
Abstract
Management of infection with hepatitis B virus (HBV) remains a global health problem. Persistence of stable covalently closed circular DNA (cccDNA) during HBV replication is responsible for modest curative efficacy of currently licensed drugs. Novel gene editing technologies, such as those based on CRISPR/Cas9, provide the means for permanently disabling cccDNA. However, efficient delivery of antiviral sequences to infected hepatocytes is challenging. A limiting factor is the large size of sequences encoding Cas9 from Streptococcus pyogenes, and resultant incompatibility with the popular single stranded adeno-associated viral vectors (ssAAVs). We thus explored the utility of ssAAVs for delivery of engineered CRISPR/Cas9 of Staphylococcus aureus (Sa), which is encoded by shorter DNA sequences. Short guide RNAs (sgRNAs) were designed with cognates in the S open reading frame of HBV and incorporated into AAVs that also encoded SaCas9. Intended targeted mutation of HBV DNA was observed after transduction of cells with the all-in-one vectors. Efficacy against HBV-infected hNTCP-HepG2 cells indicated that inactivation of cccDNA was successful. Analysis of likely off-target mutagenesis revealed no unintended sequence changes. Use of ssAAVs to deliver all components required to disable cccDNA by SaCas9 is novel and the technology has curative potential for HBV infection.
Similar content being viewed by others

Introduction
Chronic infection with hepatitis B virus (HBV) is responsible for a significant global disease burden1. There are currently about 240 million HBV chronic carriers in the world, and these individuals have an increased risk for life-threatening complications of cirrhosis and hepatocellular carcinoma. HBV gains access to hepatocytes through interaction with the human Na-taurocholate cotransporting polypeptide (hNTCP) receptor2, 3. Upon entry of virions into hepatocytes, relaxed circular DNA (rcDNA) is transported to the nucleus where it is released and repaired to form covalently closed circular DNA (cccDNA)4. Viral protein-coding mRNAs and pre-genomic RNA (pgRNA) are produced from this replication intermediate. pgRNA is the template for reverse transcription and formation of rcDNA then cccDNA. Currently licensed therapies, which include interferon-alpha derivatives, as well as nucleoside and nucleotide analogs, suppress viral replication but rarely eradicate the cccDNA reservoir. Therefore, focus has recently been placed on advancing potentially curative treatment approaches that are capable of eliminating HBV by disabling cccDNA permanently.
Several studies have explored the potential of using gene editing technologies to target cccDNA5. Zinc-finger nucleases (ZFNs)6, transcription activator-like effector nucleases (TALENs)7, and clustered regularly interspaced palindromic repeats (CRISPR) with CRISPR-associated protein (Cas9)8 have all been used in an attempt to inactivate cccDNA permanently. The rationale underlying use of gene editing to inactivate cccDNA is based on activation of the error-prone non-homologous end joining (NHEJ) repair pathway. Repeated cleavage of cccDNA by the gene editors followed by NHEJ eventually leads to targeted formation of insertions and deletions (indels) at the cognate site of the gene editors. The simplicity of employing RNA-guided CRISPR/Cas9 has allowed for its use in numerous applications, which include targeted disruption of cccDNA. There is growing evidence indicating that cccDNA may be mutated and in some cases eradicated using CRISPR/Cas99,10,11. Although application of CRISPR/Cas9 to treating chronic HBV infection has shown promise, efficient and safe delivery of the RNA-guided nucleases (RGNs) to HBV-infected hepatocytes remains a significant challenge.
Recombinant single stranded adeno-associated viruses (ssAAVs) are attractive vectors for gene therapy of liver diseases12. Typically there is a low immune response to AAVs following their administration in vivo. Pathology in humans caused by the parent parvoviruses has not been reported and their AAV derivatives have little toxicity. The molecular biology underlying generation of AAVs is now well understood. It is possible to alter capsid sequences of the vectors to confer desirable biological properties, such as tropism for particular tissues. A drawback of ssAAVs is that the size of the transgene that may be accommodated is limited to approximately 4.5 kb, and this is further reduced in self-complementary AAVs (scAAVs). This is particularly important for development of potentially therapeutic vectors that deliver sequences comprising gene editors derived from engineered CRISPR/Cas9. Cas9 from Streptococcus pyogenes (SpCas9) has been the most widely used and has been shown to inactivate HBV sequences effectively13. However, the open reading frame (ORF) of SpCas9 comprises approximately 4.1 kb. Together with regulatory sequences and a short guide RNA (sgRNA) cassette, elements comprising the Sp system are too large to be accommodated by one ssAAV. Various approaches have been employed to address this limitation of ssAAVs. These include exploiting the bilobed structure of SpCas9 and use of two vectors that together express a ‘scarless’ SpCas914. Generic methods of increasing ssAAV capacity, such as by using homologous recombination between overlapping sequences of different ssAAVs (reviewed in ref. 15), may also be useful. Although the approaches have been employed successfully, the requirement for more than one ssAAV complicates the methodology.
The recently described orthologous Cas9 that is derived from Staphylococcus aureus (SaCas9) is approximately 1 kb smaller than SpCas916. Sequences encoding SaCas9 as well as regulatory elements and the sgRNA cassette have been packaged into an ssAAV and used to edit endogenous genes in the liver. This ‘all-in-one’ system, which overcomes the need for using more than one AAV, has potential for application to therapeutic gene editing and may be useful for inactivation of genes of HBV. The approach has been used successfully to mutate CCR5, a host dependency factor that is required for HIV-1 infection by CCR5-tropic viruses17. To advance use of CRISPR/Cas9-based technology for treatment of HBV infection, we incorporated cassettes encoding SaCas9 and sgRNAs into ssAAVs. Targeting the HBV S open reading frame (ORF) resulted in efficient inactivation of HBV replication and mutation of cccDNA in cultured cells.
