
Abstract
Increased expression of Interleukin (IL)-33 has been detected in intestinal samples of patients with ulcerative colitis, a condition associated with increased risk for colon cancer, but its role in the development of colorectal cancer has yet to be fully examined. Here, we investigated the role of epithelial expressed IL-33 during development of intestinal tumors. IL-33 expression was detected in epithelial cells in colorectal cancer specimens and in the Apc Min/+ mice. To better understand the role of epithelial-derived IL-33 in the intestinal tumorigenesis, we generated transgenic mice expressing IL-33 in intestinal epithelial cells (V33 mice). V33 Apc Min/+ mice, resulting from the cross of V33 with Apc Min/+ mice, had increased intestinal tumor burden compared with littermate Apc Min/+ mice. Consistently, Apc Min/+ mice deficient for IL-33 receptor (ST2), had reduced polyp burden. Mechanistically, overexpression of IL-33 promoted expansion of ST2+ regulatory T cells, increased Th2 cytokine milieu, and induced alternatively activated macrophages in the gut. IL-33 promoted marked changes in the expression of antimicrobial peptides, and antibiotic treatment of V33 Apc Min/+ mice abrogated the tumor promoting-effects of IL-33 in the colon. In conclusion, elevated IL-33 signaling increases tumor development in the Apc Min/+ mice.
Similar content being viewed by others


Introduction
Interleukin (IL)-33 was discovered in 2003 as a nuclear factor expressed by high endothelial venules1 and subsequently described as a pro-inflammatory cytokine belonging to the IL-1 cytokine family2. IL-33 is primarily expressed in non-hematopoietic cells, including fibroblasts, epithelial cells and endothelial cells, and by cells of hematopoietic origin, particularly in macrophages and dendritic cells (DCs)2. IL-33 is a dual-function protein, functioning as conventional cytokine via its extracellular form (mature-IL-33) or as a transcriptional regulator via its nuclear form (full length-IL-33). The former is mediated by its binding to the heterodimeric receptor complex consisting of the ST2 receptor (ST2L) and the widely expressed IL-1R accessory protein3, 4. ST2 is widely expressed on immune cells including eosinophils, basophils, macrophages, T helper 2 cells (Th2 cells), regulatory T cells, NK cells, B cells and group 2 innate lymphoid cells (ILC2)5,6,7. The broad distribution of the ST2 receptor in immune cells suggests the involvement of IL-33 in the pathogenesis of a wide range of diseases8, including asthma, rheumatoid, arthritis and atherosclerosis.
Several studies document upregulation of IL-33 in the inflamed colonic mucosa of patients with inflammatory bowel disease (IBD)9,10,11,12,13,14. Recent findings suggest that enterocyte-derived IL-33 is induced and maintained by inflammatory mediators, because IL-33 is detected in the nuclei of epithelial cells of colonic crypts in acute ulcerative colitis, but undetectable during remission15. It is known that IBD patients have an increased risk for development of colitis-associated colorectal cancer (CAC), but the role of IL-33 in the development of colorectal cancer has yet to be fully examined.
Colorectal cancer (CRC) is the second most prevalent cause of death from cancer in the Western world16. The majority of CRC are sporadic, arising from dysplastic adenomatous polyps. The etiology of human CRC has been linked to genetic variables, such as adenomatous polyposis coli (APC) proteins, inflammatory processes, and gut microbiota. A multi-step process leads to the accumulation of genetic alterations that confer a selective growth advantage to the colonic epithelial cells and drive the transformation from normal epithelium to adenomatous polyp and finally to invasive colorectal cancer17. Loss of heterozygosity for APC in intestinal epithelial cells (IECs) activates Wnt signaling through stabilization of β-catenin, which is sufficient to initiate polyp formation. The Apc Min/+ mice, an animal model of human familial adenomatous polyposis18, carry a mutation in this tumor suppressor gene and have a predisposition to multiple intestinal neoplasia (Min)19.
Recent evidence suggests that IL-33 can function as a novel epithelial “alarmin”20, because it is released as a danger signal by damaged, stressed, or necrotic cells to alert the immune system of a local threat. Indeed, increased expression of full-length IL-33 is detected in ulcerative colitis epithelium10, 11, 13. However, the role of IL-33 in intestinal inflammation is incompletely understood, with both pro- and anti- inflammatory effects being reported21,22,23,24. Similarly, the role of IL-33/ST2 signaling in intestinal tumorigenesis is also unclear. Using IL-33 deficient mice, Maywald et al.25 have shown that IL-33 derived from the tumor epithelium promotes polyposis through the coordinated activation of stromal cells in Apc Min/+ mice. Ablation of IL-33/ST2 signaling by using ST2 deficient mice significantly prevents tumor formation in the azoxymethane (AOM)/dextran sodium sulphate (DSS) model of CRC26. These results are in contrast to those by Malik et al.27 who have found that IL-33 deletion increases tumor incidence in the AOM/DSS model.
Here we report that IL-33 is expressed by epithelial cells within human and mouse intestinal tumors. To study the relevance of increased IL-33 expression to tumor development we generated IL-33 transgenic mice and mice deficient for its receptor (ST2). Increased expression of IL-33 in the gut epithelium of Apc Min/+ mice promoted intestinal tumorigenesis, with expansion of ST2+ regulatory T cells and induction of a Th2 cytokine milieu in the colon. The increased tumorigenesis elicited by IL-33 was independent of additional activation of the β-catenin pathway, but appeared to be dependent on ST2 and the microbiota. IL-33 promoted marked changes in the expression of antimicrobial peptides and antibiotic treatment of V33 Apc Min/+ mice abrogated the tumor promoting-effects of IL-33 in the colon.
Results
Elevated expression of IL-33 in tumor epithelial cells of CRC patients and Apc Min/+ mice
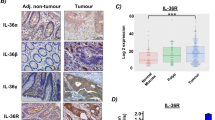
Given that IL-33 expression is significantly up-regulated in UC10, 11, we asked if expression of IL-33 was increased in UC-associated tumors. Analysis of a tumor tissue microarray of UC-CRC patients (n = 47) obtained from our MSSM Biorepository showed that there was expression of IL-33 in the epithelium in 9 out of 47 samples (19.2%). We also found epithelial expression of IL-33 in 15 out of 42 non-IBD CRC tumors (35.7%). Taken together, our data indicate that IL-33 is expressed by tumor epithelial cells in approximately 30% of all samples (n = 89). IL-33 immunostaining was detected mostly in epithelial cells and localized to the nucleus (Fig. 1A). Of note, we found expression of IL-33 in approximately 15% of well-differentiated CRC cells (n = 21), and in almost 40% of moderate- and poorly-differentiated CRC cells (n = 68) (Fig. 1B). Therefore, higher expression of IL-33 in tumor epithelial cells was observed in moderate- and poorly-differentiated CRC patients.

IL-33 expression in human colorectal cancer patients and in Apc Min/+ mice model. (A) Paraffin sections from a tissue microarray of human colorectal cancer (CRC) patients were stained with IL-33 (red) and Pan keratin (green). Representative photographs show expression of IL-33 by intestinal epithelial cells (IECs) in poorly differentiated adenocarcinomas. Scale bars, 100 μm. (B) The percentage of IL-33 immunolabeling in the IECs from 21 well-differentiated CRC patients (W) and 68 moderate- and poor-differentiated CRC patients (M/P). *P < 0.05, Fisher exact test. (C) Relative expression levels of IL-33 mRNA were analyzed by qPCR in the gut of Apc Min/+ mice at various ages. Data were normalized to the expression levels of the Ubiquitin transcript. Means ± s.e.m., n = 4 per group. **P < 0.01, nonparametric Mann-Whitney test. (D) Immunofluorescence staining for IL-33 (red) and Pan keratin (green) in Apc Min/+ mice at different ages. Zoomed-in boxed area shows IL-33 immunolabeling in the IECs in Apc Min/+ mice. Scale bars, 50 μm.
