
Abstract
The Phospholipase D (PLD) superfamily is linked to neurological disease, cancer, and fertility, and a recent report correlated a potential loss-of-function PLD2 polymorphism with hypotension. Surprisingly, PLD2 −/− mice exhibit elevated blood pressure accompanied by associated changes in cardiac performance and molecular markers, but do not have findings consistent with the metabolic syndrome. Instead, expression of endothelial nitric oxide synthase (eNOS), which generates the potent vasodilator nitric oxide (NO), is decreased. An eNOS inhibitor phenocopied PLD2 loss and had no further effect on PLD2 −/− mice, confirming the functional relationship. Using a human endothelial cell line, PLD2 loss of function was shown to lower intracellular free cholesterol, causing upregulation of HMG Co-A reductase, the rate-limiting enzyme in cholesterol synthesis. HMG Co-A reductase negatively regulates eNOS, and the PLD2-deficiency phenotype of decreased eNOS expression and activity could be rescued by cholesterol supplementation and HMG Co-A reductase inhibition. Together, these findings identify a novel pathway through which the lipid signaling enzyme PLD2 regulates blood pressure, creating implications for on-going therapeutic development of PLD small molecule inhibitors. Finally, we show that the human PLD2 polymorphism does not trigger eNOS loss, but rather creates another effect, suggesting altered functioning for the allele.
Similar content being viewed by others


Introduction
The classic Phospholipase D (PLD) isoforms, PLD1 and PLD2, hydrolyze phosphatidylcholine (PC), the most abundant membrane phospholipid, to generate choline and the second messenger signaling lipid phosphatidic acid (PA)1. PLD1 and PLD2 have partially distinct and partially overlapping / redundant roles in vivo, with current translational interest2 for PLD1 being focused on thrombotic disease3, 4, cancer5, and the immune system6,7,8, and for PLD2, Alzheimer’s Disease9, cancer10, and influenza virus infection11. A recent study reported a negative correlation of a polymorphism in PLD2, R172C, with hypertension12. This polymorphism is located in a lipid-binding regulatory phox consensus sequence (PX) domain that is conserved in PLD1 and PLD2. For PLD1, the PX domain binds phosphatidylinositol (3,4,5)-trisphosphate, which facilitates its localization to the plasma membrane and stimulates PLD1’s enzymatic activity13. Both the PLD1 and PLD2 PX domains have also been reported to mediate interaction with signaling proteins such as epidermal growth factor receptor (EGFR)14, facilitating its endocytosis. Arginine, as a basic amino acid, is frequently involved in the binding to negatively-charged polyphosphoinositides and to protein targets via arginine fingers; thus the substitution of cysteine for R172 could alter PLD2’s cellular function.
PLD activity has been proposed to facilitate multiple pathways that function to increase blood pressure, including formation of very low-density lipoproteins (VLDL)15, 16, which, following secretion from the liver, are processed to generate low-density lipoprotein (LDL) particles that can be further modified to form oxidized-LDL (ox-LDL), which oppose vascular relaxation and promote atherogenesis17, 18. PLD2 function has also been linked to endocytosis of the angiotensin II type 1 receptor (AT1R)19, which promotes increased vascular tone and blood pressure via intracellular signaling, and to the production and secretion of aldosterone20, 21, which increases blood pressure by stimulating renal water and salt retention. Taken together, these reports provided multiple rationales for the observation that a polymorphism that could affect PLD2 function correlates with decreased blood pressure12, 22.
Undertaking exploration of this topic, we uncovered and report here the unexpected observation that mice lacking PLD2 have increased blood pressure, and delineate the pathway through which PLD2 normally functions to lower blood pressure. Ultimately, we propose that the human R172C polymorphism creates a mutant protein with only partial loss of function or altered function, rather than PLD2 functional deficiency. Our findings have implications for therapeutic use of PLD2 inhibitors in other settings; the potential for increased blood pressure will need to be assessed and considered in terms of the risk and potential benefits.
Results
PLD2−/− mice have increased systemic blood pressure accompanied by decreased cardiac function
Tail-cuff measurements were used to determine the blood pressure of wild-type (WT) and PLD2 −/− mice (Fig. 1A). Unexpectedly, the PLD2 −/− mice were found to have increased systolic (∆27 mm Hg; P = 0.00052) and diastolic (∆21 mm Hg; P = 0.0043) BP. This increase is significant in the context of human hypertension, since increases of 20 mm Hg systolic BP/10 mm Hg diastolic BP above the normal range are associated with two- or greater-fold differences in death rates from stroke, ischemic heart disease, and other vascular causes23.

PLD2 −/− mice have increased BP and altered cardiac function. (A) A non-invasive tail-cuff instrument was used to measure systolic and diastolic blood pressure in 5-month old male mice (n = 6 mice of each genotype; each data point represents the average of 15 measurements) as described in Methods. (B) Heart-to-body weight ratio for PLD2 −/− and WT mice (n = 3 mice of each genotype). (C–D) Echocardiogram sonogram was used to determine fractional shortening (FS) and percent ejection fraction (EF) as described in Methods (n = 3). Mean ± SEM; *p < 0.05; **p < 0.01; ***p ≤ 0.001 (Student’s t-test).
One of the major symptoms of hypertension is development of cardiac hypertrophy, since additional ejection force is needed to overcome the increased aortic blood pressure. The heart-to-body weight ratio in PLD2 −/− mice was not significantly increased compared to WT mice (Fig. 1B, P = 0.31). However, cardiac function in PLD2 −/− mice as assessed by echocardiogram revealed a significant decrease in the % fractional shortening and ejection fraction (Fig. 1C, D; P = 0.012, 0.0071), albeit the levels are still within the normal range. Similarly, the left ventricular diastolic volume was decreased by 15% in the PLD2 −/− mice (n = 3, P = 0.025). There were no significant differences in the left ventricular posterior wall thickness, the left ventricular interior diameter, or the interventricular septal thickness; however, the interventricular septal thickness at the end of diastole trended towards being increased (15% increased, P = 0.074). These decreases, while not indicative of cardiac hypertrophy or failure, are significant and likely reflect a compensatory response (mild adaptive hypertrophy) to the increased blood pressure in the PLD2 −/− mice.