Results
Identification of an effective anti-HBV SaCas9 sgRNA
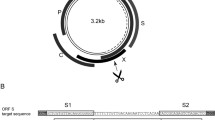
A panel of sgRNAs was designed to target the small surface (S) ORF of HBV that overlaps the polymerase (P) gene of HBV (Fig. 1a). The S protein is a viral envelope glycoprotein that is essential for viral entry and infectious virion formation. P carries out a vital role in reverse transcription of pgRNA and replication of the viral DNA. Target sites were selected on the basis of conserved status amongst HBV isolates and their being distinct from human genomic sequences. The 20 bp cognates of the sgRNAs were each common to all HBV genotypes and analysis using the Off-Spotter tool18 showed that each guide had a minimum of two mismatches with possible off-target sites of the human genome. Ten target sites were selected and guide cassettes were constructed to express sgRNAs as a single transcript comprising the SaCas9 tracrRNA and sequence of 20 nucleotides complementary to the HBV target sequence. To evaluate anti-HBV efficacy, DNA encoding SaCas9, sgRNAs and an HBV replication-competent plasmid were initially transfected into Huh7 cells19. Eight of the ten sgRNAs reduced concentrations of HBV S antigen (HBsAg) in the culture supernatants by 50–95% when compared to controls, which were a non-specific sgRNA or an sgRNA targeted to HIV (Fig. 1b). Since nuclease activity is required for potent and subsequent mutation of HBV cccDNA, catalytically dead SaCas9 (dSaCas9) was included as an additional control. Transfection of plasmids encoding each of sgRNA-6, -7, -9 and -10 reduced HBsAg in the culture supernatants when co-transfected with the dSaCas9 sequence. These data suggest that some of the repressive activity resulted from transcriptional interference, rather than from being caused by DNA cleavage and formation of indels following error-prone NHEJ. Conversely sgRNA-8 was inactive with dSaCas9 but effectively reduced HBsAg concentrations in the presence of nuclease-competent SaCas9. This indicated that the inhibitory effect of sgRNA-8 on release of the HBV replication marker from cultured cells resulted from cleavage-mediated mutagenesis. An assay using T7 endonuclease 1 (T7E1) to analyze re-annealed amplicons covering the sgRNA-8 target confirmed indel formation by SaCas9 with this guide (Fig. 1c). sgRNA-8 was thus identified as the lead and was selected for further investigation.

Screening of panel of anti-HBV SaCas9-sgRNAs. (a) Schematic illustration of the HBV genome with indicated open reading frames (ORFs). The viral rcDNA, with regulatory cis elements, is indicated at the center and coordinates are given relative to the single EcoRI site. The four HBV ORFs are shown as arrows immediately surrounding the genome and the four major viral transcripts are indicated as outermost arrows. The sgRNA target sites are depicted as arrows encompassing most of the sequence of the surface (S) region of the virus. (b) Huh7 cells were co-transfected with plasmids encoding i) a replication competent molecular clone of HBV (pCH-9/3091), ii) each of a panel of cassettes expressing S-targeting single guide RNAs (sgRNAs) and iii) either SaCas9 or catalytically dead SaCas9 (dSaCas9) expressed from a CMV promoter/enhancer element. The concentrations of HBV S antigen were measured using an ELISA and data are presented relative to the values for the mean for a non-specific sgRNA sequence. A sgRNA targeted to HIV (HIV-sgRNA) was included as an additional negative control. Data are represented as the means and the error bars indicate standard deviation. (c) HEK293T cells were transfected with SaCas9 and sgRNA-8 with pCH-9/3091. A T7E1 assay was performed on extrachromosomal cellular DNA. Arrows indicate the larger intact PCR product (~550 bp) and smaller digested fragment (~280 bp). The measured target disruption is indicated below as a percentage. (d) Huh7 cells were transfected with SaCas9 and sgRNA-8 targeting a greater than genome length molecular clone (1.3 × ) for HBV sub-genotypes A1, A2 and D3. Data are presented as in b.
To evaluate whether sgRNA-8 could target multiple sub-genotypes of HBV, DNA encoding the cassette was co-transfected with replication-competent clones of sub-genotypes A1-a, A1-b, A2 and D320 (Fig. 1d and Supp. Fig. 1). Suppression of S antigen was efficient and dependent on presence of catalytically active saCas9. The efficacy was similar to that achieved when sequences encoding the RGN were co-transfected with pCH-9/3091, a genotype D-derived sequence (Fig. 1b and d). The target of sgRNA-8 is also conserved in other HBV isolates (Supp. Table 1) and indicates that the gene editor should be active against most variants of the virus.
Evaluating inhibition of HBV replication in cultured HepG2.2.15 cells using an ssAAV to deliver cassettes encoding SaCas9 and sgRNA-8
Both cassettes of SaCas9 and sgRNA-8 were incorporated into an ssAAV to assess functionality of the all-in-one vector. The sequences comprising the RGN were packaged into an AAV2 vector, termed ssAAV-SaCas9-sgRNA-8, and used to transduce HepG2.2.15 cells21, which replicates HBV from a stable integrant. With these all-in-one vectors, the sgRNAs were each transcribed from a U6 Pol III promoter. The SaCas9 sequence was expressed from a separate cassette regulated by a CMV Pol II enhancer and promoter element. Similar concentrations of SaCas9 mRNA and different sgRNAs were verified using qRT-PCR (Fig. 2a), which indicates efficient expression of each component of the RGNs when produced from various combinations in individual ssAAV2s. After treatment of HepG2.2.15 cells with ssAAV-SaCas9-sgRNA-8, there was a rapid reduction in the concentration of the HBsAg in the culture supernatant. Baseline levels were reached over the 27 day period of the investigation (Fig. 2b). Conversely the control sgRNAs did not cause inhibition of secretion of the marker of HBV replication. Similarly, secretion of HBV viral particle equivalents (VPEs), as measured using qPCR to quantitate viral DNA in the culture supernatants, was undetectable in the ssAAV-SaCas9-sgRNA-8-treated cells (Fig. 2c). Analysis of HBV RNA revealed a reduction of approximately 80% of viral S and pgRNA transcripts (Fig. 2d).

