
Abstract
Aphidoletes aphidimyza is widely recognized as an effective predator of aphids in agricultural systems. However, there is limited understanding of its predation mechanisms. In this study, we generated a high-quality chromosome level of the A. aphidimyza genome by combining PacBio, Illumina, and Hi-C data. The genome has a size of 192.08 Mb, with a scaffold N50 size of 46.85 Mb, and 99.08% (190.35 Mb) of the assembly is located on four chromosomes. The BUSCO analysis of our assembly indicates a completeness of 97.8% (n = 1,367), including 1,307 (95.6%) single-copy BUSCOs and 30 (2.2%) duplicated BUSCOs. Additionally, we annotated a total of 13,073 protein-coding genes, 18.43% (35.40 Mb) repetitive elements, and 376 non-coding RNAs. Our study is the first time to report the chromosome-scale genome for the species of A. aphidimyza. It provides a valuable genomic resource for the molecular study of A. aphidimyza.
Similar content being viewed by others

Background & Summary
Aphids (Hemiptera: Aphididae) are prevalent insect pests that affect crops worldwide. They cause substantial economic losses by directly feeding on plants, spreading plant viruses, and producing honeydew1,2,3. Pesticides are mainly used to control aphids4,5. However, overuse of chemical pesticides can lead to drug resistance in aphids6,7,8 and may also kill various beneficial insects9. Therefore, alternative methods of pest control should be explored. The biological control method leverages living organisms to control pests and diseases. The use of natural enemies (such as birds, fungi, etc) to modulate the reproduction and transmission of pests10,11,12.
Aphidoletes aphidimyza Rondani (Diptera: Cecidomyiidae) is widely used to control aphids in agricultural systems13. It is an oligophagous insect that displays remarkable voracity and targets more than 80 species of aphids, including the major pests, namely Aphis craccivora14, Aphis gossypii15, Myzus persicae16, and others17. Owing to the limited dispersal ability of larvae, adults primarily depend on oviposition near aphid colonies to facilitate the predation of their progeny and the establishment of their population13,18,19,20. This is possibly based on chemosensory mechanisms, such as olfaction and gustation, which are also important for host selection. Olfaction is important for host orientation, while gustation is crucial in host selection21,22,23,24. Previous studies have shown that adults mainly rely on odor cues (such as aphid body volatiles, alarm pheromones, and aphid-induced plant volatiles) to precisely locate the position of aphids and complete oviposition25,26,27,28. They also use non-volatile cues, including honeydew, as a source of nutrition and an oviposition stimulant29,30,31. However, the lack of high-quality genomic data has limited the understanding of the genetic basis of search and predation on aphids.
In this study, we obtained a high-quality genome of Aphid midge using PacBio, Illumina, and Hi-C data. We annotated essential genomic elements, such as repeat elements, non-coding RNAs (ncRNAs), and protein-coding genes. The availability of a complete and detailed genome assembly is essential to basic biological research. This paper provides a valuable genomic resource for research into molecular mechanisms and evolution.
Methods
Sample collection
The larvae of A. aphidimyza were obtained from the tobacco base in Leshan Town, Zunyi City, Guizhou Province, China, in May 2017. They were raised in an artificial climate chamber (24 ± 1 °C with a 14:10 [L:D] h photoperiod, 70% relative humidity). In this experiment, initially, an inbred strain, a single pair of siblings was first used for 30 generations of mating, and then the genome and transcriptome of the inbred line were sequenced and analyzed. The larvae were fed with Megoura japonica on bean plants, and emerging adults were provided with 10% honey. A total of 500, 200, 200, and 200 female adult individuals were used for PacBio, Illumina, Hi-C, and Iso-Seq sequencing, respectively.