The increased blood pressure in PLD2−/− mice is not associated with obesity or hyperlipidemia
Hypertension can evolve as a co-morbid disease linked to obesity, HDL cholesterol, and diabetes (the metabolic syndrome)24. However, the PLD2 −/− mice were found to weigh significantly less than age-matched WT mice (Fig. 2A, P = 0.0076), and the levels of serum LDL and HDL levels were the same or lower than in WT mice when placed on a high fat diet for 7 months (Fig. 2B [P = 0.00027 at 3 mos and 0.00011 at 7 mos], C [P = 0.000094 at 3 mos and 0.0059 at 7 mos]). The ratio of total serum cholesterol/HDL is used as a risk factor to monitor the development of heart disease25; employing this assessment method, PLD2 −/− mice would be predicted to be at lower risk of cardiovascular disease than WT mice (Fig. 2D, P = 0.00047 at 7 mos). Thus, the increased BP would appear to arise from a mechanism distinct from the ones associated with the metabolic syndrome.

PLD2 −/− mice are neither obese nor hyperlipidemic. (A) weight at 5 months, n = 7 mice of each genotype. LDL (B), HDL (C), and total serum cholesterol/HDL ratio (D) after 0, 3 and 7 months on a high fat diet (n = 3 mice of each genotype). Mean ± SEM; **p < 0.01; ***p ≤ 0.001 (Student’s t-test).
PLD2−/− aortas have decreased levels of endothelial nitric oxide synthase (eNOS), an effector of vasodilation
In surveying potential causes for the increased BP, we discovered, using immunofluorescent staining, that eNOS protein expression was dramatically reduced in the aorta of PLD2 −/− mice (Fig. 3A). Quantitatively, the eNOS fluorescent signal was reduced by 62% (±2.9%, n = 4, P = 0.00049). Moreover, virtually all of the remaining fluorescence was localized diffusely in the cell rather than in punctate form at the plasma membrane. The activity of eNOS is tightly regulated via several mechanisms including control of its subcellular localization26, 27. eNOS is thought to be active primarily when present in specialized plasma membrane domains rich in sphingomyelin, cholesterol, and caveolin, called caveolae28,29,30; cytoplasmic and Golgi-localized eNOS generate relatively little NO and are not activated by signaling events. This finding suggested that aortic endothelial cells in PLD2 −/− mice might be failing to generate NO, a potent vasodilator. To test this hypothesis, we treated WT and PLD2 −/− mice with the nitric oxide synthase inhibitor N-ω-nitro-L-arginine methyl ester (L-NAME) for a week, which suffices to elicit a significant increase in blood pressure due to eNOS inhibition31, and reassessed BP. Systolic and diastolic BP in the WT mice rose by 18% (∆ 20 mm Hg) and 17% (∆ 14 mm Hg), respectively, whereas no increase was seen for the PLD2 −/− mice (Fig. 3B vs Fig. 1A). Within one week of L-NAME treatment (Fig. 3B), BP was nearly identical in both the WT and PLD2 −/− mice, suggesting that the PLD2 deficiency-induced loss of eNOS and endothelial cell NO production underlies the increased BP in the PLD2 −/− mice. Similar outcomes were observed after an additional week of L-NAME treatment. Similarly, L-NAME treatment decreased the % fractional shortening, the % ejection fraction, and the left ventricular diastolic volume in the WT mice but not in the PLD2 −/− mice, eliminating the difference between the mouse strains (not significant (n.s.), n = 3). We next examined levels of VEGF in the lung and heart, since VEGF levels have been reported to decrease in some models of hypertension32, 33. Consistent with these reports, VEGF levels fell in WT mice after 15 days of L-NAME treatment and the resulting sustained hypertension (Fig. 3C–F; the decrease was highly significant in the lung (P = 0.0002) and almost achieved significance (p = 0.069) in the heart). In contrast, VEGF levels were initially at reduced levels in the PLD2 −/− mice and were not affected by L-NAME treatment, suggesting that the PLD2-deficiency induced reduction in VEGF is occurring through an eNOS-dependent mechanism.

Decreased BP in PLD2 −/− mice results from reduced levels of eNOS protein. (A) Immunofluorescent staining of eNOS in sections of aortas from WT and PLD2 −/− mice. Bar, 7.5 μM. Representative image from 4 mice of each genotype. (B) Systolic and diastolic blood pressure following treatment with L-NAME for one week (n = 4). (C–F) Measurements of VEGF protein levels by western blotting of PLD2 −/− and WT mice lungs (C,D) and heart (E,F) were normalized to actin expression levels (n = 4 mice of each genotype). A representative blot is shown. The regions of the Western blots containing the VEGF and actin immunoreactive bands were scanned and the relative abundance of the individual samples quantified using an Odyssey CLx imaging system. Mean ± SEM; ***p ≤ 0.001; B: Student’s t-test; D, F: One-way ANOVA with Bonferroni’s Multiple Comparison Test.
PLD2 knock-down in the human umbilical vein cell line EA.hy926 decreases eNOS expression and NO production
Since the decrease in eNOS expression in the PLD2−/− mice could have arisen from a long-term compensatory effect or from loss of a scaffolding role due to lack of expression of the protein, we next sought to examine short-term protein loss or inhibition. In vivo selective inhibition of PLD2 is not currently practical, since the existing PLD2-selective small molecule compounds have short half-lives11, 34 and methods for their continuous delivery have not been established. To address these issues, delineate the mechanism through which PLD2 regulates eNOS expression and to examine eNOS activity directly, we used a commercial lentiviral shRNA approach that employs three different shRNA sequences to establish a human umbilical vein cell line with greatly diminished PLD2 expression (shPLD2). A control lentivirus expressing scrambled shRNA sequences was used to generate the control “Scramble” cell line. The resulting stable shPLD2 pooled cell line exhibited a > 90% knockdown of PLD2 mRNA as assessed by semi-quantitative qRT-PCR (Fig. 4A), and PLD2 protein was undetectable (Fig. 4B). Immunofluorescent confocal microscopy then revealed that PLD2 knockdown substantially decreased eNOS expression, especially on the plasma membrane (Fig. 4C), and quantitative western blotting indicated that the eNOS expression was 62% reduced in the shPLD2 cell pool (P = 0.0010, Fig. 4D,E). Finally, we examined NO production by the Scramble and shPLD2 endothelial cell lines. The shPLD2 cell line generated only 38% as much NO as the Scramble cell line (P = 0.0000021, Fig. 4F). Treatment of the Scramble cells with a small molecule PLD2-selective inhibitor, NFOT35, similarly reduced NO production (39% of the control value, as assessed by release of nitrite (NO2 −) into the cell culture media; P = 0.00000034), but NFOT had no further effect on the shPLD2 cells, confirming that NFOT mediates its effect through inhibition of PLD2 activity. These findings demonstrate that PLD2 regulates eNOS expression in an acute manner, rather than through long-term compensatory changes, and also demonstrate that PLD2 mediates its regulation of eNOS activity through PLD2 enzymatic activity rather than via a scaffolding mechanism, since ablation of the PLD2 protein and simple inhibition of its activity yield the same outcome on NO production. Finally, the effect we demonstrate of the PLD2 inhibitor NFOT to lower eNOS expression levels in endothelial cells suggests that pharmacological inhibition of PLD2 in vivo should increase blood pressure and induce compensatory cardiac changes in contractility.