Inhibition of markers of HBV replication following delivery of SaCas9-sgRNA-8 with ssAAVs. (a) HepG2.2.15 cells were transduced with ssAAVs that encoded HBV-targeting sgRNA-8, a non-specific guide, HIV-targeting sgRNA or a GFP reporter. RNA was extracted 21 days after transduction and subjected to qRT-PCR. Primers were complementary to (i) sgRNA-8, (ii) the non-specific control sgRNA, (iii) all sgRNAs or (iv) SaCas9 sequences. (b) Concentrations of HBsAg were measured in the culture supernatants of cells that had been transduced with ssAAVs encoding SaCas9 and sgRNA-8, sgRNA targeting HIV or a non-specific sequence. Analysis was carried out over a time course of 27 days after transduction. (c) Assay of viral particle equivalents (VPEs) in culture supernatants of HepG2.2.15 cells after transduction with HBV-targeting and control ssAAVs. Cells were washed and measurements were based on qPCR carried out on extracts of culture supernatants taken from days 12 to 21 after transduction. (d) HBV RNA was measured in transduced cells at day 21 after transduction with the indicated ssAAVs. qRT-PCR was carried out with primers specific for surface (S) or pregenomic HBV RNA sequences.
Formation of indels in the target viral DNA was evaluated using a T7E1 assay and drop-off droplet digital PCR (ddPCRTM)22. The T7E1 assay revealed that 46% of targets had indels by day 15 after treatment with ssAAV-SaCas9-sgRNA-8, and the proportion of mutated sequences increased to 61% by day 21 (Fig. 3a). The drop-off ddPCRTM analysis demonstrated indel formation in approximately 30% of the targets by day 21 (Fig. 3b). The difference between results obtained for the T7E1 and ddPCRTM assays may be a result of a more stringent but less sensitive nature of the ddPCRTM assay. Tracking of indels by decomposition (TIDE), which uses a decompression algorithm to decode automated sequencing, was employed to analyze the types of mutations at the target site23. Results showed a strong bias towards single or double base pair deletions and single insertions (Fig. 3c). These mutations continued to increase from day 15 to day 21. Importantly, such sequence changes would cause frame shift mutations and major disruption to expression of HBV genes. To evaluate effects on cccDNA, extracts from HepG2.2.15 cells were treated with plasmid-safe DNase to eliminate cellular DNA, rcDNA and linear DNA containing viral sequences7, 24. As previously described, control murine samples lacking cccDNA, but abundant in other forms of HBV DNA, were used to verify elimination of non-cccDNA. Indels were detected in these preparations of cccDNA, although at lower levels than in the DNA extracts that were not treated with plasmid-safe. The T7E1 assay revealed mutation of 28% of targets of viral sequences at day 15 and indel formation was 33% by day 21 (Fig. 3a). These results were corroborated by using the drop-off ddPCRTM analysis. Mutations may be caused by action of CRISPR/SaCas9 on viral integrants, and transmission of the changed sequences to pgRNA, rcDNA then cccDNA. Alternatively mutations detected in cccDNA may result from direct action of sgRNA-8 and saCas9 on this viral replication intermediate. Evidence of mutation of cccDNA prompted investigation of efficacy of the ssAAV-SaCas9-sgRNA-8 in cultured liver-derived cells that are infectable by HBV.

Targeted mutation of HBV DNA by AAV2-SaCas9-sgRNA-8. (a) Total DNA extracted from ssAAV-transduced cells with and without plasmid-safe (PS) treatment was subjected to T7E1 analysis to detect insertions and deletions (indels) in viral DNA. The percentages of HBV DNA sequences containing targeted mutations at 15 and 21 days after transduction are indicated for sgRNA-8 and the two control guides. (b) Drop-off ddPCR™ assay to detect indels in ssAAV-transduced cells. Analysis was performed 15 and 21 days after transduction with AAV2-SaCas9-sgRNA-8 or the HIV-targeting control vector. (c) Tracking of indels by decomposition (TIDE) was carried out on raw Sanger sequencing chromatograms that were generated using amplicons of viral sequences as the template. Sizes of the predicted indels, together with the proportions of the total that they represent, are presented. Analysis was carried out on samples extracted from cells that had been transduced at 15 and 21 days after transduction with ssAAVs.
Eradication of cccDNA from HBV-infected hNTCP-HepG2
To evaluate an effect of the combination of SaCas9 and sgRNA-8 on cccDNA more precisely, the hNTCP-HepG2 cell line was used as a model. Stable expression of NTCP in the HepG2-derived cells enables them to be infected with HBV2. The importance of using the model to evaluate efficacy of ssAAV-SaCas9-sgRNA-8 is that HBV gene expression in the infected cells is derived from cccDNA2, and not an integrated sequence as is the case with the HepG2.2.15 line. Inhibition of HBV replication following transduction of hNTCP-HepG2 cells by the RGN encoding ssAAV may thus be directly ascribed to an effect on cccDNA. hNTCP-HepG2 cells were thus infected with HBV prior to treatment with the ssAAV-SaCas9-sgRNA vectors. By day 27, concentrations of HBsAg steadily decreased to 55% of that measured at the start (Fig. 4a and Supp. Fig. 2), secreted VPEs were reduced by approximately 60% (Fig. 4b) and there was an approximate fourfold decline in total intracellular HBV DNA (Fig. 4c).

