
Abstract
Prorocentrum shikokuense (formerly P. donghaiense) is a pivotal dinoflagellate species associating with the HABs in the East China Sea. The complexity of its large nuclear genome hindered us from understanding its genomic characteristics. Full-length transcriptome sequencing offers a practical solution to decipher the physiological mechanisms of a species without the reference genome. In this study, we employed single-molecule real-time (SMRT) sequencing technology to sequence the full-length transcriptome of Prorocentrum shikokuense. We successfully generated 41.73 Gb of clean SMRT sequencing reads and isolated 105,249 non-redundant full-length non-chimeric reads. Our trial has led to the identification of 11,917 long non-coding RNA transcripts, 514 alternative splicing events, 437 putative transcription factor genes from 17 TF gene families, and 34,723 simple sequence repeats. Additionally, a total of 78,265 open reading frames were identified, of them 15,501 were the protein coding sequences. This dataset is valuable for annotating P. shikokuense genome, and will contribute significantly to the in-depth studies on the molecular mechanisms underlining the dinoflagellate bloom formation.
Similar content being viewed by others


Background & Summary
Dinoflagellates are a group of unique unicellular microorganisms known for their distinct characteristics such as flagellar insertion, pigmentation, organelles, and nuclear structure1,2. They belong to the infrakingdom Alveolata which also includes the phyla Ciliophora and Apicomplexa. Unlike their relatives, dinoflagellates possess some of the most unusual genome structures among eukaryotes, characterized by large nuclear genomes with permanently condensed liquid-crystalline chromosomes and the absence of nucleosomes3,4. Moreover, the tandem gene arrays, trans-spliced mRNAs, and the paucity of transcriptional regulation compared to other eukaryotes have been revealed in dinoflagellate genome researches5,6,7. It is noteworthy that despite having larger nuclear genomes among eukaryotes, their organellar genomes contain less genes8. These unique cellular and molecular characteristics of dinoflagellates break the dogmas established from studying other eukaryotes, making them a peculiar and significant group within the eukaryotic world. Additionally, dinoflagellates have gained increasing attentions due to their crucial roles in natural ecosystems and their importance in human food production. They are primarily responsible for HABs; they cause about 80% of the total9.
Prorocentrum shikokuense (formerly Prorocentrum donghaiense)10 is a key species responsible for HABs in the East China Sea, especially those in Yangtze River Estuary and its adjacent sea area11,12 which have experienced recurrent extensive blooms caused mainly by this species13. In addition, HABs caused by P. shikokuense have been documented in the coastal waters of Japan and Korea14, the southwest coast of India15, the Mediterranean Sea of Italy16, and the southern coast of Myanmar17. It is remarkable that P. shikokuense is able to form large-scale and sustaining blooms under inorganic phosphorus nutrient-limited conditions18,19. Multiple studies have indicated that phosphate limitation is the major factor driving the succession of microalgae in blooms from diatoms to dinoflagellates in the East China Sea20,21,22. The critical role of phosphorus availability in regulating bloom formation has been well recognized20,21,22,23,24. Additionally, phosphorus indispensable for vital life activities such as cell membrane and nucleic acid synthesis, energy storage, cell signal transduction and metabolic process regulation25,26,27 is an important nutrient for the growth and reproduction of marine phytoplankton. Thus, determining the dinoflagellate adaptation to varying phosphorus conditions is of a great importance.
Up to now, transcriptomic, proteomic and metabolomic technologies have been extensively employed to unravel the mechanisms underlining P. shikokuense bloom formation28,29,30,31,32,33,34,35. However, the physiologically and metabolically understanding P. shikokuense always appreciates its reference genome. Presently, the dinoflagellates with available genome assemblies are usually symbiotic or parasitic species with relatively small genome sizes compared to free-living dinoflagellates36,37,38,39,40,41. Assembling the high-quality genomes of free-living dinoflagellates is restricted by their larger and more complex genomes and associating expanse. Fortunately, the studying community has been tolerating such scarcity; their researches focus on the physiological aspects such as stress response, success physiology among others28,29,30,31,32,33,34,35. As the optimal solution, a full-length transcriptome should meet the studying demand, and simultaneously provides an avenue out of the budget dilemma.