Genome sequencing
Genomic DNA and RNA were extracted from the specimen using the FastPure® Blood/Cell/Tissue/Bacteria DNA Isolation Mini Kit (Vazyme Biotech Co., Ltd, Nanjing, China) and TRIzol reagent (YiFeiXue Tech, Nanjing, China), respectively. The quality and quantity of both total DNA and RNA were assessed through 1% agarose gel electrophoresis, the NanoDrop 2000 by Thermo Fisher, and the Qubit 3.0 fluorometer (Invitrogen, USA). PacBio library of a 30 kb insert size was created using the SMRTbell Template Prep Kit 2.0 from Pacific Biosciences of California, based in Menlo Park, USA. For Illumina sequencing, a short library with 150 bp paired-end reads and a 350 bp insert size was generated using the TruSeq DNA PCR-free kit. Furthermore, an Iso-Seq library with a 2 kb insert size was established using the SMRTbell prep kit 3.0 (Pacific Biosciences of California, Menlo Park, USA). Short RNA-seq libraries were also constructed for RNA sequencing on the BGIMGISEQ-500 platform (Shenzhen, China). The Hi-C sequencing was carried out by digesting extracted DNA with the Mbol restriction enzyme. We utilized the Illumina NovaSeq. 6000 platform to sequence all short-read libraries. PacBio sequencing was carried out using the PacBio Sequence RSII platforms employing the CLR mode. All these libraries were created and sequenced by Berry Genomics (Beijing, China). Our sequencing efforts yielded a total of 101.38 Gb of clean data, comprising 31.76 Gb from PacBio (168×), 26.64 Gb from Illumina (139×), 35.05 Gb from Hi-C (185×), and 7.93 Gb from RNA (6.28 Gb from Illumina and 1.65 Gb from Iso-Seq), as detailed in Table 1.
Genome survey and assembly
We used BBTools v38.8232 to perform quality control on raw Illumina data, and then eliminated duplicate reads using “clumpify.sh”. Furthermore, “bbduk.sh” was used to trim sequences with quality scores below 20, sequences containing more than 5 Ns, and reads shorter than 15 bp. Polymer trimming (>10 bp) and correction of overlapping paired reads were also performed. In addition, a 21-mer was selected for k-mer analysis and the k-mer distribution was estimated using “khist.sh” (BBTools). The 21-mer depth frequency distribution was calculated using GenomeScope v2.033 and the maximum k-mer coverage cut-off was set to 10,000. A k-mer analysis indicated that the number of unique k-mer spoke at 21 and predicted a genome assembly size of 192.09 Mb, with a heterozygosity of 0.189% and a repeat content proportion of approximately 15.4% (Fig. 1).

Genome survey at 21-mer of A. aphidimyza estimated by GenomeScope. The vertical dotted lines represent the peaks of different coverages for the heterozygous, the homozygous, and the duplicated sequences, separately.
Primary assembly from PacBio reads was performed using Flye v2.8.334, which involves one round of self-polishing with a minimum overlap of 3,000 (-i 1 -m 3000). The resulting assembly was polished with two rounds of short reads using NextPolish v1.3.135. Heterozygous regions were eliminated using Purge_Dups v1.2.536 with a 70% cut-off for identifying contigs as haplotigs. Minimap2 v2.2337 was used as the read mapper to remove redundancy and polish assembly. Hi-C reads were aligned to the assembly using Juicer v1.6.238. Then, 3D-DNA v18092239 was used to anchor the contigs onto the chromosomes. Hi-C heatmaps were manually inspected and corrected using Juicebox v1.11.0839 to identify potential errors. Possibilities of contaminants were detected using MMseqs. 2 v1140, which performed Basic Local Alignment Search Tool (BLASTN)-like searches based on the NCBI nucleotide and UniVec databases with a sequence identity of 0.8 (“-min-seq-id 0.8”). To further examine vector contaminants, we used blastn (BLAST+ v2.11.041) against the UniVec database. We considered that sequences with over 90% hits in the aforementioned database likely contained contaminants. Online BLASTN analysis in the NCBI nucleotide database was used to double-check sequences with above 80% hits. Following that, we removed any possible bacterial contamination from the assembled scaffolds. Our final genome assembly encompassed 192.08 Mb and comprised 70 scaffolds along with 444 contigs. It featured a scaffold N50 length of 46.85 Mb and a contig N50 size of 1.22 Mb (Fig. 2). The final assembly is close to the size of the genome survey (192.09 Mb) analysis. A remarkable 99.08% (190.35 Mb) of the genome was anchored into four chromosomes, as illustrated in Fig. 3 and detailed in Table 2. The assembled genome size closely resembled that of Contarinia nasturtii42(185.89 Mb).