Evaluation of targeted mutation of cccDNA in HBV-infected hNTCP-HepG2 cells. Infection with HBV was carried out 24 hours prior to transduction of hNTCP-HepG2 cells with ssAAVs. Concentrations of (a) HBsAg and (b) viral particle equivalents (VPEs) in the culture supernatant were determined as described in Fig. 2b and c, respectively. Data are represented as the means and the error bars indicate standard deviation. Statistical differences are indicated by an asterisk (*p < 0.05). (c) Intracellular HBV DNA was measured on day 27 after transduction of cells with the indicated ssAAVs. (d) DNA from culture supernatants of hNTCP-HepG2 cells was extracted with or without treating with plasmid-safe DNase for the indicated times and subjected to analysis using qPCR with primers specific for HBV DNA. (e) Representative agarose gel electrophoresis to detect HBV DNA from cell extracts and supernatants with or without treatment with plasmid-safe DNase. (f) Total DNA was extracted from HBV-infected hNTCP-HepG2 cells at day 27 after transduction with ssAAVs. cccDNA was quantified using qPCR and values are presented relative to the mean for the control non-specific guide RNA. Detection of indels in cccDNA using (g) T7E1 or (h) drop-off ddPCRTM assays. Comparisons were made using a two-tailed Student’s t-test, and significant statistical differences to the control samples treated with the non-specific guide are indicated (*p < 0.05).
To analyze mutagenic effects on cccDNA from HBV-infected hNTCP-HepG2 cells, DNA was extracted from these cells after transduction. Extended digestion with plasmid-safe nuclease eliminated the detectable rcDNA and cellular genomic DNA, indicating that the remaining HBV DNA comprised cccDNA (Fig. 4d and e). Quantification of cccDNA at day 27 showed an approximately 80% reduction in this DNA species after treatment of cells with ssAAV-SaCas9-sgRNA-8 (Fig. 4f). A decrease in intracellular cccDNA concentrations has previously been reported after treatment of HBV replicating cells with virus-targeting SpCas9 and guide sequences25, 26. Interestingly, analysis using T7E1 and drop-off ddPCRTM revealed no or little indel formation (Fig. 4g and h). Collectively the data suggest that the Sa-derived RGNs preferentially cause degradation of cccDNA. Substrates for the T7E1 and drop-off ddPCRTM assays may comprise the remaining population of wild type sequences, which would account for the low indel detection.
Analysis of off-target effects of SaCas9-sgRNA-8
Mutation at untargeted sites within the host genome may have serious consequences for potentially therapeutic RGNs. Analysis of unintended sequence changes at sites that have homology to the sgRNA cognates is thus important to enable clinical translation of the gene editing technology. Possible off-target sites were thus identified for SaCas9-sgRNA-8 using the Off-spotter online tool18. Ten sites were predicted as being the most likely sequences where unintended action of SaCas9-sgRNA-8 would occur. The top three sites were verified by additional predictive tools, namely COSMID27 and Cas-Offinder tools28 (Supp. Fig. 3). After transfection of HEK293T cells with plasmids encoding SaCas9 and the sgRNA-8 cassette, the T7E1 assay failed to detect indels at the identified off-target sites (Fig. 5). To expand the analysis of non-specific action of the anti-HBV RGN, we also took into account the importance of the seed sequence29. This element comprises nucleotides at the 3’ end of the sgRNA and is located adjacent to the protospacer adjacent motif (PAM). Previous work using an objective off-target assay that captured all possible break sites within the genome showed that the majority of sites maintained complementarity with the first 3 bp of the seed16. An additional ten most likely off-target sites, which maintained the 3 bp seed, were thus identified using the Off-Spotter tool (Supp. Fig. 3). As before, no indels were observed at these sites using the T7E1 nuclease assay (Fig. 5a). Next generation sequencing (NGS) was carried out to achieve more sensitive detection of mutagenesis by sgRNA-8. Sites 1, 2 and 3 (OT1, OT2 and OT3), which were predicted by the three bioinformatics tools to be the most likely off-target sites, were subjected to the analysis (Fig. 5b). Evaluation of sequences extracted from HepG2.2.15 cells was performed on day 21 after transduction with AAVs. Data showed presence of indels at approximately 23% of HBV target sites, which is similar to the quantification using ddPCRTM (Fig. 3b). Conversely NGS showed mutagenesis at less than 0.05% of the off-target sites (Fig. 5b). Low level mutagenesis of HBV DNA extracted from HBV-infected hNTCP cells corroborated analysis using the T7E1 and ddPCRTM assays (Fig. 4g and h). Limited detectable sequence changes at the most likely predicted off-target sites of sgRNA-8 suggest that the guide functions specifically when used in conjunction with SaCas9.

Evaluation of SaCas9-sgRNA8 off-target activity. (a) The top ten off-target sites were identified using the Off-spotter online tool with either a no seed prediction or with a fixed 3 bp seed panel. HEK293 cells were transfected with the SaCas9 vector and sgRNA-8 or non-specific guide control. Genomic DNA was extracted 48 hours after transfection, and a T7E1 assay was performed on the off-target sites. pCH-9/3091 was included to verify an HBV on-target signal. (b) Next generation sequencing analysis to detect insertions and deletions at on-target and off-target sites. The number of reads with insertions and deletions are indicated and the percentage of indels (% Indel) is calculated from the total reads.
Discussion
Chronic infection with HBV remains a significant global health problem. Although current treatments provide some benefits to carriers of the virus, licensed drugs rarely eliminate the stable viral replication intermediate comprising cccDNA. Gene-editing technologies potentially provide the means to inactivate cccDNA permanently. However, current efforts to develop gene-editing technologies for HBV treatment are hampered by difficulties with adequate delivery efficiency. ssAAVs are popular vectors that show promise for many applications of gene therapy, but a drawback of these vectors is the limited size of the transgenes that they may accommodate. DNA sequences encoding dimeric ZFNs6, TALENs7 and Sp RGN components exceed the limit of approximately 4.5 kb. A combination of ssAAVs, which is not ideal, is thus usually required to deliver the complete gene editors. In this study, we have demonstrated the feasibility of using well-characterized ssAAVs for delivery of two cassettes comprising a complete HBV-targeting SaCas9 and guide assembly.