To further investigate a link between tumorigenesis and IL-33 expression we examined IL-33 expression in Apc Min/+ mice. We found that the expression of IL-33 mRNA was significantly increased throughout tumor development (Fig. 1C). Immunostaining experiments showed that IL-33 was expressed by epithelial cell in adenomatous areas, compared with adjacent normal tissue (Fig. 1D). IL-33 was detected in epithelial cells within microadenomas by d40. Tumor progression results in the expansion of microadenomas and an increase in tumor size. With time, the number of epithelial cells expressing IL-33 increased significantly (Fig. 1D). Together, these results document expression of IL-33 in intestinal tumors of humans and mice and suggest that IL-33 is involved in their development.
Generation of transgenic mice expressing IL-33 in the intestinal epithelium
Next we examined if epithelial-cell derived IL-33 contributes to tumor development. IL-33 is a dual-function protein, functioning as a conventional cytokine and/or as a transcriptional regulator. To reduce the complexity and focus on the cytokine function of IL-33, we cloned the cDNA encoding the mature form of IL-33 (amino acids 95–267, lacking the nuclear localization signal) downstream of the villin promoter28. The transgene was injected into mouse eggs and 4 transgenic lines were derived from founder mice. These animals are referred to as V33 mice (Fig. 2A). The V33 mice were healthy, reproduced normally, and did not develop intestinal tumors. To examine IL-33 expression in the intestine of these transgenic mice we performed qPCR in RNA extracted form the small and large intestine. As expected, we detected increased expression of IL-33 mRNA in the small intestine and large intestine in the V33 transgenic mice compared with their littermate control wild-type (WT) mice (Fig. 2B). To examine expression of IL-33 protein, we performed enzyme linked immunosorbent assay in the gut extracts and found that IL-33 was elevated in the intestine of transgenic mice compared to WT mice (Fig. 2C). Finally, we examined the cellular expression of IL-33. Because we expressed the mature form of IL-33, we expected that it should be located in the cytoplasm rather than the nucleus. Immunostaining of intestinal sections showed that IL-33 immunoreactivity was indeed detected in the cytoplasm of transgenic, but not control intestinal epithelial cells (Fig. 2D). In sum, we confirmed appropriate tissue and cellular expression of the V33 transgene.

Generation of transgenic mice expressing IL-33 in the gut epithelium. (A) Scheme for generation of V33 mice. A transgene encoding IL-33 mature form (m-IL-33) under the control of the murine villin promoter (9 kb) was used to generate V33 mice. (B) Relative expression levels of transgenic IL-33 mRNA were analyzed by qPCR in the small intestine (SI) and large intestine (LI) of wild-type (WT) and V33 mice. Data were normalized to the expression levels of the Ubiquitin transcript. Means ± s.e.m., n = 6 per group. ***P < 0.001, one-way ANOVA. (C) Enzyme linked immunosorbent assay of IL-33 in the gut explants from WT and V33 mice. Data were normalized to the weight of the intestine explant. Means ± s.e.m., n = 4 per group. **P < 0.01, ***P < 0.001, one-way ANOVA. (D) Immunofluorescence staining for IL-33 (red) in the gut of WT and V33 mice. Cell nuclei were counterstained with DAPI (blue). Notice that transgenic expression of IL-33 in the cytoplasm of intestinal epithelial cells in V33 mice. Scale bars, 50 μm.
IL-33/ST2 signaling promotes polyp growth in Apc Min/+ mice
To explore the potential impact of epithelial-specific IL-33 on IEC tumors, we crossed V33 mice with Apc Min/+ mice to generate V33 Apc Min/+ mice. Next we measured the number of tumors (polyps) in V33 Apc Min/+ mice and their littermate control Apc Min/+ mice at 120 days of age. We found that V33 Apc Min/+ mice had more polyps in the small intestine than Apc Min/+ mice (Fig. 3A), which was confirmed by histological analysis (Fig. 3B). Because the Villin promoter drives transgene expression in the entire intestinal epithelium (Fig. 2D), we also investigated the effect of tumor burden of V33 Apc Min/+ mice in the large intestine. Importantly, we found a higher mean number and increased mean size of colonic polyps in V33 Apc Min/+ mice compared to their Apc Min/+ littermates (Fig. 3C,D). Histological analysis confirmed increased tumor burden, and increased tumor size in the large intestine of V33 Apc Min/+ mice (Fig. 3E).

IL-33 promotes intestinal polyp growth in Apc Min/+ mice. (A) V33 Apc Min/+ mice carried a significantly higher number of polyps in the SI than Apc Min/+ mice at d120. (B) Representative hematoxylin and eosin (H&E) staining of ileum of Apc Min/+ mice and V33 Apc Min/+ mice at d120. Individual tumors are highlighted by black lines. (C) Tumor number (left) and tumor size (right) of LI in Apc Min/+ mice and V33 Apc Min/+ mice at d120. (D) Representative gross specimens from the indicated genotype analyzed at d120. (E) Representative H&E staining of LI of Apc Min/+ mice and V33 Apc Min/+ mice at d120. Individual tumors are highlighted by black lines. Graphs show means ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, nonparametric Mann-Whitney test.
We next tested whether IL-33/ST2 signaling deficiency affected the development of tumors. To do so, we first generated ST2-deficient mice using the CRISPR/Cas9n technology29. Three ST2-deficient lines were derived from founder mice. In these animals, the exons 4 and 5 of the ST2 gene, which correspond to the IL-33 binding site30, were deleted (Fig. 4A). To test if the genetic changes induced functional inactivation of ST2, we examined ST2 expression in ILC2 of ST2−/− mice. Flow cytometric analysis showed that ST2 expression was completely abrogated in ILC2 of the ST2−/− mice, verifying that the engineered mutation is a null allele (Fig. 4B). We then crossed ST2 −/− mice with Apc Min/+ mice to produce Apc Min/+ deficient for ST2 (Apc Min/+ ST2 −/−) mice and littermate controls (Apc Min/+ ST2 +/−, Apc Min/+ ST2 +/+). Gross analysis of the intestine of these animals at 120 d of age indicated a reduction in the number of polyps in the small intestine of Apc Min/+ ST2 −/− mice compared with Apc Min/+ ST2 +/− and Apc Min/+ ST2 +/+ mice (Fig. 4C). However, there was no effect on the tumor development in the large intestine in these sets of experiments (Fig. 4D).

Genetic ablation of ST2 reduces polyp burden in Apc Min/+ mice. (A) Scheme for generation of ST2 −/− mice by CRISPR/Cas9n technology. The two g-RNA targeting sequences are underlined in red and blue. Sequencing confirmed the deletion of exons 4 and 5. (B) Flow cytometric analysis of the expression of ST2 on ILC2 (CD45+ Lin− IL7R+ KLRG1+) isolated from the mesenteric fat of WT and ST2 −/− mice. Notice the absence of ST2 expression in the ILC2 isolated from ST2 −/− mice. (C,D) Quantification of the number of tumors in the small (C) and large (D) intestine of Apc Min/+ ST2 +/+, Apc Min/+ ST2 +/− and Apc Min/+ ST2 −/− mice. Graphs show means ± s.e.m. *P < 0.05, **P < 0.01, N.S. means not significant, nonparametric Mann-Whitney test.
Taken together these results confirm a positive role for IL-33 signaling in tumor development in the Apc Min/+ background.