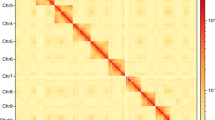
Genomic heatmap. Genome-scale chromosome heatmap of A. aphidimyza, with individual chromosomes outlined in blue.

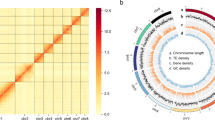
Genomic features. Circos plot with a window size of 100 kb. Each circle from inside to outside represents simple repeats, LTR, LINE, SINE, DNA, gene density, GC content, and chromosome length.
Genome annotation
The annotated A. aphidimyza genome included the following three important genomic components: repetitive elements, ncRNAs, and protein-coding genes. The de novo repeat library was established by RepeatModeler v2.0.343 with the parameter “-LTRStruct”. We then combined Dfam 3.544 and RepBase-20181026 databases45 to generate a custom library, which was employed to mask repeat elements using RepeatMasker v4.1.2-p146. To summarize, RepeatMasker analysis revealed that the A. aphidimyza genome contains approximately 18.43% (35.40 Mb) repeat elements, i.e. long terminal repeat elements (LTR, 3.61%), DNA transposons (1.50%), long interspersed nuclear elements (LINE, 1.02%), and short interspersed nuclear elements (SINE, 0.01%), and other elements (Table 2). The annotations of rRNA, snRNA, and miRNA were compared with the Rfam v14.10 database using Infernal v1.1.447 and tRNAscan-SE v2.0.948. We identified 376 ncRNAs in the genome of A. aphidimyza, including 84 ribosomal RNAs, 52 miRNAs, 38 small nuclear RNAs, and 202 tRNAs (Table 3).
Protein-coding genes were annotated by combining results from ab initio, transcriptomic data, and protein homology using the MAKER pipeline v3.01.0349. BRAKER v2.1.650 and GeMoMa v1.7.151 predictions were combined as the ab initio input for MAKER, which combined transcriptome and protein evidence. Transcriptome data was used for annotation using a mixed assembly of Iso-seq and RNA-seq data. Transcriptome alignment was performed using HISAT2 v2.2.152 and then assembled into transcripts using Stringtie v2.1.653. BRAKER, employing Augustus v3.4.054 and GeneMark-ES/ET/EP v4.68_lic55, was used to automatically train prediction models. This mode was based on RNA-seq alignments and reference proteins obtained from the OrthoDB10 v1 database56. GeMoMa predicted genes using protein homology, intron conservation, and transcripts. GeMoMa was used with the parameters “GeMoMa.c = 0.5 GeMoMa.p = 8” and protein sequences from five species, namely Contarinia nasturtii42 (GCF_009176525.2), Bradysia coprophila57 (GCF_014529535.1), Anopheles arabiensis58 (GCF_016920715.1), Drosophila melanogaster59 (GCF_000001215.4), and Bombyx mori60 (GCF_014905235.1). In addition, the protein sequences obtained from the same set of five species used in the GeMoMa analysis were included in the MAKER pipeline as supporting evidence for protein homology. To summarize, 13,073 protein-coding genes were annotated in A. aphidimyza. The number of PCGs in A. aphidimyza was fewer than that of C. nasturtii (14,889 genes) (Table 3). The completeness of 98.6% of A. aphidimyza was confirmed by Benchmarking Universal Single-Copy Orthologs (BUSCO), which was much higher than that of C. nasturtii (92.9%) (Table 3). Functional annotation of PCGs was performed using Diamond v2.0.861, which used the sensitive mode and an e-value of 1e-5 to explore the UniProtKB database. Furthermore, we used EggNOG-mapper v2.1.562 and InterProScan 5.48–83.063 software to explore Gene Ontology (GO), enzyme codes (EC), Kyoto Encyclopedia of Genes and Genomes (KEGG) orthologous groups, clusters of orthologous groups (COG), and KEGG pathway annotations. Structural domains of genes were predicted using InterProScan, including the following five databases: Pfam64, Simple Modular Architecture Research Tool (SMART65), Superfamily66, Gene3D67, and Conserved Domain Database (CDD68). Finally, Genes with 9,798 GO terms, 8,646 KEGG pathways, 2,594 Enzyme Codes, 9,799 Reactome pathways, and 10,774 COG categories were identified by combining the eggNOG and InterProScan annotation results (Table 4).