In an unusually compact arrangement of the HBV genome, protein-coding sequences overlap with each other and encompass the entire 3.2 kb that make up the viral genome (Fig. 1a). Use of different reading frames enables viral DNA sequences to encode more than one protein. Moreover, cis elements that are responsible for regulating gene expression are embedded within the protein coding sequences, adding to the multifunctional nature of the viral genome. This highly economical use of its DNA constrains sequence plasticity of HBV. The sequence of HBV that was targeted in this study spanned the S ORF, which overlaps P in its entirety. Mutations introduced through action of gene editors will therefore change both Surface and Polymerase proteins to effect highly efficient inhibition of viral replication. Analysis using T7E1 and drop-off ddPCRTM assays confirmed mutation of the viral DNA with concomitant decrease in HBV replication in HepG2.2.15 cells. Interestingly the finding in hNTCP-HepG2 cells was different. Rather than targeted mutation, a marked decrease in the concentration of cccDNA was demonstrated. This bias towards eradication, and low indel detection, of episomal DNA has been noted by others when using SpCas9 to target HBV30. The effect has been attributed to delayed repair of cleaved circular DNA, thereby enhancing its degradation10. Although indels have been demonstrated in cccDNA, the effect is thought to occur when CRISPR/Cas9-treated cells contain high concentrations of cccDNA11. The fate of mutated cccDNA within a cell is not established. If the disabled cccDNA is eliminated, the risk of genotoxic integration into the host genome would be diminished.
Specificity of gene editors is crucial to their safety and clinical utility. Evaluating off-target action and deciding on acceptable, if any, unintended action is difficult. Use of sensitive unbiased methodology is essential and assays such as BLESS16 and Guide-seq.31 may be capable of identifying global genomic off-target effects with sufficient depth to inform decisions about safety of particular gene editors. Analyses of SpCas9 have shown that the first generation gene editors may be prone to unintended off-target cleavage (reviewed in ref. 32). This prompted investigation aimed at improving specificity, which amongst other methods entailed truncation of guides33 and rational engineering of the SpCas9 endonuclease34. The SaCas9 has an improved on-target profile when compared to that of SpCas916. Combination of SaCas9 with its guide may be intrinsically more specific for an intended target than the RGN derived from S. pyogenes, and reasons for this include the following: 1) sgRNAs derived from S. aureus are less tolerant of mismatches; 2) an extended PAM sequence needed for SaCas9 binding reduces non-specific interactions; and 3) there is a requirement for a longer seed sequence. Furthermore, modifications have recently been made to SaCas9 to reduce non-specific interactions with DNA, which increases the dependence of cleavage on interaction of the guide with its cognate34. These features indicate that the good fidelity of repurposed SaCas9 may make it an RGN with therapeutic potential.
ssAAVs are being developed to treat several hereditary liver diseases, and they show promise in a clinical trial35. Accommodating sequences encoding all components of S. aureus - derived CRISPR/Cas9 in a recombinant ssAAVs is very convenient. The good anti-HBV efficacy demonstrated here suggests that these vectors are suitable for evaluation in vivo and may also be applicable to permanently eradicating chronic HBV infection
Materials and Methods
Plasmids encoding SaCas9 and HBV target sequences
A sgRNA cloning cassette was generated from a gBLOCK (IDT, Coralville, IA, USA) containing a BbsI site for sgRNA insertion (Supp. Table 2). The gBLOCK was amplified using a U6-F and tracrRNA-R primer set (Supp. Table 3) with KAPA Taq ReadyMix (Roche, Basel, Switzerland) and cloned into pTZ57R/T using the insTAclone PCR cloning Kit according to manufacturer’s instructions (Thermo Fisher, MA, USA). DNA encoding targeting sequences of the sgRNAs were synthesized as oligonucleotides and inserted into the BbsI site of pTZ-SaU6-sgRNA. Sequences were verified using standard Sanger sequencing procedures with the U6-F primer (Supp. Table 2).
The pAAV-CMV-SaCas9-Bgh-U6sgRNA expression vector was kindly donated by Dr Feng Zhang16. The pAAV-CMV-dSaCas9 vector was generated using Gibson Assembly® (NEB, MA, USA) to insert synthesized DNA gBLOCKs (IDT, IA, USA) harboring the D10A and H557A mutations (Supp. Table 3). Nickase intermediates were used to generate a catalytically dead double-mutant dSaCas9. To insert HBV sgRNA-8 into the ssAAV vector, the pTZ-U6-sgRNA-HBV was digested with KpnI and NotI and the purified fragments were ligated into a similarly digested pAAV-CMV-SaCas9-Bgh vector to generate the pAAV-CMV-SaCas9-Bgh-U6-sgRNA-HBV. The HBV replication competent plasmid, pCH-9/3091, has been described previously19. HBV-1.3 × A1-a, HBV-1.3 × A2 and HBV-1.3 × D3 were kindly donated by Prof. Anna Kramvis36 and HBV-1.3 × A-b was generated using similar standard procedures. Preparation of pCMV-GFP has been previously described37.
Cell culture and transfections
Human embryonic kidney (HEK293T), Huh7 and HepG2.2.15 cell lines were maintained in Dulbecco’s modified Eagle’s medium (DMEM; BioWhittaker, MD, USA) supplemented with 10% fetal calf serum (FCS) (Thermo Fisher, MA, USA). The hNTCP-HepG2 cells38 were maintained in DMEM/F12 + GlutaMAX™-I medium (Thermo Fisher, MA, USA) supplemented with 10% FCS, 10 mM HEPES, 0.05 mM Hydrocortisone, 0.005 mg/ml insulin, and 0.4 mg/ml G418 (DMEM/F12-complete medium). Unless otherwise stated, the cell lines were cultured at 37 °C with 5% CO2.