Stable shRNA knockdown of PLD2 in EA.hy926 endothelial cells decreases eNOS protein expression levels and NO production. (A,B) Stable PLD2 knockdown cells (shPLD2 cells) were generated using shRNA lentiviral particles. In parallel, control shRNA sequences were used to generate a control cell line (Scramble cells). PLD2 knockdown was confirmed by qRT-PCR (A) and western blotting as shown in the cropped gel and blot images, respectively (B). Bar, 5 μM. The region of the blot containing the PLD2 immunoreactive band was scanned using an Odyssey CLx imaging system. (C) Confocal microscopy of eNOS protein expression as detected by immunofluorescent staining in Scramble and shPLD2 cells. Representative image of 3 experiments. (D) Western blotting of eNOS in Scramble and shPLD2 cells, representative blot (n = 3 experiments). The regions of the Western blots (Suppl. Figure 1) containing the eNOS and actin immunoreactive bands were scanned and the relative abundance of the individual samples quantified using an Odyssey CLx imaging system. (E) Quantification of western blot with eNOS levels normalized to actin (n = 3 experiments). (F) Measurement of nitrate production using a Griess Reaction Kit to quantify eNOS activity with and without treatment with a small molecule PLD2 inhibitor (NFOT) at 10 μM for 24 hours (n = 7 experiments). Mean ± SEM; ***p ≤ 0.001; E, Student’s t-test; F, one-way ANOVA with Bonferroni’s Multiple Comparison Test.
PLD2 knock-down increases expression of HMG-CoA reductase, a negative regulator of eNOS expression levels
HMG-CoA reductase, the rate-limiting enzyme in cholesterol synthesis, has been well studied as a negative regulator of eNOS function, through its production of mevalonate, which decreases eNOS mRNA stability36. Statins, which inhibit HMG-CoA reductase activity, have been shown widely to increase eNOS mRNA and protein levels and NO production37. Here, using western blotting, we found that HMG-CoA reductase levels were increased by 25% in the shPLD2 endothelial cells (P = 0.0032) and in Scramble cells treated with the PLD2 inhibitor NFOT in comparison to Scramble cells (Fig. 5A,B). N-SIM (structured illumination) microscopy similarly revealed an increase in shPLD2 endothelial cell HMG-CoA reductase expression (Fig. 5C ), and qRT-PCR demonstrated increased levels of HMG-CoA reductase mRNA in the NFOT-treated Scramble endothelial cells (P = 0.013) and shPLD2 endothelial cells (P = 0.0048, Fig. 5D).

Upregulation of HMG-CoA reductase in shPLD2 cells decreases eNOS expression. (A) Representative western blot of HMG-CoA reductase in Scramble and shPLD2 cells. The regions of the Western blots containing the HMG-CoA reductase and actin immunoreactive bands were scanned and the relative abundance of the individual samples quantified using an Odyssey CLx imaging system (B) Quantification of western blots with HMG-CoA reductase normalized to actin (n = 4 experiments). (C) N-SIM microscopy of HMG-CoA reductase; Bar, 7.5 μM. (D) RT-PCR of HMG-CoA reductase in Scramble and shPLD2 cells (n = 3). (E) Western blot of eNOS after treatment with NFOT and Simvastatin. Representative image of 3 experiments. The regions of the Western blots (Suppl. Figure 2) containing the eNOS and actin immunoreactive bands were scanned and the relative abundance of the individual samples quantified using an Odyssey CLx imaging system. (F) Quantification of western blots with eNOS levels normalized to actin (n = 3). Comparisons without bars made to untreated Scramble values. (G) eNOS activity after treatment with Simvastatin (n = 7). Mean ± SEM; *p < 0.05; **p < 0.01; ***p ≤ 0.001; One-way ANOVA with Bonferroni’s Multiple Comparison Test.
Next, we tested whether an HMG CoA reductase inhibitor could overcome the PLD2-mediated decrease in eNOS protein expression levels and activity. Scramble endothelial cells treated with the PLD2 inhibitor NFOT had decreased expression levels of eNOS protein (Fig. 5E,F, lanes 1 and 2, P = 0.012), as described above, whereas treatment with the HMG-CoA reductase inhibitor, Simvastatin, strongly increased eNOS protein expression (lane 3, P = 0.0000071). The combined treatment was intermediate in outcome (lane 4), with expression being elevated above the Scramble control, but not as high as Simvastatin alone. Similarly, eNOS protein expression was decreased in shPLD2 cells as described above (lane 5, P = 0.0010), and increased, but not fully, with exposure to Simvastatin (lane 6, P = 0.0027). Nonetheless, these findings suggest that the PLD2-deficiency-stimulated increase in HMG-CoA reductase expression level and activity underlies the decrease in eNOS protein expression. Finally, we observed a similar outcome through examining NO production: as before, shPLD2 cells generated less NO than Scramble endothelial cells (Fig. 5G, lanes 1 vs 3, P = 0.000000035), and Simvastatin increased the amounts of NO production in both the control (P = 0.012) and PLD2-knockdown cells (lanes 2 and 4, P = 0.036), identifying a role for HMG-CoA reductase in the PLD2-deficiency-mediated inhibition.