RNA-seq (RNA sequencing) has been pivotal in advancing our understanding of marine dinoflagellates, and shedding light on their transcript information and genetic basis28,29,30,31,32,34,35. Although RNA-seq is widely used, it has the limitation of short reads, which challenges the accurate acquisition of full-length transcripts, especially in regions with repeats and in complex genomes42,43. In contrast, single-molecule real-time (SMRT) sequencing, a third-generation technology developed by Pacific Biosciences, overcomes these limitations by providing longer reads and faster sequencing44,45. This enables the direct acquisition of full-length cDNA sequences without the assembly, facilitating more precise identification of gene isoforms and the discovery of novel genes. While SMRT sequencing has drawbacks, such as higher error rates and lower throughput compared to NGS, these can be mitigated through hybrid sequencing strategies or self-correction with circular-consensus reads44.
In this study, we conducted a full-length transcriptomic analysis of P. shikokuense under various phosphorus (P) nutrition conditions and at different developmental stages. This analysis was performed using SMRT sequencing on the Pacific Biosciences Sequel Platform (Fig. 1). Totally, 41.73 Gb SMAT sequencing clean reads were generated, 573,159 circular consensus (CCS) reads with average lengths of 1,435 bp were produced. Among them, 78.70% (451,077) were identified as full length non-chimeric (FLNC) reads. Furthermore, a total of 154,441 high quality sequences were obtained by clustering full-length non-chimeric sequences. After removing the redundant, 105,249 non-redundant FLNC reads were obtained, with 50,338 (47.83%) successfully annotated against the five public databases. In addition, a total of 78,265 open reading frames (ORF) were identified, of them 15,501 were the protein coding sequences. Furthermore, a total of 11,917 long non-coding RNA (lncRNA) transcripts, 514 alternative splicing (AS) events, 437 putative transcription factor (TF) members from 17 TF families, and 34,723 simple sequence repeats (SSRs) were identified, respectively. Our results offered a valuable set of the full-length cDNAs of P. shikokuense, which was significant for advancing the studies on its molecular mechanisms, particularly those associating with the bloom formation. This data set is also useful for the future genome annotation of P. shikokuense, and the enhancement of our understanding its genetic makeup.

Flow diagram shows the overview of the study design.
Methods
Sample collection and RNA preparation
P. shikokuense, provided by the Center for Collection of Marine Algae, College of Ocean and Earth Sciences, Xiamen University, was cultured at a controlled temperature of 20 ± 1°C. The culture condition was 12 h light and 12 h dark as a cycle, with a light intensity maintained between 50 and 100 μmol photons m−2 s−1. To eliminate the effects of phosphorus from seawater, the culture medium was L1 medium prepared with artificial seawater. The samples were collected at three different developmental stages: exponential growth phase, stationary phase and declining phase, and three different treatments: P-sufficient, P-deficient, and inorganic P-resupplied. For three different treatments, medium with 36.3 μM and 0 μM Na2HPO4 were used for the P-sufficient and P-deficient cultures, respectively. On the seventh day, P-deficient cultures were resupplied with Na2HPO4 to the final concentration of 36.3 μM, forming the DIP-resupplied group.
The collected samples were concentrated via concentration (1800 g for 10 minutes at 20°C), immediately frozen in liquid nitrogen, and then stored at −80°C until RNA extraction. Total RNA from each sample (100 mg) was extracted using the RNeasy Plus Mini Kit (Qiagen, Valencia, CA, USA). The purity and concentration of RNA extracted by the above process were assessed by Nanodrop ND-1000 (Thermo Fisher Scientific, USA). Additionally, Agilent 2100 Bioanalyzer system (Agilent Technologies, USA) was used to measure RNA samples integrity.