Screening of anti-HBV sgRNAs
For HBV knockdown and off-target analysis, HEK293T or Huh7 cells were seeded at 40,000 cells per well in 48-well plates, 24 hours prior to transfection. The cells were transfected with 200 ng of SaCas9 or dSaCas9 vector with an equal amount of pTZ-U6-sgRNA using Lipofectamine® 3000 according to the manufacturer’s instructions (Invitrogen™, Thermo Fisher, MA, USA). Forty eight hours after transfection HBsAg was measured in the supernatant using the Monolisa™ HBs Ag ULTRA kit (Bio-Rad, CA, USA). DNA was extracted using the KAPA Express Extract Kit and the genomic target sites amplified from 1 μl of lysate using primers described in Supp. Table 4. The KAPA2G Robust HotStart ReadyMix (Roche, Basel, Switzerland) was used for PCR. A T7E1 assay was performed on the amplicons as described below.
ssAAV production
ssAAVs were produced using an in-house protocol. Briefly, five 15 cm2 plates were seeded with 3.75 × 106 HEK293T cells 72 hours prior to transfection. The cells were transfected with 10 μg of Adenohelper plasmid, 6 μg pAAV-CMV-SaCas9-Bgh-U6-sgRNA vector and 8 μg of Rep-Cap2 plasmid (AAV2) with PEI MAX at a 1:5 ratio of DNA to PEI. The Adenohelper and Rep-Cap2 plasmids were kindly provided by Dr. Dirk Grimm. The pAAV-CAG-GFP control vector was obtained from Addgene (#11150). The supernatants were collected on days 1, 3, 5 and 7 after transfection, clarified through 0.22 micron filters and the ssAAVs then precipitated using PEG 8000 after overnight incubation at 4 °C. The precipitated virus was pelleted by centrifugation. In parallel, the AAV-containing cells were lysed using freeze-thaw cycles and sonication in viral lysis solution. After centrifugation to clear the lysate, the supernatant was used to resuspend the PEG-precipitated ssAAV pellet. ssAAVs were separated on an Iodixanol gradient and the fraction containing the ssAAVs (40%) was collected and stored at −80 °C until needed.
Concentrating infectious HBV particles
HepG2.2.15 cells were seeded at 50% confluence in 175 cm2 flasks and the supernatant thereafter collected on days 3 and 5. The supernatant was clarified through a 0.2 micron filter and the HBV particles were PEG-precipitated after overnight incubation at 4 °C. The precipitated virions were pelleted by centrifugation at 4 °C at 4,000 × g for 30 minutes and resuspended in 200 µl of virus concentrating solution. The concentrated virus was quantified by qPCR using ssoFAST Eva Green supermix (Bio-Rad, CA, USA) with primers complementary to the S region of HBV (Supp. Table 4). Copies of VPEs were determined using the Acrometrix™ HBV panel to generate a standard curve (Thermo Fisher, MA, USA).
ssAAV transduction of HBV-producing cell lines
The HepG2.2.15 line was seeded at 40,000 cells per well in a 48-well plate and transduced with 1 × 105 genome copies (GC) of ssAAV per cell. Cells were maintained at 30 °C and 5% CO2. The hNTCP-HepG2 cells were seeded in DMEM/F12 complete medium at 20,000 cells per well in a 96-well plate. The medium was replaced after 24 hours with complete media containing 3% dimethyl sulfoxide (DMSO) and incubated for 6 hours. Infection was carried out using 3 × 103 GC of HBV per cell in DMEM/F12 complete medium containing 3% DMSO and 4% PEG-8000 (Sigma, MO, USA). Sixteen hours after HBV infection the cells were washed three times with PBS and medium containing 4 × 105 GC of ssAAV-SaCas9-U6sgRNA-8 per cell added. AAV transductions were repeated on days 3 and 6 with both the HepG2.2.15 and hNTCP-HepG2 cells.
Quantification of HBV viral particle equivalents
HBV genomes were extracted from 50 μl of culture supernatant using the QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany). Five microliters of extracted HBV DNA was analyzed using qPCR with FastStart Universal SYBR qPCR Master (Roche, Basel, Switzerland). Quantification of VPEs was carried out as described above.
Detection and quantification of RNA
Total RNA was extracted with TRIzol (Thermo Fisher, MA, USA) following procedures recommended by the manufacturer. One microgram of RNA was reverse transcribed using the Quantitect Reverse Transcription Kit (Qiagen, Hilden, Germany), and 1 μl of the cDNA was used in a qPCR with the FastStart Universal SYBR Master (Roche, Basel, Switzerland). Concentrations of SaCas9 and HBV RNA were determined relative to mRNA of GAPDH, while the sgRNAs were measured relative to hU6snRNA using primers described in Supp. Table 4.
Preparation and quantification of HBV cccDNA
Total cellular DNA was extracted using QIAamp DNA Blood Mini Kit (Qiagen, Hilden, Germany) and 200 ng of extracted DNA was subjected to plasmid-safe digestion (Epicenter, WI, USA) in a total reaction volume of 20 μl. The extraction procedure, with DNA from HBV transgenic mice as control for elimination of non-cccDNA, has been described previously7, 24. Digestions were carried out for 16 hours at 37 °C and the enzyme was inactivated following incubation at 70 °C for 30 min. Five microliters of treated DNA was amplified using FastStart Universal SYBR Master (Roche, Basel, Switzerland) with S primers (Supp. Table 4) and HBV DNA quantified as described earlier.