IL-33 increases colonic ST2+ regulatory T cells
IL-33 influences various immune cells during differentiation, immune responses, and homeostasis. Its receptor ST2 is widely expressed on immune cells including eosinophils, basophils, macrophages, Th2 cells, regulatory T cells, NK cells, and ILC25,6,7. To start examining possible cellular differences in our mice, we analyzed these cell populations. We found that there were no differences in the relative and absolute number of colonic eosinophils, macrophages, monocytes, DC, neutrophils, NK cells, NKT cells, B cells (Supplementary Figure 1) and ILC2 (Supplementary Figure 2) between the V33 Apc Min/+ mice and Apc Min/+ mice. However, there was an increase in the CD3+ population in V33 Apc Min/+ mice compared to Apc Min/+ mice (Supplementary Figure 1).
Next we examined whether expression of IL-33 affected the number of Treg, especially ST2+ Treg in the intestine of V33 mice. We found that expression of IL-33 in the epithelium led to an increase the relative number of colonic Treg cells (Fig. 5A). Importantly, V33 mice also had more colonic ST2+ Treg cells compared with WT control in both relative and absolute number (Fig. 5A). Because V33 Apc Min/+ mice had higher tumor burden than Apc Min/+ mice, we asked whether promotion of intestinal tumorigenesis was correlated with increased number of ST2+ Treg cells in the gut. Flow-cytometric analysis demonstrated that there was no difference in the frequency and total number of colonic Treg cells between the V33 Apc Min/+ mice and Apc Min/+ mice (Fig. 5B). However, V33 Apc Min/+ mice had more colonic ST2+ Treg cells than Apc Min/+ mice (relative and absolute number) (Fig. 5B). Since epithelium-expressed IL-33 promoted tumor growth not only in the LI (Fig. 3C) but also in the SI (Fig. 3A), we also investigated the ST2+ Treg cells in the SI. First, we analyzed ST2+ Treg cells between Apc Min/+ mice and WT mice. We found that Apc Min/+ mice had more Treg cells (relative and absolute number) in the SI than WT controls (Supplementary Figure 3A and B). Importantly, Apc Min/+ mice had more ST2+ Treg cells in the SI than WT mice (Supplementary Figure 3C and D). Similarly, V33 Apc Min/+ mice had more ST2+ Treg cells in the SI than Apc Min/+ mice (Supplementary Figure 3E). Therefore, we conclude that IL-33 increases the numbers of intestinal ST2+ Treg cells in both small and large intestine.

Epithelial-derived IL-33 signaling expands colonic ST2+ regulatory T (Treg) cells and induces a Th2 cytokine profile. (A) Relative and absolute number of Treg cells (CD4+Foxp3+) in the colon of Apc Min/+ mice and V33 Apc Min/+ mice at d120. Left, representative flow cytometry plots gated on CD45+ cells; right, statistical data show means ± s.e.m. (B) Relative and absolute number of ST2+ Treg cells in the colon of Apc Min/+ mice and V33 Apc Min/+ mice at d120. Left, representative flow cytometry plots gated on CD45+ CD4+ cells; right, statistical data show means ± s.e.m. (C) Relative expression levels of ST2+Treg and Th2 signature genes were analyzed by qPCR in sorted T cells of colonic lamina propria of WT, V33, Apc Min/+ mice and V33 Apc Min/+ mice at d120. Graphs show means ± s.e.m., n = 4–5/group. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA.
To better evaluate the phenotype of colonic lamina propria T cells, we next sorted colonic T cells (CD45+ CD3+ cells) from mice of all genotypes and performed qPCR. In line with flow cytometric results, mRNA levels of ST2 and Foxp3 were upregulated in colonic T cells of V33 mice compared with WT mice (Fig. 5C). Of note, IL-4, IL-5, IL-13 and Gata3 mRNAs were also upregulated in colonic T cells of V33 mice (Fig. 5C), suggesting that V33 colonic T cells showed a Th2-predominant phenotype. Accordingly, qPCR analysis of sorted T cells also confirmed increased expression of ST2, Foxp3 and Th2 cytokines in V33 Apc Min/+ mice when compared with Apc Min/+ mice (Fig. 5C). Together these results demonstrate that epithelial-derived IL-33 induces expansion of ST2+ Treg and promotes a Th2-predominant cytokine milieu.
IL-33 induces colonic alternatively activated macrophages in Apc Min/+ mice
Macrophages are a major component of the leukocyte infiltrates in various tumor stroma. ST2 is constitutively expressed at the surface of mouse macrophages and IL-33 has been implicated in their activation31, 32. IL-33 promotes or amplifies the expression of chemokines by M1 or M2-polarized macrophages33. We first determined if overexpression of IL-33 could change the relative number of macrophages in the colon. Flow-cytometric results demonstrated that there was no difference in the relative number of colonic macrophages among the WT, V33, Apc Min/+ mice and V33 Apc Min/+ mice (Fig. 6A), indicating that epithelium-derived IL-33 did not affect the number of colonic macrophages. To determine whether epithelium-derived IL-33 had a role in induction of colonic macrophages, we investigated mRNA expression of representative M1 and M2 genes on the sorted macrophages (CD45+CD11b+F4/80+ cells) from the colonic lamina propria leucocytes (LPL) in WT, V33, Apc Min/+ and V33 Apc Min/+ mice. qPCR analysis data, normalized to fold change from age-matched WT littermate control mice, showed that expression of the M2 markers arginase 1 (Arg-1), Mrc-1, Fizz1, Ccl17, Ccl22 and Ccl24, was higher in colonic macrophages of V33 mice than in those of WT mice (Fig. 6B). Expression of the M1 markers iNOS, IL-12p35, Tnf-a and Cd86 was not different between V33 mice and WT mice, but expression of Cxcl9, Cxcl10 and Cxcl11, key chemokines produced by M1 macrophages, was lower in the colonic macrophages of V33 mice compared with WT mice (Fig. 6B). Thus, overexpression of IL-33 in the gut activated colonic macrophages to M2 phenotypes. Of note, we also found increased expression of Arg-1, Chi313 and Ccl24 in the colonic macrophages of V33 Apc Min/+ mice when compared with WT mice (Fig. 6C). Similarly, when compared with Apc Min/+ mice, V33 Apc Min/+ mice had colonic macrophages with increased expression of M2 markers (Fig. 6C). Taken together, the results suggest that IL-33 induces colonic alternatively activated macrophages in Apc Min/+ mice.

IL-33 regulates alternative activation of colonic macrophages. (A) Relative number of macrophages (CD45+CD11b+F480+) in the colon of WT, V33, Apc Min/+ mice and V33 Apc Min/+ mice at d120. Graphs show means ± s.e.m., n = 3–5/group. (B) Relative expression levels of M1 and M2 macrophage signature genes were analyzed by qPCR in sorted macrophages of colonic lamina propria of WT mice and V33 mice at d120. (C) Relative expression levels of M2 macrophage signature genes were analyzed by qPCR in sorted macrophages of colonic lamina propria of WT mice, Apc Min/+ mice and V33 Apc Min/+ mice at d120. Data were normalized to fold change from age-matched WT littermate control mice. Graphs show means ± s.e.m., n = 3–5/group. *P < 0.05, **P < 0.01, ***P < 0.001, versus WT group. # P < 0.05, ## P < 0.01, versus Apc Min/+ group.
IL-33 promotes tumorigenesis independent of the β-catenin pathway in IEC
Loss of APC protein leads to a deregulated WNT/β-catenin pathway34, as APC is part of the β-catenin destruction complex18, 34. Therefore, β-catenin plays a central role in intestinal proliferation and neoplastic transformation. To determine whether activation of the Wnt pathway plays a role in the increased polyp burden observed in V33 Apc Min/+ mice, we performed β-catenin immunostaining within size-matched tumors from V33 Apc Min/+ mice and Apc Min/+ mice. Polyps from both genotypes showed an equivalent increase in cytoplasmic and nuclear accumulation of β-catenin and frequencies of cells with nuclear β-catenin (an indicator of activated β-catenin) (Supplementary Figure 4A). To further validate these results we examined expression of the Wnt target genes in the IEC isolated from tumor areas and adjacent normal area in V33 Apc Min/+ mice and Apc Min/+ mice. The mRNA levels of c-myc, Birc5, Cd44, Lgr5, Tcf7, Mmp7, and Ephb3 were significantly increased in tumor area compared with non-tumor area (Supplementary Figure 4B). However, there was no difference in the expression of these genes when we compared tumors from V33 Apc Min/+ mice and Apc Min/+ mice (Supplementary Figure 4B). These results suggest that IL-33 does not affect β-catenin nuclear translocation in dysplastic epithelium.