Intracellular free cholesterol levels are decreased in PLD2-deficient endothelial cells, and when restored, bypass PLD2 deficiency to elevate eNOS expression
HMG-CoA reductase is regulated both transcriptionally and with respect to activation of the protein by the levels of intracellular free cholesterol38. We found that free cholesterol levels are 9% reduced in shPLD2 cells (p < 0.001, Fig. 6A). To establish whether this reduction is biologically significant, we supplemented the shPLD2 cells with 25 μM cholesterol for 24 hours, which increased the intracellular levels by 7%, bringing it almost back to normal levels (P = 0.00092, Fig. 6A). We then examined eNOS protein expression levels, and found that the cholesterol supplementation resulted in a 42% increase (Fig. 6B,C, P = 0.012). Placing all of these observations together, our findings reveal a novel pathway in which PLD2 deficiency decreases intracellular cholesterol, leading to increased HMG-CoA reductase protein expression and activity, which results in decreased eNOS expression, deceased production of the vasodilator NO, and, finally, hypertension (Fig. 6D).

Recovery of eNOS levels in shPLD2 cells following exogenous cholesterol supplementation. (A) Free cholesterol levels monitored by cholesterol oxidase assay after culture of cells with 25 μM exogenous cholesterol (in media) for 24 hours (n = 4). (B) Representative western blot of eNOS protein in shPLD2 cells with and without cholesterol supplementation. The regions of the Western blots (Suppl. Figure 3) containing the eNOS and actin immunoreactive bands were scanned and the relative abundance of the individual samples quantified using an Odyssey CLx imaging system. (C) Quantification of western blot with eNOS levels normalized to actin (n = 5). (D) Schematic summary of model. Dotted line indicates that the mechanism involved is unknown at this time. Mean ± SEM; *p < 0.05; **p ≤ 0.001; C, Student’s t-test; A, one-way ANOVA with Bonferroni’s Multiple Comparison Test.
R172C confers selective loss of function on PLD2
Returning to the human polymorphism linked to reduced blood pressure12, we sought to address whether it functions differently than the wild-type PLD2 allele, since it clearly does not phenocopy PLD2 loss-of-function in mice. Human PLD2 overexpression in cells stimulates F-actin reorganization39, 40, as illustrated in Fig. 7A, where both filopodia (arrowhead) and peripheral (chevron) and dorsal (*) membrane ruffles can be observed. In contrast, overexpression of R172C-hPLD2 did not provoke dramatic F-actin reorganization (Fig. 7B). The majority of the R172C-hPLD2 protein appeared to localize to subcortical actin (arrow), in contrast to hPLD2 which localized primarily to the cell cortex. Overexpression of hPLD2 (P = 0.038) and R172C-hPLD2 (n.s., P = 0.27) increased eNOS protein expression levels in shPLD2 cells (Fig. 7C,D), and more importantly, restored NO production (Fig. 7E). Finally, we examined eNOS and caveolin-1 expression using N-SIM super-resolution microscopy. As before, eNOS protein levels were greatly diminished at the plasma membrane in the shPLD2 cells (Fig. 7F,G), and decreased expression was also observed for caveolin-1, a protein that interacts with PLD2 in lipid rafts in a lipid-dependent manner41,42,43. Overexpression of hPLD2 restored WT levels of expression for both eNOS and caveolin-1 (Fig. 7H). However, overexpression of R172C-hPLD2 rescued only the eNOS expression; no increase in caveolin-1 was observed (Fig. 7I). Together, these findings suggest that the R172C polymorphism does not alter eNOS function; hence, it would not lead to increased BP via this pathway. However, its contrasting effect on caveolin-1 is intriguing, as discussed below.

The human PLD2 polymorphism R172C does not alter eNOS signaling but does decrease caveolin-1 protein levels. Overexpression of HA-tagged hPLD2 (A) and HA-R172C-PLD2 (B) in HeLa cells, visualized using anti-HA immunofluorescent staining (green). Arrowhead, PLD2 localization in filopodia; chevron, in peripheral actin ruffles; *, in dorsal actin ruffles; arrow, in subcortical actin network. Bar, 7.5 μM. Representative image of multiple experiments. (C) Representative western blot of eNOS after transfection of HA-hPLD2 or HA-R172C-PLD2 into shPLD2 cells. The regions of the Western blots containing the eNOS and actin immunoreactive bands were scanned and the relative abundance of the individual samples quantified using an Odyssey CLx imaging system. (D) Quantification of western blotting with eNOS levels normalized to actin (n = 3). (E) eNOS activity as measured by nitrate formation (n = 7). Lane 1 vs 2, P = 0.0021; lane 2 vs 3, P = 0.00035; lane 2 vs 4, P = 0.00012. (F–I) N-SIM microscopy of plasma membrane eNOS and caveolin-1. Bar, 7.5 μM. Representative image of multiple Scramble or shPLD2 cells imaged. Cells in H and I were selected for imaging based on expression of HA-hPLD2 or HA-R172C-PLD2 as visualized by anti-HA immunofluorescence in a separate channel (not shown). Mean ± SEM; *p < 0.05; **p < 0.01; ***p ≤ 0.001; One-way ANOVA with Bonferroni’s Multiple Comparison Test.
Discussion
The regulation of BP is complex, involving numerous physiological and cellular pathways. Supporting roles for PLD2 in this process have been proposed, based on its contribution to the secretion of aldosterone20, 21, via facilitation of AT1R signaling19, and through promotion of VLDL formation15, 16. Based on these studies, the report that a polymorphism in human PLD2 negatively correlates with the development of hypertension appeared to support the hypothesis that PLD2 promotes increased blood pressure and that loss of PLD2 function should result in hypotension. Unexpectedly, we report here that mice lacking PLD2 exhibit hypertension (Fig. 1) as a consequence of reduced levels of eNOS (Fig. 3), which accordingly decreases production of NO (Fig. 4), an important physiological vasodilatory factor.
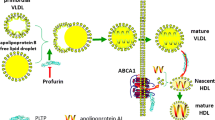
The proximal cause of PLD2-deficiency-induced eNOS reduction was identified to be a decrease in intracellular free cholesterol (Fig. 6), which stimulates upregulation and activation of HMG CoA-reductase, a well-known negative regulator of eNOS (Fig. 5). The relationship of PLD2 function to intracellular cholesterol levels is poorly understood, but an intriguing possibility arises from a report that PLD2 may be involved in a pathway that destabilizes ABCA144, which mediates the transport of cholesterol and phospholipids from cells to HDL apolipoproteins. PLD2 loss, in this model, would lead to increased ABCA1 protein levels and activity, and consequently, reduced intracellular free cholesterol. This possibility represents an area for future exploration.