Data Records
The raw sequencing data and genome assembly of Aphidoletes aphidimyza have been deposited at the National Center for Biotechnology Information (NCBI). The Illumina, Iso-Seq, Hi-C, PacBio, and RNA-seq data can be found under identification numbers SRR1333379069, SRR1333378970, SRR132366638071, SRR1322240772, SRR1323672573, respectively. The assembled genome has been deposited in the NCBI assembly with the accession number GCA_030463065.174. Additionally, the results of annotation for repeated sequences, gene structure, and functional prediction have been deposited in the figshare75.
Technical Validation
Two independent methods were used to assess the completeness and quality of our genome assembly. We first used BUSCO v5.4476 with the “insecta_odb10” database (n = 1,367) to examine the completeness of the final assembled genome. In our BUSCO analysis, a commendable 97.8% of complete BUSCOs were identified, which included 95.6% of single-copy genes and 2.2% of duplicated BUSCOs (Table 2). To evaluate mapping success, we employed Minimap2 and SAMtools v1.977 to align the clean reads obtained from both Illumina and PacBio sequencing with the final assembly. Impressively, we accomplished a mapping rate of 94.78% for Illumina reads, 98.09% for PacBio reads, 94.26% for Iso-seq reads, and 87.73% for RNA-seq reads, respectively. Overall, these assessments reflect the high quality of the genomic assembly.
Code availability
No specific script was used in this work. All commands and pipelines used in data processing were executed according to the manual and protocols of the corresponding bioinformatic software.
References
Zhang, G. X. & Zhong, T. S. Economic insect fauna of China. Beijing: Science Press. 25, 1–65 (1983).
Blackman, R. L. & Eastop, V. F. Aphids on the world’s crops: an identification and information guide. John Wiley & Sons Ltd (2000).
Ng, J. C. & Perry, K. L. Transmission of plant viruses by aphid vectors. Mol Plant Pathol. 5, 505–511 (2004).
Kift, N. B. et al. The impact of insecticide resistance in the currant‐lettuce aphid, Nasonovia ribisnigri, on pest management in lettuce. Agric for Entomol. 6, 295–309 (2004).
Guo, K., Yang, P., Chen, J., Lu, H. & Cui, F. Transcriptomic responses of three aphid species to chemical insecticide stress. Sci China Life Sci. 60, 931–934 (2017).
Herron, G. A., Powis, K. & Rophail, J. Insecticide resistance in Aphis gossypii Glover (Hemiptera: Aphididae), a serious threat to Australian cotton. Austral Entomol. 40, 85–91 (2001).
Koo, H., An, J., Park, S., Kim, J. & Kim, G. Regional susceptibilities to 12 insecticides of melon and cotton aphid, Aphis gossypii (Hemiptera: Aphididae) and a point mutation associated with imidacloprid resistance. Crop Prot. 55, 91–97 (2014).
Bass, C., Denholm, I., Williamson, M. S. & Nauen, R. The global status of insect resistance to neonicotinoid insecticides. Pestic Biochem Physiol. 121, 78–87 (2015).