IL-33 changes antimicrobial gene expression in the epithelium
Next, we examined if epithelium expressed IL-33 affected IEC function. Injection of IL-33 has been shown to impair epithelial barrier function13. We therefore examined epithelial permeability in V33 mice. There were no significant difference in intestinal permeability in WT and V33 mice assessed by measuring serum FITC-Dextran levels 5 h after administration (Supplementary Figure 5).
Antimicrobial peptides produced by epithelial cells protect the host intestinal mucosa against microorganisms35. Since IL-33 is expressed by epithelial cells in adenomatous areas compared with adjacent normal tissue in Apc Min/+ mice (Fig. 1D), we decided to examine whether IL-33 affected antimicrobial gene expression. We found that cells within the adenomas of Apc Min/+ mice expressed less Retnlb and Muc2 mRNA (Fig. 7A) than cells in normal adjacent tissue. Furthermore, compared with Apc Min/+ mice, V33 Apc Min/+ mice expressed less Reg3b and Reg3g in the colonic mucosa, and more Retnlb, Ang4 and Muc2 mRNA (Fig. 7A). These changes, however, were not observed when comparing tumor areas from both genotypes (Fig. 7A).

Epithelial-derived IL-33 alters antimicrobial genes expression in the colon. (A) Relative expression levels of Reg3b, Reg3g, Ang4, Retnlb and Muc2 were analyzed by qPCR in the colon of size-matched tumor area (T) and adjacent non-tumor normal area (NT) of Apc Min/+ mice and V33 Apc Min/+ mice at d120. Data were normalized to the expression levels of the Ubiquitin transcript. Graphs show means ± s.e.m. n = 4–5/group. *P < 0.05, **P < 0.01, ***P < 0.001, one-way ANOVA. (B) Number of polyps in the intestine of Apc Min/+ mice and V33 Apc Min/+ mice receiving water or antibiotic continuously from weaning to 120 days post-birth. Graphs show means ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, nonparametric Mann-Whitney test.
Pro-tumorigenic activities of IL-33 are partially dependent on the microbiota in the colon
Antimicrobial factors are critical in controlling the microbiota, which in turn affects CRC development. To examine a potential contribution of the microbiota to development of tumors in our model system, we treated V33 Apc Min/+ mice and Apc Min/+ mice with antibiotics (ampicillin metronidazole, neomycin, vancomycin)28, 36. Antibiotics were provided in the drinking water continuously, from weaning to 120 days post-birth. Antibiotic treatment significantly decreased polyp load in the colon of Apc Min/+ mice and V33 Apc Min/+ mice, but did not affect tumorigenesis in the small intestine (Fig. 7B). Consistent with previous report37, colonic polyps are highly sensitive to antibiotic treatment rather than small intestine. Importantly, after antibiotic treatment, there was no difference in the number of tumors between Apc Min/+ mice and V33 Apc Min/+ mice (Fig. 7B). These observations suggest that the protumorigenic activities of IL-33 at least in the large intestine may be partly mediated by changes in the microbiota.
Discussion
We show here that expression of IL-33 is increased within epithelial cells of intestinal tumors in humans and mice and that constitutive expression of IL-33 in the context of the Apc mutation leads to increased tumorigenesis. Together these results strongly suggest that IL-33 is a factor involved in intestinal tumorigenesis.
Inflammation is a risk factor for CRC development. Increased expression of IL-33 has been detected in intestinal samples of patients with ulcerative colitis9,10,11,12,13,14. A recent study shows that IL-33 is present in the nuclei of enterocytes in scattered colonic crypts in acute ulcerative colitis, but is not present in these cells at remission15. Genetic studies have suggested a role for the Il33 gene in the risk of developing IBD38. However, the precise effect of epithelial-derived IL-33 to the gut inflammatory conditions has remained unclear. Experimental data obtained using different animal models of intestinal inflammation have produced conflicting results21,22,23,24, with IL-33 having both pro- and anti- inflammatory effects. Increased expression of IL-33 is detected in SAMP mice, a spontaneous model of chronic intestinal inflammation characterized by a mixed Th1/Th2 immune phenotype. In this model, IL-33 was shown to potently increase the production of the pro-inflammatory cytokines, such as IL-5, IL-6 and IL-1711. IL-33 appears to have a protective effect in trinitrobenzene sulfonic acid-induced colitis, mostly driven by a Th1 immune response39. In addition, IL-33 signaling protects from murine oxazolone colitis by supporting intestinal epithelial function40. Disparate results also exist regarding the role of IL-33 in the DSS-induced colitis model. Following epithelial barrier disruption caused by DSS administration, IL-33 injection worsened colitis, inducing the recruitment of neutrophils to the site of inflammation41,42,43 and induction of type 2 cytokines44,45,46, whereas, during the recovery phase, it showed a prominent effect in promoting mucosal healing41, 43 and inducing goblet cell proliferation42, eventually restoring epithelial barrier function. IL-33 has also been reported to increase expression of the growth factor amphiregulin to enhance colonic mucin responses in DSS-induced colitis model47. In the present study, we used the villin promoter to drive IL-33 expression in mice, which targeted IL-33 expression specifically to the gut epithelium. Expression of IL-33 in this setting did not cause significant inflammation (data not shown). Therefore, we conclude that epithelial-derived IL-33 promoted intestinal tumorigenesis in Apc min/+ mice independent of its pro-inflammatory properties.
As discussed above, the role of IL-33 in tumor biology is still controversial. Some studies show that IL-33 inhibits tumor growth by stimulating production of IFN-γ by CD8+ T cells and Th1 cells48, 49. Systemic over-expression of IL-33 in transgenic mice promotes NK and CD8+ T cell function and inhibits tumor growth and metastasis50. Overexpression of IL-33 in tumor cells strongly inhibited tumor growth by increasing the numbers of tumor-infiltrating NK cells and CD8+ T cells, and their production of IFN-γ51. However, other studies report that IL-33 promotes tumor growth. Jovanovic et al.52, using a breast cancer model, have shown that IL-33 promotes cancer progression through increased intratumoral accumulation of immunosuppressive cells and by diminishing innate antitumor immunity.
In our study, we examined expression of IL-33 in humans and in mice. We show here that the expression of IL-33 is higher in the epithelium of moderate- and poorly-differentiated CRC than in well-differentiated tumors suggesting a positive correlation of IL-33 expression in the epithelium and tumor grade. Our data are consistent with an earlier report that IL-33/ST2 pathway contributes to human colorectal cancer53. Our study provides evidence for dynamic changes in IL-33 expression in polyps of Apc Min/+ mice. Epithelial expression of mature IL-33 correlated positively with tumor growth, and deletion of ST2 decreased tumor incidence. Since CRC associated IL-33 is predominantly nuclear, we also generated transgenic mice that expressed full-length IL-33 in the epithelium to test its role on the tumor development. Overexpression of full-length IL-33 in the context of the Apc mutation also led to increased tumorigenesis (data not shown), which is consistent with the phenotype driven by epithelial expression of mature IL-33.
