED: A peer-to-peer environment for genome annotation. Commun. ACM 47, 47–51 (2004).
Mohiuddin, M. & Schellhorn, H. E. Spatial and temporal dynamics of virus occurrence in two freshwater lakes captured through metagenomic analysis. Front. Microbiol. 6, 960 (2015).
Breitbart, M. et al. Genomic analysis of uncultured marine viral communities. Proc. Natl. Acad. Sci. US.A. 99, 14250–5 (2002).
Cann, A. J., Fandrich, S. E. & Heaphy, S. Analysis of the virus population present in equine faeces indicates the presence of hundreds of uncharacterized virus genomes. Virus Genes 30, 151–156 (2005).
Gascuel, O. et al. BIONJ: an improved version of the NJ algorithm based on a simple model of sequence data. Mol. Biol. Evol. 14, 685–695 (1997).
Ghedin, E. & Claverie, J.-M. M. Mimivirus relatives in the Sargasso sea. Virol. J. 2, 62 (2005).
Short, S. M. & Short, C. M. Diversity of algal viruses in various North American freshwater environments. Aquat. Microb. Ecol. 51, 13–21 (2008).
Angly, F. E. et al. The marine viromes of four oceanic regions. PLoS Biol. 4, e368 (2006).
Dinsdale, E. A. et al. Functional metagenomic profiling of nine biomes. Nature 452, 629–632 (2008).
de Wit, R. & Bouvier, T. ‘Everything is everywhere, but, the environment selects’; what did Baas Becking and Beijerinck really say? Environ. Microbiol. 8, 755–758 (2006).
Häring, M., Rachel, R., Peng, X., Garrett, R. A. & Prangishvili, D. Viral Diversity in Hot Springs of Pozzuoli, Italy, and Characterization of a Unique Archaeal Virus, Acidianus Bottle-Shaped Virus, from a New Family, the Ampullaviridae. J Virol 79, 9904–9911 (2005).
Watkins, S. C. et al. Assessment of a metaviromic dataset generated from nearshore Lake Michigan. Mar. Freshw. Res. 67, 1700 (2016).
Ashelford, K. E., Day, M. J. & Fry, J. C. Elevated abundance of bacteriophage infecting bacteria in soil. Appl. Environ. Microbiol. 69, 285–289 (2003).
Raoult, D., La Scola, B. & Birtles, R. The discovery and characterization of Mimivirus, the largest known virus and putative pneumonia agent. Clin. Infect. Dis. 45, 95–102 (2007).
Adriaenssens, E. M. & Cowan, D. A. Using signature genes as tools to assess environmental viral ecology and diversity. Appl. Environ. Microbiol. 80, 4470–4480 (2014).
Adriaenssens, E. M. et al. Bacteriophages LIMElight and LIMEzero of Pantoea agglomerans, belonging to the ‘phiKMV-Like Viruses’. Appl. Environ. Microbiol. 77, 3443–3450 (2011).
Roux, S. et al. Analysis of metagenomic data reveals common features of halophilic viral communities across continents. Environ. Microbiol. 18, 889–903 (2015).
Hulo, C. et al. ViralZone: A knowledge resource to understand virus diversity. Nucleic Acids Res. 39, 576–582 (2011).
Penadés, J. R., Chen, J., Quiles-Puchalt, N., Carpena, N. & Novick, R. P. Bacteriophage-mediated spread of bacterial virulence genes. Curr. Opin. Microbiol. 23, 171–178 (2015).
Wittmann, J., Gartemann, K.-H., Eichenlaub, R. & Dreiseikelmann, B. Genomic and molecular analysis of phage CMP1 from Clavibacter michiganensis subspecies michiganensis. 1, 6–14 (2011).
Rybniker, J., Plum, G., Robinson, N., Small, P. L. & Hartmann, P. Identification of three cytotoxic early proteins of mycobacteriophage L5 leading to growth inhibition in Mycobacterium smegmatis. Microbiology 154, 2304–2314 (2008).
Letunic, I. et al. SMART 4.0: towards genomic data integration. Nucleic Acids Res. 32, 142D–144 (2004).
Dziewit, L., Oscik, K., Bartosik, D. & Radlinska, M. Molecular characterization of a novel temperate sinorhizobium bacteriophage, ФLM21, encoding DNA methyltransferase with CcrM-like specificity. J. Virol. 88, 13111–24 (2014).