Sánchez-Bayo, F. Indirect Effect of Pesticides on Insects and Other Arthropods. Toxics. 9, 177 (2021).
Hoddle, M. S. & Van Driesche, R. G. Biological control of insect pests. Encyclopedia of insects. pp. 91–101 (2009).
Nazir, T., Khan, S. & Qiu, D. Biological control of insect pest. Pests Control and Acarology. 21, 78–87 (2019).
Abdulle, Y. A., Nazir, T., Keerio, A. U., Ali, H. & Qiu, D. In vitro virulence of three Lecanicillium lecanii strains against the whitefly, Bemisia tabaci (Genn.) (Hemiptera Aleyrodidae). Egypt J Biol Pest Co. 30, 1–6 (2020).
Boulanger, F. X., Jandricic, S., Bolckmans, K., Wäckers, F. L. & Pekas, A. Optimizing aphid biocontrol with the predator Aphidoletes aphidimyza, based on biology and ecology. Pest Manag Sci. 75, 1479–1493 (2019).
Madahi, K., Sahragard, A. & Hosseini, R. Predation rate and numerical response of Aphidoletes aphidimyza feeding on different densities of Aphis craccivora. Biocontrol Sci Technol. 25, 72–83 (2015).
Mottaghinia, L., Hassanpour, M., Razmjou, J., Hosseini, M. & Chamani, E. Functional Response of Aphidoletes aphidimyza Rondani (Diptera: Cecidomyiidae) to Aphis gossypii Glover (Hemiptera: Aphididae): Effects of Vermicompost and Host Plant Cultivar. Neotrop Entomol. 45, 88–95 (2016).
Khadijeh, M., Ahad, S., Reza, H. & Valiollah, B. Prey-Stage Preference and Comparing Reproductive Performance of Aphidoletes aphidimyza (Diptera: Cecidomyiidae) Feeding on Aphis gossypii (Hemiptera: Aphididae) and Myzus persicae. J Econ Entomol. 112, 1073–1080 (2019).
Hosseini, M. et al. Plant quality effects on intraguild predation between Orius laevigatus and Aphidoletes aphidimyza. Entomol Exp Appl. 135, 208–216 (2010).
Laurema, S., Husberg, G. B. & Markkula, M. Composition and functions of the salivary gland of the larvae of the aphid midge Aphidoletes aphidimyza. Ecology of Aphidophaga. pp. 113–118 (1986).
Němec, V., Havelka, J. & Šula, J. Enzymatic activities in the saliva of larvae of the gall midge Aphidoletes aphidimyza (Diptera, Cecidomyiidae). Eur J Entomol. 95, 211–216 (1992).
Higashida, K. et al. Reproduction and oviposition selection by Aphidoletes aphidimyza (Diptera: Cecidomyiidae) on the banker plants with alternative prey aphids or crop plants with pest aphids. Appl Entomol Zool. 51, 445–456 (2016).
Lahondere, C. & Lazzari, C. R. Thermal effect of blood feeding in the telmophagous fly Glossina morsitans morsitans. J Therm Biol. 48, 45–50 (2015).
Leung, K. et al. Next-generation biological control: the need for integrating genetics and genomics. Biol Rev Camb Philos Soc. 95, 1838–1854 (2020).
Erb, M., Zust, T. & Robert, C. Using plant chemistry to improve interactions between plants, herbivores and their natural enemies: challenges and opportunities. Curr Opin Biotechnol. 70, 262–265 (2021).
Yuan, H. et al. Genome of the hoverfly Eupeodes corollae provides insights into the evolution of predation and pollination in insects. BMC Biol. 20, 1–16 (2022).
Mansour, M. H. The role of plants as a factor affecting oviposition by Aphidoletes aphidimyza (Diptera: Cecidomyiidae). Entomol Exp Appl. 18, 173–179 (1975).
Sentis, A., Lucas, É. & Vickery, W. L. Prey Abundance, Intraguild Predators, Ants and the Optimal Egg-laying Strategy of a Furtive Predator. J Insect Behav. 25, 529–542 (2012).