Reductions in intracellular cholesterol stimulate both transcriptional upregulation and activation of HMG-CoA reductase, which negatively regulates eNOS mRNA stability and hence protein expression. Simvastatin, an HMG-CoA reductase inhibitor, partially but not fully rescued the decreased eNOS phenotype induced by PLD2 deficiency (Fig. 5), suggesting that HMG-CoA reductase upregulation is only one of the mechanisms affecting eNOS levels in this setting. It is well established that reductions in intracellular cholesterol also suppress eNOS activity27, 30, 45, 46 through decreasing the association of eNOS with the plasma membrane in caveolae, where it can be activated by signaling events. Thus, while inhibition of HMG-CoA reductase blocks destabilization of eNOS mRNA, it does not address the underlying cholesterol deficiency, which also impacts eNOS activity.
Conversely, while statins lower blood pressure, and would presumably lower blood pressure in PLD2 −/− mice at least in part through increasing eNOS protein expression levels37, statins actually mediate numerous effects, including inhibiting the production of specific prenylated proteins, and this may partially underlie the improvement in endothelial function, modulation of immune function, and other pleiotropic cardiovascular benefits associated with statin use. Hence, observing a reduction in blood pressure in statin-treated PLD2 −/− mice would not be conclusive evidence for the specific pathway we describe here.
The marked decrease in caveolin-1 in shPLD2 endothelial cells transfected with R172C-hPLD2 is intriguing in that caveolin-1 is a well-known negative regulator of eNOS activity47. Reduced caveolin-1 could lead to increased eNOS activity, increased NO production, and vasodilation, consistent with the resistance to hypertension reported to be associated with the human allele12.
In vivo roles and potential opportunities for therapeutic interventions for PLD2 have been explored in recent years using PLD2−/− mice. PLD2 ablation has been shown to improve outcomes in a model of Alzheimer’s disease9, to suppress cancer growth and metastasis in a heterotopic implant model10, and to improve outcomes in thrombotic disease models in combination with PLD1 ablation4. Finally, PLD2 inhibition has been reported to suppress influenza virus infection of a lung cell line in vitro and mouse lungs in an in vivo model11. While concerns regarding increased blood pressure are of lesser concern in the context of transient inhibition, such as would be performed for management of viral disease and acute thrombotic events, this issue may affect enthusiasm for pursuing PLD2 inhibition in the setting of long-term cancer or neurodegenerative treatment. In particular for cancer, some therapeutics, for example, VEGFR tyrosine kinase inhibitors, are independently associated with a substantially elevated risk of hypertension development48, and thus use of a PLD2 inhibitor in parallel with them could result in synergistic adverse effects. Nonetheless, murine studies do not always predict the outcomes of complex signaling pathways in humans. Given the potential utility of PLD2 inhibitors for cancer and acute thrombotic disease, in particular in combination with PLD1 inhibitors2, an effect on blood pressure represents a topic that should be assessed carefully if and when PLD2 inhibitors enter the clinic.
Materials and Methods
Materials
Simvastatin (Cayman Chemical) was used in culture for 24 hrs at 10 μM, N-[2-[1-(3-Fluorophenyl)-4-oxo-1,3,8-triazaspiro[4.5]dec-8-yl]ethyl]-2-naphthalenecarboxamide (NFOT) (Tocris Bioscience) at 10 μM, and cholesterol (Sigma-Aldrich C8667) at 25 μM.
Animals
PLD2 −/− mice (C57BL/6)9, generously provided by Dr. Gilbert di Paolo (Columbia University), were bred in the Stony Brook University animal facilities. Mice were fed standard chow or high-fat diet chow and water ad libitum and kept on a 12:12-h dark-light cycle. All experimental procedures were performed in accordance with relevant guidelines and regulations and were reviewed and approved by the Stony Brook University Institutional Animal Care and Use Committee (IACUC). Unless otherwise stated, male mice at 5 mos. of age were studied. Animals were injected with L-NAME (80210; Cayman Chemical) at a dose of 400 µg/g IP once a day for 15 days31.
Non-Invasive Blood Pressure Measurements
Tail-cuff blood pressure (BP) readings were taken at days 0, 7 and 15 of L-NAME treatment using the CODA Multi-Channel, Computerized, and Non-invasive Blood Pressure System for Mice (Kent Scientific). BP measurements were taken twice during the two days leading to each acquisition date to acclimate the mice to the procedure. Blood pressure measurements were obtained by performing 5 acclimation cycles (a cycle = a BP reading) followed by 3 sets of 5 cycles per set.
Echocardiography
Echocardiogram measurements were taken after 15 days of L-NAME treatment using a Vevo 770 ultrasound device (VisualSonics) with a 30-MHz transducer. Mice were anesthetized with 1% isoflurane and transthoracic echocardiography performed as previously described49.
Cell line creation and qRT-PCR
Human umbilical vein cell line EA.hy926 (CRL-2922; ATCC) was cultured in Dulbecco’s modified Eagle’s medium supplemented with L-glutamine (2 mM), 10% fetal calf serum (FCS), 100 U/mL penicillin and 100 µg/mL streptomycin. The cells were transduced with either Control shRNA Lentiviral Particles-A or with PC-PLD2 shRNA (h) Lentiviral Particles (Santa Cruz Biotechnology), which contain a puromycin-resistance gene to enable selection of stable cell lines or pools. The culture medium was changed 24 hrs following transduction and the cells cultured another day. Puromycin (0.5 μg/ml) was then added for positive selection. The resulting stably-transduced EA.hy926 cell pools were denoted “scramble” and shPLD2, respectively.
qRT-PCR
Total RNA was extracted with Trizol (Invitrogen). 200–300 ng of total cellular RNA was used with the qScript One-Step SYBR Green qRT-PCR Kit (Quanta Biosciences). Primers used for human HMG-CoA Reductase were: TGTGTGTGGGACCGTAATGG and ACCAAGTGGCTGTCTCAGTG; and for human GAPDH: ACAGTCAGCCGCATCTTCTT and GCATCGCCCCACTTGATTTT.