IL-33 has been shown to affect various immune cells during the inflammation and cancer development. Here we demonstrate that two main targets of IL-33 (T cells and macrophages) are affected in vivo. We demonstrate changes in the number and function of T cells. More specifically, we document changes in the number of ST2+ Tregs. This effect is likely to be caused by expansion of ST2+ Tregs, because IL-33 does not appear to promote their differentiation from ST2- Treg54. Tregs from IL-33-treated mice display enhanced capacities to suppress IFN-γ production by effector T cells55, and thus contribute to their pro-tumorigenic activities in Apc Min/+ mice. In addition, our results show increased expression of IL-4, IL-5, IL-13, Foxp3 and Gata3 in colonic T cells of V33 mice (Fig. 5). ST2+ Tregs exhibit a Th2-biased character, express GATA-3 and produce the Th2 cytokines IL-5 and IL-13 in vitro 56. Expression of IL-33 in our model did not affect the numbers of other IL-33 target cells such as eosinophils, monocytes, DC, neutrophils, NK cells, NKT cells, B cells and ILC2 cells (Supplementary Figure 1 and Supplementary Figure 2). Whether IL-33 modifies their function to promote tumorigenesis requires additional evaluation.
The ability of macrophages to affect tumor development is well described in the literature57. Two general mechanisms could be invoked to explain how IL-33 could affect macrophage function in our model. First, IL-33 could affect the macrophage phenotype. It is well documented that IL-33 induces macrophages to switch from the M1 to the M2 phenotype in vivo 58, 59. In agreement with these results, we found in our study that IL-33 expression was associated with polarized M2 phenotype. Mechanistically this effect could be due to Th2 cytokines, since it has been demonstrated that Th2 cytokines are drivers of M2 polarization58. We suggest that the increased expression of Th2-type cytokines by ST2+ Tregs in our model may be critical for the development of the M2 phenotype. Second, IL-33 could promote expression of protumorigenic factors such as prostaglandin E2, as recently reported60.
In addition to immune cells, we also find that IL-33 may act on epithelial cells to promote tumor growth. Intestinal epithelial cells express ST2 in both mice and humans13. We found that expression of IL-33 in the gut epithelium increases Ang4, Retnlb and Muc2 transcription levels. We suggest that IL-33 promotes the colonic goblet cells to secrete these factors. IL-33 induces goblet cell hyperplasia, and stimulates mucous production at mucosal surfaces2, 42. Ang4 is an antimicrobial peptide important in epithelial host defence in the small intestine61, and is produced by goblet cells in the large intestine62. The expression of Retnlb is tightly restricted to intestinal goblet cells, from where it is secreted apically into the intestinal lumen63. Th2 cytokines can induce expression of Retnlb by goblet cells64. Reduced Retnlb transcriptional levels are associated with fewer IL-13 mRNA transcripts in the intestines of IL-33−/− compared with WT mice42, 65. MUC2, a major goblet cell mucin, has been used as the goblet cell marker. Reg3β and Reg3γ are expressed by different epithelial cell types in colon including enterocytes and goblet cells. Absence of Muc2 in Muc2 −/− mice results in up-regulation of Reg3β and Reg3γ expression, suggesting altered bacterial-epithelial signaling and an innate defense response66. Although it is unknown whether IL-33 affects enterocytes, we observed that expression of IL-33 in the epithelium increased Muc2 expression together with decreased the Reg3β and Reg3γ expression in the colon.
Changes in gut microbiota have been associated with tumor growth67. The growing tumors can selectively alter the microbial community causing expansion of pathogenic bacteria that promote tumor development37. Immune system components can also regulate the host microbiota that modulate the susceptibility to tumor27. As shown here, epithelium expressed IL-33 changes antimicrobial gene expression in the epithelium. Abnormal expression of these antimicrobial genes could potentially modify the microbiota and promote dysbiosis, and favor tumor development68. In addition, the changes in the immune parameters induced by IL-33 expression (increase in ST2+ Tregs, induction of Th2 cytokines and M2 macrophages) could also contribute to regulation of the commensal bacteria that modulate the susceptibility to tumor. To test if the microbiota could potentially affect IL-33 induced tumorigenesis, we treated mice with a cocktail of broad-spectrum antibiotics that produces a profound shift in the composition and abundance of intestinal microbiota in the murine intestine36. Our results show that V33 Apc Min/+ mice have more polyps in the small intestine and large intestine than Apc Min/+ mice. However, after antibiotic treatment, there was no difference in the number of colonic tumors between Apc Min/+ mice and V33 Apc Min/+ mice, suggesting that some of the protumorigenic activities of IL-33 in the colon may be mediated by changes in the microbiota. This effect could be mediated via direct regulation of antimicrobial gene expression, or by other factors associated with immune system. Indeed, a recent study by Malik et al.27 suggest that IL-33 protects against colitis and CRC by regulating intestinal IgA production, which in turn affects the composition of the microbiota.
In summary, we show here that expression of IL-33 is increased within epithelial cells of intestinal tumors in humans and mice; that IL-33 promotes tumor development when overexpressed in intestinal cells of Apc Min/+ mice; and that deletion of IL-33 signaling by deletion of ST2, significantly reduces tumorigenesis in these animals. IL-33 overexpression did not increase Wnt signaling but promoted expansion of ST2+ Treg cells and induction of colonic alternatively activated macrophages. IL-33 also promoted marked changes in the expression of antimicrobial peptides and antibiotic treatment of V33 Apc Min/+ mice abrogated the tumor promoting-effects of IL-33 in the colon. Therefore, the results presented here, reveal a previously unappreciated ability of epithelial expressed-IL-33 to control tumor progression in the intestine through effects on both immune and epithelial cells (Supplementary Figure 6).
Materials and Methods
Ethics approval
All animal experiments in this study were approved by the Institutional Animal Care and Use Committee of Icahn School of Medicine at Mount Sinai, and were performed in accordance with the approved guidelines for animal experimentation at the Icahn School of Medicine at Mount Sinai (IACUC-2015-0004).
Mice
C57BL/6 mice and Apc Min/+ mice were purchased from The Jackson laboratory (Bar Harbor, ME). Mice were maintained under specific pathogen-free conditions.
Generation of transgenic mice expressing IL-33 in the intestinal epithelium
The cDNA of IL-33 mature form (95–267 amino acids) was cloned into a pBS-Villin vector that contained a 9 kb segment of the mouse villin promoter69. The pBS-Villin/m-IL-33 plasmid was verified by sequencing, and the transgene was isolated from the plasmid by restriction enzyme digestion and gel purification. To generate transgenic mice, the transgene was microinjected into C57BL/6 mouse eggs28. Identification of the transgenic V33 mice was done by PCR amplification using the following primers: 5′- ggctgtgatagcacacagga-3′ and 5′- gttccttggatgctcaatgtgt -3′.
Generation of ST2-deficient mice
We used the CRISPR/Cas9n system to delete exons 4 and 5 of the ST230. To do so we designed two pairs of sgRNA flanking these exons, as described before29. Plasmids expressing each sgRNA were prepared by ligating oligos into BbsI site of pX460 (http://www.addgene.org/48873/)29. To construct a single plasmid for injection, four PCR-amplified sgRNA driven by U6 promoter were inserted into the plasmid (pX460) containing hCas9n. All these reagents were validated by sequencing. C57BL/6 eggs were injected with the pX460 plasmid containing 4 sgRNAs and hCas9n at 1 ng/ul as described previously70. Identification of mice carrying the deletion of exons 4 and 5 was done by PCR amplification using the primers: 5′- cccatgtatttgacagttacgg-3′ and 5′- tggcttttatatccaggaactca-3′.
Tissue microarray of human colorectal cancer patients
A colorectal cancer tissue microarray was generated as previously described71. Briefly, sections were obtained from de-identified paraffin blocks and HE stained. A certified pathologist defined representative tumor regions. Small tissue cylinders with a diameter of 0.6 mm were taken from selected areas of each donor block using a tissue chip micro-arrayer (Beecher Instruments, Silver Spring, MD, USA) and transferred to a recipient paraffin block. The recipient paraffin block was cut in 2 μm paraffin sections using standard techniques. The use of the pathologic samples was approved and reviewed by the Ethics Committee of the Icahn School of Medicine at Mount Sinai.