Jandricic, S. E., Wraight, S. P., Gillespie, D. R. & Sanderson, J. P. Oviposition behavior of the biological control agent Aphidoletes aphidimyza (Diptera: Cecidomyiidae) in environments with multiple pest aphid species (Hemiptera: Aphididae). Biol Control. 65, 234–245 (2013).
Higashida et al. Volatiles from eggplants infested by Aphis gossypii induce oviposition behavior in the aphidophagous gall midge Aphidoletes aphidimyza. Arthropod Plant Interact. 16, 45–52 (2022).
Choi, M. Y., Roitberg, B. D., Shani, A., Raworth, D. A. & Lee, G. H. Olfactory response by the aphidophagous gall midge, Aphidoletes aphidimyza to honeydew from green peach aphid, Myzus persicae. Entomol Exp Appl. 111, 37–45 (2004).
Watanabe, H. et al. Effects of aphid honeydew sugars on the longevity and fecundity of the aphidophagous gall midge Aphidoletes aphidimyza. Biol Control. 78, 55–60 (2014).
Hiroshi, W. et al. An Attractant of the Aphidophagous Gall Midge Aphidoletes aphidimyza From Honeydew of Aphis gossypii. J Chem Ecol. 42, 149–155 (2016).
Bushnell, B. BBtools. Available online: https://sourceforge.net/projects/bbmap/ (accessed on 1 October 2022) (2014).
Vurture, G. W. et al. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics. 33, 2202–2204 (2017).
Kolmogorov, M., Yuan, J., Lin, Y. & Pevzner, P. A. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol. 37, 540–546 (2019).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898 (2020).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Steinegger, M. & Söding, J. MMseqs. 2 enables sensitive protein sequence searching for the analysis of massive data sets. Nat Biotechnol. 35, 1026–1028 (2017).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J Mol Biol. 215, 403–410 (1990).
Mori, B. A. et al. De Novo Whole-Genome Assembly of the Swede Midge (Contarinia nasturtii), a Specialist of Brassicaceae, Using Linked-Read Sequencing. Genome Biol. Evol. 13, evab036 (2021).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. Proc. Natl. Acad. Sci. 117, 9451–9457 (2020).
Storer, J., Hubley, R., Rosen, J., Wheeler, T. J. & Smit, A. F. The Dfam community resource of transposable element families, sequence models, and genome annotations. Mob DNA. 12, 1–14 (2021).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob DNA. 6, 1–6 (2015).
Smit, A. F. A., Hubley, R. & Green, P. RepeatMasker Open-4.0. Available online: http://www.repeatmasker.org (accessed on 1 October 2022) (2013–2015).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics. 29, 2933–2935 (2013).
Chan, P. P. & Lowe, T. M. tRNAscan-SE: searching for tRNA genes in genomic sequences. Methods Mol Biol. 1962, 1–14 (2019).
Holt, C. & Yandell, M. MAKER2: an annotation pipeline and genome-database management tool for second-generation genome projects. Bmc Bioinformatics. 12, 1–14 (2011).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. Nar Genom Bioinform. 3, lqaa108 (2021).
Keilwagen, J., Hartung, F., Paulini, M., Twardziok, S. O. & Grau, J. Combining RNA-seq data and homology-based gene prediction for plants, animals and fungi. Bmc Bioinformatics. 19, 189 (2018).
Kim, D., Langmead, B. & Salzberg, S. L. HISAT: a fast spliced aligner with low memory requirements. Nat Methods. 12, 357–360 (2015).
Kovaka, S. et al. Transcriptome assembly from long-read RNA-seq alignments with StringTie2. Genome Biol. 20, 1–13 (2019).
Stanke, M., Steinkamp, R., Waack, S. & Morgenstern, B. AUGUSTUS: a web server for gene finding in eukaryotes. Nucleic Acids Res. 32, W309–W312 (2004).