Willner, D., Thurber, R. V. & Rohwer, F. Metagenomic signatures of 86 microbial and viral metagenomes. Environ. Microbiol. 11, 1752–1766 (2009).
Zablocki, O. et al. Niche-dependent genetic diversity in Antarctic metaviromes. Bacteriophage 4, e980125 (2014).
Leigh, E. G. Structure and climate in tropical rain forest. Source Annu. Rev. Ecol. Syst. 6, 67–86 (1975).
Fry, M. A detailed characterization of soils under different Fynbos-climate-geology combinations in the south-western Cape. (1987).
Cai, L., Zhang, R., He, Y., Feng, X. & Jiao, N. Metagenomic Analysis of Virioplankton of the Subtropical Jiulong River Estuary, China. Viruses 8, 35 (2016).
Roux, S., Krupovic, M., Debroas, D. & Forterre, P. Assessment of viral community functional potential from viral metagenomes may be hampered by contamination with cellular sequences. Open Bio 3 (2013).
Casas, V. & Rohwer, F. Phage metagenomics. Methods Enzymol. 421, 259–268 (2007).
Merseguel, K. B. et al. Genetic diversity of medically important and emerging Candida species causing invasive infection. BMC Infect. Dis. 15, 57 (2015).
Ackermann, H.-W. In Bacteriophages 113–126 (Springer, 2009).
Angly, F. E. et al. The GAAS metagenomic tool and its estimations of viral and microbial average genome size in four major biomes. PLoS Comput. Biol. 5, e1000593 (2009).
Smits, S. A. & Ouverney, C. C. jsPhyloSVG: A Javascript Library for Visualizing Interactive and Vector-Based Phylogenetic Trees on the Web. PLoS One 5, e12267 (2010).
Arndt, D. et al. METAGENassist: A comprehensive web server for comparative metagenomics. Nucleic Acids Res. 40, 1–8 (2012).
Suzuki, R. & Shimodaira, H. Pvclust: An R package for assessing the uncertainty in hierarchical clustering. Bioinformatics 22, 1540–1542 (2006).
Aziz, R. K. et al. SEED Servers: High-Performance Access to the SEED Genomes, Annotations, and Metabolic Models. PLoS One 7, e48053 (2012).
Oksanen J, Blanchet FG, Kindt R, L. P. and others. ‘vegan’ 2.3.4.4— community ecology package, http://CRAN.R-project.org/package=vegan (2016).
Racine, J. S. RStudio: A Platform-Independent IDE for R and Sweave. J. Appl. Econom. 27, 167–172 (2012).
Acknowledgements
We gratefully acknowledge financial support from National Research Foundation (NRF), the Department of Science and Technology, the University of Pretoria Genomics Research Institute, the Claude Leon Foundation (to EMA), Cape Nature (Kogelberg Biosphere reserve) and the Council for Scientific and Industrial Research (CSIR). We declare no conflicts of interest.
Author information
Authors and Affiliations
Contributions
D.C., E.A., T.T. and K.R. conceived and supervised the study. J.S., K.R., and T.T. designed the experiments. J.S. and K.R. performed the experiments. J.S., E.A., and K.R. analysed data. J.S., D.C., E.A., T.T., and K.R., wrote the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Segobola, J., Adriaenssens, E., Tsekoa, T. et al. Exploring Viral Diversity in a Unique South African Soil Habitat. Sci Rep 8, 111 (2018). https://doi.org/10.1038/s41598-017-18461-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-18461-0
- Springer Nature Limited
This article is cited by
-
Metavirome mining from fjord sediments of Svalbard Archipelago
Journal of Soils and Sediments (2024)
-
Uncovering a hidden diversity: optimized protocols for the extraction of dsDNA bacteriophages from soil
Microbiome (2020)
-
Seasonal dynamics in taxonomy and function within bacterial and viral metagenomic assemblages recovered from a freshwater agricultural pond
Environmental Microbiome (2020)
-
Functional screening of a soil metagenome for DNA endonucleases by acquired resistance to bacteriophage infection
Molecular Biology Reports (2020)
-
Genetic and functional diversity of double-stranded DNA viruses in a tropical monsoonal estuary, India
Scientific Reports (2018)