Antibodies
For flow cytometry, the following fluorochrome-conjugated anti-mouse antibodies were used: CD45 (30-F11), CD11b (M1/70), CD3e (145-2C11), CD4 (GK1.5), F4/80 (BM8), Foxp3 (FJK-16s), CD127 (A7R34), KLRG1 (2F1) were from eBioscience; IL-33R-biotin (T1/S2, clone DJ8) from MD Bioproducts; Streptavidin PE-Cyanine7 was from eBioscience; FITC anti-mouse Lineage Cocktail with Isotype Ctrl was from Biolegend. For immunofluorescence staining, purified anti-mouse IL-33 polyclonal antibody (AF3626) and anti-human IL-33 polyclonal antibody (AF3625) were from R&D Systems; beta-Catenin antibody (Cat: #9587) was from Cell Signaling Technology and Pan-keratin antibody (PCK-26) was from Abcam. Alexa Fluor 488-conjugated Donkey antibody to mouse IgG (A21202), Alexa Fluor 594-conjugated Donkey antibody to goat IgG (A11058) and Alexa Fluor 594-conjugated goat antibody to rabbit IgG (A11037) were from Invitrogen.
Flow cytometry and sorting
Cell suspensions from the lamina propria were prepared as described previously36. All cells were first pre-incubated with anti-mouse CD16/CD32 for blockade of Fc γ receptors, then were washed and incubated for 40 min with the appropriate monoclonal antibody conjugates in a total volume of 200 μl PBS containing 2 mM EDTA and 2% (vol/vol) bovine serum. DAPI (Invitrogen) was used to distinguish live cells from dead cells during cell analysis and sorting. For detection of intracellular Foxp3, a Foxp3 Staining Buffer Set (eBioscience) was used for fixation and permeabilization of the cells. Stained cells were analyzed on a LSRII machine using Diva program (BD Bioscience) or purified with a MoFlo Astrios cell sorter (DakoCytomation). Cells were >98% pure after sorting. Data were analyzed with FlowJo software (TreeStar).
Enzyme-linked immunosorbent assay
Small pieces of small intestine or colon (~5 mm of mid-part) were isolated, rinsed in HBSS/BSA, weighed, and cultured overnight in 12-well tissue culture plates (Costar) in 1000 μl complete DMEM at 37 °C in an atmosphere containing 5% CO2. After centrifugation to pellet debris, culture supernatants were transferred to fresh tubes and stored at −80 °C. IL-33 was quantified in the supernatant of intestinal explant cultures from V33 and WT mice by enzyme-linked immunosorbent assay (ELISA) according to standard manufacturer’s recommendations (eBioscience) and the results were normalized to the weight of the intestinal explant.
Reverse-transcription polymerase chain reaction
Total RNA from tissues/sorted cells was extracted using the RNeasy mini/micro Kit (Qiagen) according to the manufacturer’s instructions. Complementary DNA (cDNA) was generated with Superscript III (Invitrogen). Quantitative PCR was performed using SYBR Green Dye (Roche) on the 7500 Real Time System (Applied Biosystems) machine. Thermal cycling conditions used were as follows: 50 °C for 2 min and 95 °C for 10 min, 40 cycles of 95 °C for 15 s, 60 °C for 1 min, followed by dissociation stage. Results were normalized to the housekeeping gene Ubiquitin. Relative expression levels were calculated as 2(Ct(Ubiquitin)-Ct(gene)). Primers were designed using primer express 2.0 software (System Applied Biosystems).
Histology and immunofluorescence staining
Tissues were dissected, fixed in 10% phosphate-buffered formalin, and then processed for paraffin sections. Five-micrometer sections were stained with hematoxylin and eosin for histological analyses. For immunofluorescence staining, five-micrometer sections were dewaxed by immersion in xylene (twice for 5 minutes each time) and hydrated by serial immersion in 100%, 90%, 80%, and 70% ethanol and PBS. Antigen retrieval was performed by microwaving sections for 20 minutes in Target Retrieval Solution (DAKO). Sections were washed with PBS (twice for 10 minutes each time), and blocking buffer (10% BSA in TBS) was added for 1 hour. Sections were incubated with primary antibody in blocking buffer overnight at 4 °C. After washing, conjugated secondary Abs were added and then incubated for 35 min. Cell nuclei were stained using DAPI. The slides were next washed and mounted with Fluoromount-G (Southern Biotech). Images were captured using a Nikon fluorescence microscope. Colocalization was performed with ImageJ (http://rsbweb.nih.gov/ij/) and the colocalization finder plug-in.
FITC-Dextran assay
Mice were kept without food and water for 4 hr and then FITC-dextran (#FD4-1G, Sigma) was administered by oral gavage at a concentration of 40 mg/ml in phosphate-buffered saline (PBS) in 400 μl (16 mg) per mouse (~800 mg/kg). Five hours later, plasma was collected from peripheral blood, then mixed 1:1 with PBS and analyzed on a plate reader at an excitation wavelength of 485 nm and an emission wavelength of 535 nm.
Antibiotic treatment
For reduction of the intestinal microbiota, a ‘cocktail’ of antibiotics containing ampicillin 1 g/L, metronidazole 1 g/L, neomycin 1 g/L, and vancomycin 0.5 g/L (Sigma Aldrich) in the drinking water was provided to the mice through their drinking water continuously from weaning to 120 days post-birth. Antibiotic treatment was renewed every week.
Statistics
Differences between groups were analyzed with Student’s t tests or nonparametric Mann-Whitney test. For the comparison of more than two groups a one-way ANOVA followed by a Bonferroni multiple comparison test was performed. All statistical analyses were performed with GraphPad Prism 5 software.
References
Baekkevold, E. S. et al. Molecular characterization of NF-HEV, a nuclear factor preferentially expressed in human high endothelial venules. Am J Pathol 163, 69–79, doi:10.1016/S0002-9440(10)63631-0 (2003).
Schmitz, J. et al. IL-33, an interleukin-1-like cytokine that signals via the IL-1 receptor-related protein ST2 and induces T helper type 2-associated cytokines. Immunity 23, 479–490, doi:10.1016/j.immuni.2005.09.015 (2005).
Chackerian, A. A. et al. IL-1 receptor accessory protein and ST2 comprise the IL-33 receptor complex. Journal of immunology 179, 2551–2555 (2007).
Greenfeder, S. A. et al. Molecular cloning and characterization of a second subunit of the interleukin 1 receptor complex. The Journal of biological chemistry 270, 13757–13765 (1995).
Ohno, T., Morita, H., Arae, K., Matsumoto, K. & Nakae, S. Interleukin-33 in allergy. Allergy 67, 1203–1214, doi:10.1111/all.12004 (2012).
Mirchandani, A. S., Salmond, R. J. & Liew, F. Y. Interleukin-33 and the function of innate lymphoid cells. Trends in immunology 33, 389–396, doi:10.1016/j.it.2012.04.005 (2012).
Lu, J., Kang, J., Zhang, C. & Zhang, X. The role of IL-33/ST2L signals in the immune cells. Immunol Lett 164, 11–17, doi:10.1016/j.imlet.2015.01.008 (2015).
Liew, F. Y., Pitman, N. I. & McInnes, I. B. Disease-associated functions of IL-33: the new kid in the IL-1 family. Nature reviews. Immunology 10, 103–110, doi:10.1038/nri2692 (2010).
Beltran, C. J. et al. Characterization of the novel ST2/IL-33 system in patients with inflammatory bowel disease. Inflammatory bowel diseases 16, 1097–1107, doi:10.1002/ibd.21175 (2010).
Seidelin, J. B. et al. IL-33 is upregulated in colonocytes of ulcerative colitis. Immunol Lett 128, 80–85, doi:10.1016/J.Imlet.2009.11.001 (2010).