Western Blots
In vivo samples: Tissues were homogenized in lysis buffer (150 mM NaCl, 1% Triton X-100, 100 mM Tris-HCl, pH7.4, 1 mM EDTA, 1 mM phenylmethylsulfonyl fluoride, 10 μg/ml aprotinin, and 10 μg/ml pepstatin A), ultrasonicated at 40C, microfuged, and supernatants collected. Equal amounts of lysates (20 μg/lane) were separated on SDS-PAGE, transferred to filter paper, and immunostained using mouse anti-VEGF at 1:500 (ab3109; abcam) and rabbit anti-actin at 1:100 (A2066; Sigma-Aldrich). Cell line samples: Cells were cultured in six-well plates until 80–90% confluent, trypsinized, and equal amounts of total protein resolved by SDS-PAGE and blotted onto nitrocellulose membranes (162–0115; Bio-Rad) using NuPAGE Transfer Buffer (NP0006–1; Life Technologies). Membranes were blocked for 1-h in 5% BSA at room temperature (RT) and probed overnight (O/N) with rabbit anti-eNOS at 1:500 (NB300–500; Novus Biologicals), mouse anti-caveolin-1 at 1:500 (sc-53564; Santa Cruz Biotechnology), goat anti-HMG CoA reductase at 1:100 (GTX88456; Genetex), rabbit anti-PLD2 at 1:100 (sc-25513; Santa Cruz Biotechnology), and rabbit anti-Actin at 1:100 (A2066; Sigma-Aldrich) or rabbit anti-GAPDH at 1:500 (sc-25778; Santa Cruz Biotechnology). Membranes were probed with anti-rabbit or anti-mouse secondary Ab at 1:3,000 and developed and quantified using a LI-COR Odyssey Infrared Imager.
With the exception of panel B in Fig. 4, which is shown for illustrative purposes, all of the western blot images shown are provided as representatives of the multiple experiments conducted. The LI-COR Odyssey Infrared Imager quantifies each band based on pixel intensity and generates a pseudo-autorad from the data set. The quantitation presented for all of the western blots (save Fig. 4B) was generated using the raw data returned by the Odyssey imager, not on a scan or digitalization of the autorad shown for illustrative purposes.
Immunofluorescent Staining and Microscopy
Cryosections (8 μm) of fixed aortas from age-matched male mice on a normal chow diet were stained with rabbit anti-eNOS at 1:200 O/N and for 1 hr with DAPI and Alexa 647 anti-rabbit secondary antibody. Images were captured using a Leica TCS5 Confocal Microscope. Cells were cultured in 12-well plates on glass coverslips until 80–90% confluent, washed with PBS, fixed with 4% paraformaldehyde, permeabilized with Triton X-100, blocked with 5% goat serum for 1 hr, incubated for 1 hr at RT with primary antibodies followed by secondary antibodies and DAPI for another hour, and imaged as above.
Free-cholesterol measurements
In vivo samples: 4–6-week old male mice were placed on a high fat diet chow (HFD) for a total of 7 months. At time points 0, 3 and 7 months, blood was collected retro-orbitally. Serum lipoprotein levels were assessed using the HDL & LDL/VLDL Cholesterol Quantification Kit (K613-100; BioVision) following the manufacturer’s protocol. For cell lines, Scramble and shPLD2 cells were plated in 100-mm tissue culture treated plates until 80–90% confluent, rinsed with PBS, trypsinized and stored at −80 °C until ready for cholesterol extraction. Cholesterol was extracted by lysing the cell pellets in 200 μL 7:11:0.1 chloroform:isopropanol:Nonidet P-40 and incubating at 25 °C for 10 min. The cell debris was removed by centrifugation for 10 min at 15,000 × g and the supernatant collected, dried with a rotary evaporator followed by 30 min in vacuum, and resuspended in 50 μL isopropanol. Cholesterol concentrations were measured using the enzyme cholesterol oxidase50, which converts cholesterol to cholest-4-en-3-one, the formation of which can be monitored at 240 nm. Cholesterol samples were added to assay buffer (50 mM sodium phosphate pH 7.0, 0.025% Triton X-100, 0.020% bovine serum albumin) pre-incubated for 10 min with 125 nM cholesterol oxidase at 37 °C. The total reaction was incubated for 45 min at 37 °C. The samples were loaded into a 96-well UV transparent quartz microplate and the absorbance at 240 nm measured using a BioTek Synergy 2 plate reader. A standard curve of known cholesterol concentrations was prepared and measured in parallel to convert absorbance values of each sample to cholesterol concentrations.
NO Release Analysis into Cell Media
EA.hy926 cells were cultured in 24-well plates in normal growth medium until 80–90% confluent. NO level was assessed in 20 µl of reduced serum medium using a Nitrate/Nitrite Fluorometric Assay Kit (Cayman Chemical) following the manufacturer’s protocol.
Statistical analysis
Numerical data are presented as mean ± SEM. Student’s t-test was used to compare the differences between two groups and one-way ANOVA with Bonferroni’s Multiple Comparison Test to compare the differences between three or more groups. Significance was based on a value of p < 0.05. Experiments were performed in duplicate unless otherwise noted in the Figure Legends. Statistical analysis was performed on the cumulative average values for each independent experiment (n = repeats [separate experiments]).
Data availability statement
All data generated or analyzed during this study are included in this published article. No datasets were generated or analyzed during the current study.
References
Jenkins, G. M. & Frohman, M. A. Phospholipase D: a lipid centric review. Cellular and molecular life sciences: CMLS 62, 2305–2316, doi:10.1007/s00018-005-5195-z (2005).
Frohman, M. A. The phospholipase D superfamily as therapeutic targets. Trends in pharmacological sciences 36, 137–144, doi:10.1016/j.tips.2015.01.001 (2015).
Elvers, M. et al. Impaired alpha(IIb)beta(3) integrin activation and shear-dependent thrombus formation in mice lacking phospholipase D1. Science signaling 3, ra1, doi:10.1126/scisignal.2000551 (2010).
Stegner, D. et al. Pharmacological inhibition of phospholipase D protects mice from occlusive thrombus formation and ischemic stroke–brief report. Arteriosclerosis, thrombosis, and vascular biology 33, 2212–2217, doi:10.1161/atvbaha.113.302030 (2013).
Chen, Q. et al. Key roles for the lipid signaling enzyme phospholipase d1 in the tumor microenvironment during tumor angiogenesis and metastasis. Science signaling 5, ra79, doi:10.1126/scisignal.2003257 (2012).
Ali, W. H. et al. Deficiencies of the lipid-signaling enzymes phospholipase D1 and D2 alter cytoskeletal organization, macrophage phagocytosis, and cytokine-stimulated neutrophil recruitment. PloS one 8, e55325, doi:10.1371/journal.pone.0055325 (2013).