Detection of Indels
The target site of the sgRNA was amplified using KAPA2G Robust HotStart ReadyMix (Roche, Basel, Switzerland) with S primers (Supp. Table 4) and either 200 ng of extracted DNA or 5 μl of plasmid-safe treated DNA as the template. The PCR products were purified using a GeneJET PCR purification kit (Thermo Fisher, MA, USA), and 400 ng of the purified products were subjected to the T7E1 assay as previously described7. For TIDE analysis, the PCR products were analyzed by Sanger sequencing (Inqaba Biotec, Pretoria, South Africa) and the chromatograms analyzed using the TIDE online tool23.
Drop-Off ddPCRTM
Drop-off assays were performed using the ddPCRTM instrument from Bio-Rad (Hercules, CA, USA) as described before39. Briefly, either 200 ng of total extracted DNA or 5 μl of plasmid-safe treated DNA was mixed with ddPCR™ supermix. Primers specific for the target regions were included together with a FAM-conjugated reference probe and HEX-conjugated probe for the sgRNA-8 cognate (Supp. Tables 4 and 5). The mixture was transferred to a DG8™ cartridge and droplets were generated using 70 μl of ddPCR™ oil for probes. The droplets were transferred to a 96-well plate and the plate sealed. DNA was amplified after initial denaturation at 95 °C for 10 minute, then 40 cycles of 95 °C for 30 seconds, 55 °C for 15 seconds and 72 °C for 1 minute and a final incubation at 98 °C for 10 min. The droplets were analyzed on a QX200™ droplet reader using QuantaSoft™ software. Total intracellular HBV DNA was determined as an absolute count.
Off-target site analysis
Off-target sites within the host genome were identified using the Off-spotter tool18 for a sgRNA-8 recognition sequences with or without a 3 bp seed prediction. DNA was extracted from transiently transfected HEK293T cells and the target sites amplified using Kapa2G Robust HotStart ReadyMix (Roche, Basel, Switzerland) with primers specific for the predicted sites (Supp. Table 4). The T7E1 assays were performed as described earlier.
Next-generation sequencing
The on-target and selected off-targets were amplified using Q5 hotstart high-fidelity PCR master mix (NEB, MA, USA). Amplicons were purified using the QiaQuick PCR purification kit (Qiagen, Hilden, Germany). Samples were sequenced using an Illumina instrument (HiSeq. 2500) and processing was outsourced to Inqaba Biotec (Pretoria, South Africa). The Fastq files were analyzed using the CRISPResso online indel prediction tool40.
Statistical analysis
Statistical calculations were performed using GraphPad Prism 4 software (GraphPad, CA, USA). Comparisons were determined using an unpaired two-tailed Student’s t-test and differences were considered statistically significant when p was less than 0.05.
Data Availability Statement
Materials, data and associated protocols will be made available on request.
References
Stanaway, J. D. et al. The global burden of viral hepatitis from 1990 to 2013: findings from the Global Burden of Disease Study 2013. The Lancet 388, 1081–1088 (2016).
Yan, H. et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 1, e00049 (2012).
Ni, Y. et al. Hepatitis B and D viruses exploit sodium taurocholate co-transporting polypeptide for species-specific entry into hepatocytes. Gastroenterology 146, 1070–1083 (2014).
Morikawa, K., Suda, G. & Sakamoto, N. Viral life cycle of hepatitis B virus: Host factors and druggable targets. Hepatology Research 46, 871–877 (2016).
Ely, A., Moyo, B. & Arbuthnot, P. Progress With Developing Use of Gene Editing To Cure Chronic Infection With Hepatitis B Virus. Molecular Therapy 24, 671–677 (2016).
Cradick, T. J., Keck, K., Bradshaw, S., Jamieson, A. C. & McCaffrey, A. P. Zinc-finger Nucleases as a Novel Therapeutic Strategy for Targeting Hepatitis B Virus DNAs. Molecular Therapy 18, 947–954 (2010).
Bloom, K., Ely, A., Mussolino, C., Cathomen, T. & Arbuthnot, P. Inactivation of hepatitis B virus replication in cultured cells and in vivo with engineered transcription activator-like effector nucleases. Molecular Therapy 21, 1889–1897 (2013).
Lin, Y. et al. CRISPR/Cas9 systems have off-target activity with insertions or deletions between target DNA and guide RNA sequences. Nucleic Acids Research (2014).
Dong, C. et al. Targeting hepatitis B virus cccDNA by CRISPR/Cas9 nuclease efficiently inhibits viral replication. Antiviral Research 118, 110–117 (2015).
Liu, X., Hao, R., Chen, S., Guo, D. & Chen, Y. Inhibition of hepatitis B virus by the CRISPR/Cas9 system via targeting the conserved regions of the viral genome. Journal of General Virology 96, 2252–2261 (2015).
Seeger, C. & Sohn, J. A. Targeting Hepatitis B Virus With CRISPR/Cas9. Molecular Therapy - Nucleic Acids 3, Article e216 (2014).
Kattenhorn, L. M. et al. Adeno-Associated Virus Gene Therapy for Liver Disease. Human Gene Therapy 27, 947–961 (2016).
Moyo, B., Bloom, K., Scott, T., Ely, A. & Arbuthnot, P. Advances with Using Crispr/Cas-Mediated Gene Editing to Treat Infections with Hepatitis B Virus and Hepatitis C Virus. Virus Research (2017).
Chew, W. L. et al. A multifunctional AAV-CRISPR-Cas9 and its host response. Nature Methods 13, 868–874 (2016).
Chamberlain, K., Riyad, J. M. & Weber, T. Expressing Transgenes That Exceed the Packaging Capacity of Adeno-Associated Virus Capsids. Human gene therapy methods 27, 1–12 (2016).
Ran, F. A. et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015).
Friedland, A. E. et al. Characterization of Staphylococcus aureus Cas9: a smaller Cas9 for all-in-one adeno-associated virus delivery and paired nickase applications. Genome Biol 16, 257 (2015).