Library construction
For library construction, a total of 5 μg of high-quality RNA from different samples were mixed in equal amounts. The sequencing libraries were created according to PacBio’s iso-seq sequencing protocol (Fig. 1). The process involved synthesizing full-length cDNA by NEBNext® Single Cell/Low Input cDNA Synthesis & Amplification Module, followed by PCR amplification and purification of the synthesized full-length cDNA. Then, the cDNA damage repair and terminal repair were performed. Last, SMRT hairpin adapters were ligated to the end of double-stranded cDNA molecules. In order to ensure the quality of library, the purity, concentration and insert size of the library were checked by using the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA), and the qualified library was processed for full-length transcriptome sequencing on PacBio sequencing platform.
Single molecule real-time (SMRT) sequencing and analysis pipeline
The raw subreads were analyzed following the Iso-Seq3 pipeline (https://github.com/PacificBiosciences/IsoSeq) included three initial steps: generation of CCS subreads, classification of full length (FL) reads, and clustering of full length non-chimeric (FLNC) reads. The circular consensus sequence (CCS) was obtained using the SMRTlink (v10.1) software and polished CCS subreads were generated by using ccs (v6.2.0) (https://github.com/PacificBiosciences/ccs), from the subreads bam files with a minimum quality of 0.9 (-min-rq 0.9). The default minimum number of FL subreads (n = 3) required to generate CCS for a zero-mode waveguide (ZMW) was used. FL transcripts were identified based on sequences with the poly(A) and the 5′ and 3′ cDNA primers. Lima (v2.1.0) and IsoSeq3 refine were used to remove the primers and poly(A) tails, respectively. FLNC with similar sequences (copies originated from a same transcript) was clustered by IsoSeq analysis application in SMRTLink software. The clustering algorithm Iterative Clustering for Error Correction (ICE) was used to identify consensus isoforms (high-quality transcripts, accuracy > 99%). The redundancy in high quality FL transcripts was removed using CD-HIT (identity > 0.99). The completeness of the transcriptome after de-redundancy was assessed by benchmarking universal single-copy orthologs BUSCO46 (v3.0.2) software which is based on OrthoDB database.
Functional annotation of full-length transcriptome
Sequences of non-redundant transcripts were annotated by DIAMOND software according to several databases, including NR47, Swissprot48, GO49, COG50, KOG51, Pfam52, KEGG53. KEGG Orthology of transcripts were obtained by the KEGG Automatic Annotation Server, while GO Orthology of transcripts were conducted by InterProScan, a core component of the InterPro integrated database. Protein domain annotation information was obtained by HMMER software to blast the amino acid sequences of the transcripts against Pfam database.
Analysis of alternative splice events
The transcripts after de-redundancy were used to predict alternative splicing candidate events. The alternative splice events were predicted by BLAST54 to compare every two sequences. BLAST alignments that satisfied the all of the following conditions were considered to be a candidate variable AS events: both sequences are longer than 1000 bp and there are contain 2 HSPs (high-scoring segment pairs) in the alignment; the gap of AS is over 100 bp and at least 100 bp away from the 3′/5′ end; the 5 bp overlap is permitted between transcripts. Using these stringent criteria, a total of 514 AS events were predicted. However, due to the absence of a reference genome for P. shikokuense, the types of AS events were unable to categorize.
Structure analysis of simple sequence repeats (SSRs)
The microsatellite identification tool (MISA v1.0) was used to identify simple sequence repeats (SSRs) within the non-redundancy transcriptome. The transcripts above 500 bp were screened and analyzed by MISA software.