Pastorelli, L. et al. Epithelial-derived IL-33 and its receptor ST2 are dysregulated in ulcerative colitis and in experimental Th1/Th2 driven enteritis. Proceedings of the National Academy of Sciences of the United States of America 107, 8017–8022, doi:10.1073/pnas.0912678107 (2010).
Kobori, A. et al. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J Gastroenterol 45, 999–1007, doi:10.1007/S00535-010-0245-1 (2010).
Sedhom, M. A. K. et al. Neutralisation of the interleukin-33/ST2 pathway ameliorates experimental colitis through enhancement of mucosal healing in mice. Gut 62, 1714–1723, doi:10.1136/Gutjnl-2011-301785 (2013).
Sponheim, J. et al. Inflammatory Bowel Disease-Associated Interleukin-33 Is Preferentially Expressed in Ulceration-Associated Myofibroblasts. Am J Pathol 177, 2804–2815, doi:10.2353/Ajpath.2010.100378 (2010).
Gundersen, M. D. et al. Loss of interleukin 33 expression in colonic crypts - a potential marker for disease remission in ulcerative colitis. Scientific reports 6, 35403, doi:10.1038/srep35403 (2016).
Droste, J. S. T. S. et al. Diagnostic yield of colorectal neoplasia in daily clinical practice: consequences for future colorectal cancer screening? Eur J Gastroen Hepat 21, A37–A38 (2009).
Erreni, M., Mantovani, A. & Allavena, P. Tumor-associated Macrophages (TAM) and Inflammation in Colorectal Cancer. Cancer microenvironment: official journal of the International Cancer Microenvironment Society 4, 141–154, doi:10.1007/s12307-010-0052-5 (2011).
Oshima, M. et al. Loss of Apc heterozygosity and abnormal tissue building in nascent intestinal polyps in mice carrying a truncated Apc gene. Proceedings of the National Academy of Sciences of the United States of America 92, 4482–4486 (1995).
Moser, A. R., Pitot, H. C. & Dove, W. F. A Dominant Mutation That Predisposes to Multiple Intestinal Neoplasia in the Mouse. Science 247, 322–324, doi:10.1126/Science.2296722 (1990).
Luthi, A. U. et al. Suppression of Interleukin-33 Bioactivity through Proteolysis by Apoptotic Caspases. Immunity 31, 84–98, doi:10.1016/J.Immuni.2009.05.007 (2009).
Pastorelli, L., De Salvo, C., Vecchi, M. & Pizarro, T. T. The role of IL-33 in gut mucosal inflammation. Mediators of inflammation 2013, 608187, doi:10.1155/2013/608187 (2013).
Pastorelli, L., De Salvo, C., Cominelli, M. A., Vecchi, M. & Pizarro, T. T. Novel cytokine signaling pathways in inflammatory bowel disease: insight into the dichotomous functions of IL-33 during chronic intestinal inflammation. Therapeutic advances in gastroenterology 4, 311–323, doi:10.1177/1756283X11410770 (2011).
Nunes, T., Bernardazzi, C. & de Souza, H. S. Interleukin-33 and inflammatory bowel diseases: lessons from human studies. Mediators of inflammation 2014, 423957, doi:10.1155/2014/423957 (2014).
Lopetuso, L. R., Chowdhry, S. & Pizarro, T. T. Opposing Functions of Classic and Novel IL-1 Family Members in Gut Health and Disease. Frontiers in immunology 4, 181, doi:10.3389/fimmu.2013.00181 (2013).
Maywald, R. L. et al. IL-33 activates tumor stroma to promote intestinal polyposis. Proceedings of the National Academy of Sciences of the United States of America 112, E2487–2496, doi:10.1073/pnas.1422445112 (2015).
Mertz, K. D. et al. The IL-33/ST2 pathway contributes to intestinal tumorigenesis in humans and mice. Oncoimmunology 5, e1062966, doi:10.1080/2162402X.2015.1062966 (2016).
Malik, A. et al. IL-33 regulates the IgA-microbiota axis to restrain IL-1alpha-dependent colitis and tumorigenesis. The Journal of clinical investigation, doi:10.1172/JCI88625 (2016).
Chen, L. et al. IL-23 activates innate lymphoid cells to promote neonatal intestinal pathology. Mucosal immunology 8, 390–402, doi:10.1038/mi.2014.77 (2015).
Ran, F. A. et al. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8, 2281–2308, doi:10.1038/nprot.2013.143 (2013).
Townsend, M. J., Fallon, P. G., Matthews, D. J., Jolin, H. E. & McKenzie, A. N. T1/ST2-deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. The Journal of experimental medicine 191, 1069–1076 (2000).
Espinassous, Q. et al. IL-33 enhances lipopolysaccharide-induced inflammatory cytokine production from mouse macrophages by regulating lipopolysaccharide receptor complex. Journal of immunology 183, 1446–1455, doi:10.4049/jimmunol.0803067 (2009).
Kurowska-Stolarska, M. et al. IL-33 Amplifies the Polarization of Alternatively Activated Macrophages That Contribute to Airway Inflammation. Journal of immunology 183, 6469–6477, doi:10.4049/Jimmunol.0901575 (2009).
Joshi, A. D. et al. Interleukin-33 contributes to both M1 and M2 chemokine marker expression in human macrophages. Bmc Immunol 11, doi:Artn 52, doi:10.1186/1471-2172-11-52 (2010).
Sansom, O. J. et al. Loss of Apc in vivo immediately perturbs Wnt signaling, differentiation, and migration. Genes & development 18, 1385–1390, doi:10.1101/gad.287404 (2004).
Duerkop, B. A., Vaishnava, S. & Hooper, L. V. Immune Responses to the Microbiota at the Intestinal Mucosal Surface. Immunity 31, 368–376, doi:10.1016/J.Immuni.2009.08.009 (2009).
Bongers, G. et al. Interplay of host microbiota, genetic perturbations, and inflammation promotes local development of intestinal neoplasms in mice. The Journal of experimental medicine 211, 457–472, doi:10.1084/jem.20131587 (2014).
Dennis, K. L. et al. Adenomatous polyps are driven by microbe-instigated focal inflammation and are controlled by IL-10-producing T cells. Cancer research 73, 5905–5913, doi:10.1158/0008-5472.CAN-13-1511 (2013).
Latiano, A. et al. Associations between Genetic Polymorphisms in IL-33, IL1R1 and Risk for Inflammatory Bowel Disease. PloS one 8, doi:ARTN e62144, doi:10.1371/journal.pone.0062144 (2013).
Duan, L. H. et al. Interleukin-33 Ameliorates Experimental Colitis through Promoting Th2/Foxp3(+) Regulatory T-Cell Responses in Mice. Mol Med 18, 753–761, doi:10.2119/Molmed.2011.00428 (2012).
Waddell, A. et al. IL-33 Signaling Protects from Murine Oxazolone Colitis by Supporting Intestinal Epithelial Function. Inflammatory bowel diseases 21, 2737–2746, doi:10.1097/MIB.0000000000000532 (2015).
Oboki, K. et al. IL-33 is a crucial amplifier of innate rather than acquired immunity. Proceedings of the National Academy of Sciences of the United States of America 107, 18581–18586, doi:10.1073/Pnas.1003059107 (2010).
Imaeda, H. et al. Interleukin-33 suppresses Notch ligand expression and prevents goblet cell depletion in dextran sulfate sodium-induced colitis. Int J Mol Med 28, 573–578, doi:10.3892/Ijmm.2011.718 (2011).
Gross, P., Doser, K., Falk, W., Obermeier, F. & Hofmann, C. IL-33 attenuates development and perpetuation of chronic intestinal inflammation. Inflammatory bowel diseases 18, 1900–1909, doi:10.1002/Ibd.22900 (2012).
Pushparaj, P. N. et al. Interleukin-33 exacerbates acute colitis via interleukin-4 in mice. Immunology 140, 70–77, doi:10.1111/imm.12111 (2013).