Pliatsika, V. & Rigoutsos, I. “Off-Spotter”: very fast and exhaustive enumeration of genomic lookalikes for designing CRISPR/Cas guide RNAs. Biology direct 10, 4 (2015).
Nassal, M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. Journal of virology 66, 4107–4116 (1992).
Sunbul, M. Hepatitis B virus genotypes: Global distribution and clinical importance. World Journal of Gastroenterology 20, 5427–5434 (2014).
Sells, M. A., Chen, M. L. & Acs, G. Production of hepatitis B virus particles in Hep G2 cells transfected with cloned hepatitis B virus DNA. Proceedings of the National Academy of Sciences of the United States of America 84, 1005–1009 (1987).
Hindson, C. M. et al. Absolute quantification by droplet digital PCR versus analog real-time PCR. Nature Methods 10, 1003–1005 (2013).
Brinkman, E. K., Chen, T., Amendola, M. & van Steensel, B. Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Research 42, e168 (2014).
Bloom, K., Ely, A. & Arbuthnot, P. A T7 Endonuclease I Assay to Detect Talen-Mediated Targeted Mutation of HBV cccDNA. Methods Mol Biol 1540, 85–95 (2017).
Lin, S. R. et al. The CRISPR/Cas9 System Facilitates Clearance of the Intrahepatic HBV Templates In Vivo. Molecular Therapy Nucleic Acids 3, e186 (2014).
Kennedy, E. M. et al. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology 476, 196–205 (2015).
Cradick, T. J., Qiu, P., Lee, C. M., Fine, E. J. & Bao, G. COSMID: A Web-based Tool for Identifying and Validating CRISPR/Cas Off-target Sites. Molecular Therapy Nucleic Acids 3, e214 (2014).
Bae, S., Park, J. & Kim, J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics 30, 1473–1475 (2014).
Sternberg, S. H., Redding, S., Jinek, M., Greene, E. C. & Doudna, J. A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature 507, 62–67 (2014).
Ramanan, V. et al. CRISPR/Cas9 cleavage of viral DNA efficiently suppresses hepatitis B virus. Nature Scientific Reports 5, 10833 (2015).
Tsai, S. Q. et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nature Biotechnoloy 33, 187–197 (2015).
Wu, X., Kriz, A. J. & Sharp, P. A. Target specificity of the CRISPR-Cas9 system. Quantitative biology 2, 59–70 (2014).
Fu, Y., Sander, J. D., Reyon, D., Cascio, V. M. & Joung, J. K. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nature biotechnology 32, 279–284 (2014).
Slaymaker, I. M. et al. Rationally engineered Cas9 nucleases with improved specificity. Science 351, 84–88 (2016).
Asokan, A., Schaffer, D. V. & Jude Samulski, R. The AAV Vector Toolkit: Poised at the Clinical Crossroads. Molecular Therapy 20, 699–708 (2012).
Bhoola, N. H., Reumann, K., Kew, M. C., Will, H. & Kramvis, A. Construction of replication competent plasmids of hepatitis B virus subgenotypes A1, A2 and D3 with authentic endogenous promoters. Journal of Virological Methods 203, 54–64 (2014).
Passman, M., Weinberg, M., Kew, M. & Arbuthnot, P. In situ demonstration of inhibitory effects of hammerhead ribozymes that are targeted to the hepatitis Bx sequence in cultured cells. Biochemical and biophysical research communications 268, 728–733 (2000).
Watashi, K. et al. Cyclosporin A and its analogs inhibit hepatitis B virus entry into cultured hepatocytes through targeting a membrane transporter, sodium taurocholate cotransporting polypeptide (NTCP). Hepatology 59, 1726–1737 (2014).
Findlay, S. D., Vincent, K. M., Berman, J. R. & Postovit, L.-M. A Digital PCR-Based Method for Efficient and Highly Specific Screening of Genome Edited Cells. PloS one 11, e0153901 (2016).
Pinello, L. et al. Analyzing CRISPR genome-editing experiments with CRISPResso. Nat Biotech 34, 695–697 (2016).
Acknowledgements
Financial assistance for work carried out in the authors’ laboratory, which was received from the South African National Research Foundation (NRF, GUNs 81768, 81692, 68339, 85981 & 77954), Medical Research Council, Johnson & Johnson Innovation and the Poliomyelitis Research Foundation (PRF-15/27) is gratefully acknowledged.
Author information
Authors and Affiliations
Contributions
T.S. carried out most of the experimental work and wrote the manuscript with P.A. B.M. and S.N. assisted with screening the sgRNAs and ssAAV production. M.B.M. helped with quantification of infectious genome equivalents. K.W. propagated the HBV-infectable hNTCP cell line. A.E. established the hNTCP-HepG2 infection assays and advised on cccDNA quantification. M.S.W. and P.A. oversaw the study.
Corresponding author
Ethics declarations
Competing Interests
Authors affiliated to the Wits/SAMRC Antiviral Gene Therapy Research Unit receive funding from Johnson & Johnson Innovation to develop therapy for HBV infection.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Scott, T., Moyo, B., Nicholson, S. et al. ssAAVs containing cassettes encoding SaCas9 and guides targeting hepatitis B virus inactivate replication of the virus in cultured cells. Sci Rep 7, 7401 (2017). https://doi.org/10.1038/s41598-017-07642-6
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-07642-6
- Springer Nature Limited
This article is cited by
-
Making genome editing a success story in Africa
Nature Biotechnology (2024)
-
CRISPR/Cas systems usher in a new era of disease treatment and diagnosis
Molecular Biomedicine (2022)
-
Prospects of viral vector-mediated delivery of sequences encoding anti-HBV designer endonucleases
Gene Therapy (2022)
-
Advances in designing Adeno-associated viral vectors for development of anti-HBV gene therapeutics
Virology Journal (2021)
-
Optimization of AAV vectors to target persistent viral reservoirs
Virology Journal (2021)