Gobel, K. et al. Phospholipase D1 mediates lymphocyte adhesion and migration in experimental autoimmune encephalomyelitis. European journal of immunology 44, 2295–2305, doi:10.1002/eji.201344107 (2014).
Schonberger, T. et al. Pivotal role of phospholipase D1 in tumor necrosis factor-alpha-mediated inflammation and scar formation after myocardial ischemia and reperfusion in mice. The American journal of pathology 184, 2450–2464, doi:10.1016/j.ajpath.2014.06.005 (2014).
Oliveira, T. G. et al. Phospholipase d2 ablation ameliorates Alzheimer’s disease-linked synaptic dysfunction and cognitive deficits. J Neurosci 30, 16419–16428, doi:10.1523/JNEUROSCI.3317-10.2010 (2010).
Ghim, J. et al. Endothelial deletion of phospholipase D2 reduces hypoxic response and pathological angiogenesis. Arteriosclerosis, thrombosis, and vascular biology 34, 1697–1703, doi:10.1161/atvbaha.114.303416 (2014).
Oguin, T. H. 3rd et al. Phospholipase D facilitates efficient entry of influenza virus, allowing escape from innate immune inhibition. The Journal of biological chemistry 289, 25405–25417, doi:10.1074/jbc.M114.558817 (2014).
Hong, K. W. et al. Non-synonymous single-nucleotide polymorphisms associated with blood pressure and hypertension. Journal of human hypertension 24, 763–774, doi:10.1038/jhh.2010.9 (2010).
Lee, J. S. et al. Phosphatidylinositol (3,4,5)-trisphosphate specifically interacts with the phox homology domain of phospholipase D1 and stimulates its activity. Journal of cell science 118, 4405–4413, doi:10.1242/jcs.02564 (2005).
Lee, C. S. et al. The phox homology domain of phospholipase D activates dynamin GTPase activity and accelerates EGFR endocytosis. Nature cell biology 8, 477–484, doi:10.1038/ncb1401 (2006).
Asp, L., Claesson, C., Boren, J. & Olofsson, S. O. ADP-ribosylation factor 1 and its activation of phospholipase D are important for the assembly of very low density lipoproteins. The Journal of biological chemistry 275, 26285–26292, doi:10.1074/jbc.M003520200 (2000).
Olofsson, S. O., Asp, L. & Boren, J. The assembly and secretion of apolipoprotein B-containing lipoproteins. Curr Opin Lipidol 10, 341–346 (1999).
Quinn, M. T., Parthasarathy, S., Fong, L. G. & Steinberg, D. Oxidatively modified low density lipoproteins: a potential role in recruitment and retention of monocyte/macrophages during atherogenesis. Proceedings of the National Academy of Sciences of the United States of America 84, 2995–2998 (1987).
Tanner, F. C., Noll, G., Boulanger, C. M. & Luscher, T. F. Oxidized low density lipoproteins inhibit relaxations of porcine coronary arteries. Role of scavenger receptor and endothelium-derived nitric oxide. Circulation 83, 2012–2020 (1991).
Zheng, X. L., Gui, Y., Du, G. W., Frohman, M. A. & Peng, D. Q. Calphostin-C induction of vascular smooth muscle cell apoptosis proceeds through phospholipase D and microtubule inhibition. Journal of Biological Chemistry 279, 7112–7118, doi:10.1074/jbc.M310721200 (2004).
Qin, H., Frohman, M. A. & Bollag, W. B. Phospholipase D2 mediates acute aldosterone secretion in response to angiotensin II in adrenal glomerulosa cells. Endocrinology 151, 2162–2170, doi:10.1210/en.2009-1159 (2010).
Tsai, Y. Y. et al. Phospholipase D activity underlies very-low-density lipoprotein (VLDL)-induced aldosterone production in adrenal glomerulosa cells. Endocrinology 155, 3550–3560, doi:10.1210/en.2014-1159 (2014).
Kundu, A., Ramakrishnan, P., Rajendran, A., Dharwar, N. V. & Anbarasu, A. Analysis of non-synonymous single-nucleotide polymorphisms and population variability of PLD2 gene associated with hypertension. International journal of bioinformatics research and applications 9, 227–241, doi:10.1504/ijbra.2013.053604 (2013).
Lewington, S., Clarke, R., Qizilbash, N., Peto, R. & Collins, R. Age-specific relevance of usual blood pressure to vascular mortality: a meta-analysis of individual data for one million adults in 61 prospective studies. Lancet (London, England) 360, 1903–1913 (2002).
Vassallo, P., Driver, S. L. & Stone, N. J. Metabolic Syndrome: An Evolving Clinical Construct. Progress in cardiovascular diseases. doi:10.1016/j.pcad.2016.07.012 (2016).
Linn, S. et al. Serum total cholesterol: HDL cholesterol ratios in US white and black adults by selected demographic and socioeconomic variables (HANES II). American journal of public health 81, 1038–1043 (1991).
McCormick, M. E. et al. Platelet-endothelial cell adhesion molecule-1 regulates endothelial NO synthase activity and localization through signal transducers and activators of transcription 3-dependent NOSTRIN expression. Arteriosclerosis, thrombosis, and vascular biology 31, 643–649, doi:10.1161/atvbaha.110.216200 (2011).
Zhang, Q. et al. Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in reconstituted endothelial cells. Arteriosclerosis, thrombosis, and vascular biology 26, 1015–1021, doi:10.1161/01.ATV.0000216044.49494.c4 (2006).
Boje, K. M. & Fung, H. L. Endothelial nitric oxide generating enzyme(s) in the bovine aorta: subcellular location and metabolic characterization. J Pharmacol Exp Ther 253, 20–26 (1990).
Forstermann, U., Pollock, J. S., Schmidt, H. H., Heller, M. & Murad, F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proceedings of the National Academy of Sciences of the United States of America 88, 1788–1792 (1991).
Shaul, P. W. Endothelial nitric oxide synthase, caveolae and the development of atherosclerosis. J Physiol 547, 21–33, doi:10.1113/jphysiol.2002.031534 (2003).
Peotta, V. A., Vasquez, E. C. & Meyrelles, S. S. Cardiovascular neural reflexes in L-NAME-induced hypertension in mice. Hypertension (Dallas, Tex.: 1979) 38, 555–559 (2001).