Brůna, T., Lomsadze, A. & Borodovsky, M. GeneMark-EP+: eukaryotic gene prediction with self-training in the space of genes and proteins. Nar Genom Bioinform. 2, lqaa26 (2020).
Kriventseva, E. V. et al. OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811 (2019).
Urban JM, Gerbi SA, Spradling AC. Chromosome-scale scaffolding of the fungus gnat genome (Diptera: Bradysia coprophila). bioRxiv. 11.03.515061 (2022).
Zamyatin, A. et al. Chromosome-level genome assemblies of the malaria vectors Anopheles coluzzii and Anopheles arabiensis. Gigascience. 10, giab017 (2021).
Hoskins, R. A. et al. The Release 6 reference sequence of the Drosophila melanogaster genome. Genome Res. 25, 445–458 (2015).
Kim, S. W. et al. Whole-genome sequences of 37 breeding line Bombyx mori strains and their phenotypes established since 1960s. Sci Data. 189, 1–8 (2022).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat Methods. 12, 59–60 (2015).
Huerta-Cepas, J. et al. Fast genome-wide functional annotation through orthology assignment by eggNOG-mapper. Mol Biol Evol. 34, 2115–2122 (2017).
Finn, R. D. et al. InterPro in 2017—beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199 (2017).
El-Gebali, S. et al. The Pfam protein families database in 2019. Nucleic Acids Res. 47, D427–D432 (2019).
Letunic, I. & Bork, P. 20 years of the SMART protein domain annotation resource. Nucleic Acids Res. 46, D493–D496 (2018).
Wilson, D. et al. SUPERFAMILY—sophisticated comparative genomics, data mining, visualization and phylogeny. Nucleic Acids Res. 37, D380–D386 (2009).
Lewis, T. E. et al. Gene3D: extensive prediction of globular domains in proteins. Nucleic Acids Res. 46, D435–D439 (2018).
Marchler-Bauer, A. et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 45, D200–D203 (2017).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR13333790 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR13333789 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR13236663 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR13222407 (2023).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR13236725 (2023).
NCBI Assembly https://identifiers.org/ncbi/insdc.gca:GCA_030463065.1 (2023).
Zhang, F. Genome assembly and annotations of Aphidoletes aphidimyza (Diptera: Cecidomyiidae). Figshare, https://doi.org/10.6084/m9.figshare.24077871 (2024).
Manni, M., Berkeley, M. R., Seppey, M., Simão, F. A. & Zdobnov, E. M. BUSCO update: novel and streamlined workflows along with broader and deeper phylogenetic coverage for scoring of eukaryotic, prokaryotic, and viral genomes. Mol Biol Evol. 38, 4647–4654 (2021).
Danecek, P. et al. Twelve years of SAMtools and BCFtools. GigaScience. 10, giab008 (2021).
Acknowledgements
This work was funded by the Science and Technology Project of Zunyi Branch of Guizhou Tobacco Company (2022XM11; 2020XM05) and the Guizhou Province Science and Technology Innovation Talent Team Project (Qian Ke He Pingtai Rencai-CXTD [2021]004).
Author information
Authors and Affiliations
Contributions
M.Y., H.W., and F.Z. supervised the project. X.S., X.Y., and B.Y. contributed to the research design. X.S., J.J., B.Y., and G.Z. collected the samples for PacBio, Illumina, Hi-C, and RNA sequencing. F.Z., J.J., H.W., and G.Z. performed the genome assembly and annotation. X.S., H.W., and G.Z performed transcriptome analysis. X.S., J.J., X.Y., B.Y., H.W., G.Z., F.Z., and M.Y. wrote the manuscript. All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Shen, X., Jin, J., Zhang, G. et al. The chromosome-level genome assembly of Aphidoletes aphidimyza Rondani (Diptera: Cecidomyiidae). Sci Data 11, 785 (2024). https://doi.org/10.1038/s41597-024-03614-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03614-4
- Springer Nature Limited