Yang, Z. et al. IL-33-induced alterations in murine intestinal function and cytokine responses are MyD88, STAT6, and IL-13 dependent. American journal of physiology. Gastrointestinal and liver physiology 304, G381–389, doi:10.1152/ajpgi.00357.2012 (2013).
Seidelin, J. B. et al. IL-33 promotes GATA-3 polarization of gut-derived T cells in experimental and ulcerative colitis. J Gastroenterol 50, 180–190, doi:10.1007/s00535-014-0982-7 (2015).
Monticelli, L. A. et al. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proceedings of the National Academy of Sciences of the United States of America 112, 10762–10767, doi:10.1073/pnas.1509070112 (2015).
Yang, Q. et al. IL-33 synergizes with TCR and IL-12 signaling to promote the effector function of CD8+ T cells. European journal of immunology 41, 3351–3360, doi:10.1002/eji.201141629 (2011).
Ngoi, S. M. et al. Presensitizing with a Toll-like receptor 3 ligand impairs CD8 T-cell effector differentiation and IL-33 responsiveness. Proceedings of the National Academy of Sciences of the United States of America 109, 10486–10491, doi:10.1073/pnas.1202607109 (2012).
Gao, K. et al. Transgenic expression of IL-33 activates CD8(+) T cells and NK cells and inhibits tumor growth and metastasis in mice. Cancer letters 335, 463–471, doi:10.1016/j.canlet.2013.03.002 (2013).
Gao, X. et al. Tumoral expression of IL-33 inhibits tumor growth and modifies the tumor microenvironment through CD8+ T and NK cells. Journal of immunology 194, 438–445, doi:10.4049/jimmunol.1401344 (2015).
Jovanovic, I. P. et al. Interleukin-33/ST2 axis promotes breast cancer growth and metastases by facilitating intratumoral accumulation of immunosuppressive and innate lymphoid cells. International journal of cancer. Journal international du cancer 134, 1669–1682, doi:10.1002/ijc.28481 (2014).
Liu, X. et al. IL-33/ST2 pathway contributes to metastasis of human colorectal cancer. Biochemical and biophysical research communications 453, 486–492, doi:10.1016/j.bbrc.2014.09.106 (2014).
Meinicke, H. et al. Tumor-associated changes in intestinal epithelial cells cause local accumulation of KLRG1+ GATA3+ regulatory T cells in mice. Immunology, doi:10.1111/imm.12750 (2017).
Biton, J. et al. In Vivo Expansion of Activated Foxp3+ Regulatory T Cells and Establishment of a Type 2 Immune Response upon IL-33 Treatment Protect against Experimental Arthritis. J Immunol 197, 1708–1719, doi:10.4049/jimmunol.1502124 (2016).
Siede, J. et al. IL-33 Receptor-Expressing Regulatory T Cells Are Highly Activated, Th2 Biased and Suppress CD4 T Cell Proliferation through IL-10 and TGFbeta Release. PloS one 11, e0161507, doi:10.1371/journal.pone.0161507 (2016).
Isidro, R. A. & Appleyard, C. B. Colonic macrophage polarization in homeostasis, inflammation, and cancer. American journal of physiology. Gastrointestinal and liver physiology 311, G59–73, doi:10.1152/ajpgi.00123.2016 (2016).
Seo, D. H. et al. Interleukin-33 regulates intestinal inflammation by modulating macrophages in inflammatory bowel disease. Scientific reports 7, 851, doi:10.1038/s41598-017-00840-2 (2017).
Tu, L. et al. IL-33-induced alternatively activated macrophage attenuates the development of TNBS-induced colitis. Oncotarget 8, 27704–27714, doi:10.18632/oncotarget.15984 (2017).
Fang, M. et al. IL-33 promotes colon cancer cell stemness via JNK activation and macrophage recruitment. Cancer research. doi:10.1158/0008-5472.CAN-16-1602 (2017).
Hooper, L. V., Stappenbeck, T. S., Hong, C. V. & Gordon, J. I. Angiogenins: a new class of microbicidal proteins involved in innate immunity. Nature immunology 4, 269–273, doi:10.1038/ni888 (2003).
Forman, R. A. et al. The Goblet Cell Is the Cellular Source of the Anti-Microbial Angiogenin 4 in the Large Intestine Post Trichuris muris Infection. PloS one 7, doi:ARTN e42248, doi:10.1371/journal.pone.0042248 (2012).
Steppan, C. M. et al. A family of tissue-specific resistin-like molecules. Proceedings of the National Academy of Sciences of the United States of America 98, 502–506, doi:10.1073/pnas.98.2.502 (2001).
Artis, D. et al. RELM beta/FIZZ2 is a goblet cell-specific immune-effector molecule in the gastrointestinal tract. Proceedings of the National Academy of Sciences of the United States of America 101, 13596–13600, doi:10.1073/pnas.0404034101 (2004).
Hung, L. Y. et al. IL-33 drives biphasic IL-13 production for noncanonical Type 2 immunity against hookworms. Proceedings of the National Academy of Sciences of the United States of America 110, 282–287, doi:10.1073/pnas.1206587110 (2013).
Burger-van Paassen, N. et al. Mucin Muc2 deficiency and weaning influences the expression of the innate defense genes Reg3beta, Reg3gamma and angiogenin-4. PloS one 7, e38798, doi:10.1371/journal.pone.0038798 (2012).
Gounaris, E. et al. T-regulatory cells shift from a protective anti-inflammatory to a cancer-promoting proinflammatory phenotype in polyposis. Cancer research 69, 5490–5497, doi:10.1158/0008-5472.CAN-09-0304 (2009).
Arthur, J. C. et al. Intestinal inflammation targets cancer-inducing activity of the microbiota. Science 338, 120–123, doi:10.1126/science.1224820 (2012).
Pinto, D., Robine, S., Jaisser, F., El Marjou, F. & Louvard, D. Regulatory sequences of the mouse villin gene that efficiently drive transgenic expression in immature and differentiated epithelial cells of small and large intestines. Journal of Biological Chemistry 274, 6476–6482, doi:10.1074/Jbc.274.10.6476 (1999).
Mashiko, D. et al. Generation of mutant mice by pronuclear injection of circular plasmid expressing Cas9 and single guided RNA. Scientific reports 3, 3355, doi:10.1038/srep03355 (2013).
Kononen, J. et al. Tissue microarrays for high-throughput molecular profiling of tumor specimens. Nature medicine 4, 844–847 (1998).
Acknowledgements
This work was supported in part by the SUCCESS (Sinai Ulcerative Colitis Clinical, Experimental and System Studies) grant, GCO14-0560. Funds for the generation of the V33 mice were provided by Janssen Pharmaceuticals. Lili Chen received a Research Fellowship Award from the Crohn’s & Colitis Foundation of America (CCFA). We thank the Mouse Genetics and Gene Targeting CORE, and the Flow Cytometry and Histology CORES at Mount Sinai for technical support.
Author information
Authors and Affiliations
Contributions
L.C. and Z.H. designed study, did experiments, analyzed data and wrote the manuscript; F.O.S., M.D., contributed to histology and immunostaining; H.B.K., and N.H. contributed with the tissue microarrays and pathological analyses; C.C. helped with mice genotyping; K.K. assisted with generation of transgene and deficient mice. G.B. assisted with experimental design; G.C.F. and S.A.L. designed study, analyzed data and wrote the manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Competing Interests
The authors declare that they have no competing interests.
Additional information
Publisher's note: Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
He, Z., Chen, L., Souto, F.O. et al. Epithelial-derived IL-33 promotes intestinal tumorigenesis in Apc Min/+ mice. Sci Rep 7, 5520 (2017). https://doi.org/10.1038/s41598-017-05716-z
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-017-05716-z
- Springer Nature Limited