Gien, J., Seedorf, G. J., Balasubramaniam, V., Markham, N. & Abman, S. H. Intrauterine pulmonary hypertension impairs angiogenesis in vitro: role of vascular endothelial growth factor nitric oxide signaling. American journal of respiratory and critical care medicine 176, 1146–1153, doi:10.1164/rccm.200705-750OC (2007).
Grover, T. R. et al. Intrauterine hypertension decreases lung VEGF expression and VEGF inhibition causes pulmonary hypertension in the ovine fetus. American journal of physiology. Lung cellular and molecular physiology 284, L508–517, doi:10.1152/ajplung.00135.2002 (2003).
Scott, S. A. et al. in Probe Reports from the NIH Molecular Libraries Program (National Center for Biotechnology Information (US), 2010).
O’Reilly, M. C. et al. Development of dual PLD1/2 and PLD2 selective inhibitors from a common 1,3,8-Triazaspiro[4.5]decane Core: discovery of Ml298 and Ml299 that decrease invasive migration in U87-MG glioblastoma cells. Journal of medicinal chemistry 56, 2695–2699, doi:10.1021/jm301782e (2013).
Kalinowski, L., Dobrucki, I. T. & Malinski, T. Cerivastatin potentiates nitric oxide release and enos expression through inhibition of isoprenoids synthesis. Journal of physiology and pharmacology: an official journal of the Polish Physiological Society 53, 585–595 (2002).
Meda, C., Plank, C., Mykhaylyk, O., Schmidt, K. & Mayer, B. Effects of statins on nitric oxide/cGMP signaling in human umbilical vein endothelial cells. Pharmacological reports: PR 62, 100–112 (2010).
Mutungi, G., Torres-Gonzalez, M., McGrane, M. M., Volek, J. S. & Fernandez, M. L. Carbohydrate restriction and dietary cholesterol modulate the expression of HMG-CoA reductase and the LDL receptor in mononuclear cells from adult men. Lipids in health and disease 6, 34, doi:10.1186/1476-511x-6-34 (2007).
Colley, W. C. et al. Phospholipase D2, a distinct phospholipase D isoform with novel regulatory properties that provokes cytoskeletal reorganization. Current biology: CB 7, 191–201 (1997).
Honda, A. et al. Phosphatidylinositol 4-phosphate 5-kinase alpha is a downstream effector of the small G protein ARF6 in membrane ruffle formation. Cell 99, 521–532 (1999).
Czarny, M. et al. Phospholipase D2: functional interaction with caveolin in low-density membrane microdomains. FEBS letters 467, 326–332 (2000).
Qin, H. & Bollag, W. B. The caveolin-1 scaffolding domain peptide decreases phosphatidylglycerol levels and inhibits calcium-induced differentiation in mouse keratinocytes. PloS one 8, e80946, doi:10.1371/journal.pone.0080946 (2013).
Zheng, X. & Bollinger Bollag, W. Aquaporin 3 colocates with phospholipase d2 in caveolin-rich membrane microdomains and is downregulated upon keratinocyte differentiation. J Invest Dermatol 121, 1487–1495, doi:10.1111/j.1523-1747.2003.12614.x (2003).
Wang, Y. & Oram, J. F. Unsaturated fatty acids phosphorylate and destabilize ABCA1 through a phospholipase D2 pathway. The Journal of biological chemistry 280, 35896–35903, doi:10.1074/jbc.M506210200 (2005).
Lungu, A. O. et al. Cyclosporin A inhibits flow-mediated activation of endothelial nitric-oxide synthase by altering cholesterol content in caveolae. The Journal of biological chemistry 279, 48794–48800, doi:10.1074/jbc.M313897200 (2004).
Peterson, T. E. et al. Opposing effects of reactive oxygen species and cholesterol on endothelial nitric oxide synthase and endothelial cell caveolae. Circ Res 85, 29–37 (1999).
Zhao, Y. Y. et al. Persistent eNOS activation secondary to caveolin-1 deficiency induces pulmonary hypertension in mice and humans through PKG nitration. The Journal of clinical investigation 119, 2009–2018, doi:10.1172/jci33338 (2009).
Liu, B. et al. Incidence and risk of hypertension associated with vascular endothelial growth factor receptor tyrosine kinase inhibitors in cancer patients: a comprehensive network meta-analysis of 72 randomized controlled trials involving 30013 patients. Oncotarget. doi:10.18632/oncotarget.11813 (2016).
Lu, Z. et al. Decreased L-type Ca2 + current in cardiac myocytes of type 1 diabetic Akita mice due to reduced phosphatidylinositol 3-kinase signaling. Diabetes 56, 2780–2789, doi:10.2337/db06-1629 (2007).
Kass, I. J. & Sampson, N. S. Evaluation of the role of His447 in the reaction catalyzed by cholesterol oxidase. Biochemistry 37, 17990–18000 (1998).
Acknowledgements
This work was supported by NIH R01 GM084251 and GM100109 to MAF, NIH R01 DK108989 and VA Merit BX002263 to RZL, and an NIH F31 NRSA DK097957 to RKN. We thank G. di Paolo (Columbia University) for providing the PLD2−/− mice and W. Bollag for helpful discussions on the manuscript.
Author information
Authors and Affiliations
Contributions
R.K.N. conducted most of the experiments and initial data analysis and generated the first draft of the manuscript; J.Y.P. and R.Z.L. conducted the echocardiogram measurements, provided training on blood pressure measurement, and provided text and editorial comments; J.G. and N.S. conducted the free cholesterol measurements and provided text and editorial comments; D.A. performed genotyping, assisted R.K.N. experimentally, and provided input on the manuscript, and M.A.F. initiated the project and oversaw the experimental direction, data analysis, and final development of the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Nelson, R.K., Ya-Ping, J., Gadbery, J. et al. Phospholipase D2 loss results in increased blood pressure via inhibition of the endothelial nitric oxide synthase pathway. Sci Rep 7, 9112 (2017). https://doi.org/10.1038/s41598-017-09852-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-09852-4
- Springer Nature Limited
This article is cited by
-
Whole blood methylome-derived features to discriminate endocrine hypertension
Clinical Epigenetics (2022)
-
Phospholipase D functional ablation has a protective effect in an Alzheimer’s disease Caenorhabditis elegans model
Scientific Reports (2018)