Analysis of ORF and TF prediction
Potential coding sequence (CDS) and corresponding amino acid sequences in novel transcripts were predicted by Transdecode software (v5.0.0) based on length of ORF (Open Reading Frame), log-likelihood score, alignment of amino acid sequence against Pfam protein domain database. The iTAK software, which utilizes PlnTFDB and PlantTFBD as reference databases, was used for TF prediction.
Long non-coding RNA (LncRNAs) prediction
LncRNAs, as their name suggests, are long transcripts without coding capacity. Transcripts with coding potential were filtered using a minimum length and exon number thresholds. In addition, screening for coding potential was performed using four widely recognized methods: CPC analysis55, CNCI analysis, CPAT analysis56 and Pfam protein domain analysis. The noncoding transcripts obtained by the above four analyses were taken from their intersecting parts and considered as LncRNA. A step-by-step screening approach was used. At first, the intersection of CPAT and CPC predictions was taken. Then CNCI predictions were made based on the results of the CPAT and CPC intersection. Finally, Pfam predictions were made using the results of the CNCI predictions.
Data Record
The raw sequence data reported in this paper have been deposited in the Genome Sequence Archive57 in National Genomics Data Center58, China National Center for Bioinformation/Beijing Institute of Genomics, Chinese Academy of Sciences (GSA: CRA014836)59 that is publicly accessible at https://ngdc.cncb.ac.cn/gsa.
Technical Validation
Quality control of RNA exaction
High-quality RNA is essential for successful full-length transcriptome sequencing. To ensure the accuracy of the sequencing data, the purity, concentration and integrity of the RNA samples were examined using Nanodrop and Agilent Bioanalyzer 2100 system, respectively.
Date analysis of full-length sequencing
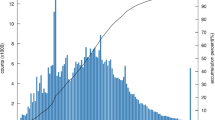
By full-length transcriptome sequencing, 41.73 Gb of SMRT sequencing data were obtained. A total of 573,159 circular consensus (CCS) reads were produced, in which 451,077 were identified as FLNC reads. FLNC reads were clustered into 154,490 consensus sequences by IsoSeq analysis application in SMRTLink software. A total of 154,441 high quality sequences were obtained by polishing consensus sequences. After removing redundant reads, 105,249 non-redundant FLNC reads were obtained (Table 1). Multiple steps of data polishing were performed during the data analysis process to improve the quality of data outputs. One of them was that the CCS sequences were derived from the original sequences according to the criteria full passes ≥3 and sequence accuracy >0.9, and then were polished by ccs software. The number of CCS sequences, the number of bases and the average length of the sequences in each library were counted to evaluate the downstream data (Table 1). The length of full-length sequences is indicative of cDNA length in library construction, and sequence length is a key metric in estimating quality of library construction (Fig. 2a). Besides, FLNC reads were clustered and removed the redundant (de-redundant) to obtain non-redundancy HQ transcripts (high-quality transcripts, accuracy >99%. The Benchmarking Universal Single-Copy Orthologs (BUSCO, v3.0.2), based on OrthoDB database was applied to assess the completeness of the transcriptome after de-redundancy. By comparing the transcriptome with closely related species, the completeness and accuracy can be quantified. The BUSCO analysis results showed that among the 303 conserved eukaryotic orthologous genes, 65.3% (198 genes) of the genes were found in the P. shikokuense transcriptome of which 54.1% (164 genes) were complete genes while 11.2% (34 genes) were fragments BUSCOs (Fig. 2b).

Data analysis of full-length sequencing. (a) Full-length non-chemical read length distribution. (b) The assessment results of transcriptome completeness.
Annotation quality of transcripts
In this study, multiple reference databases were used for functional annotation. Of the sequences analyzed, 50,338 (47.82%) were annotated against the databases including NR, Swissprot, GO, COG, KOG, Pfam, and KEGG. Among them, 43,295 (86%) sequences were annotated by NR database, making it the most source of annotation. This was followed by the GO database (37,256, 74%) and the Pfam database (33,920, 67.38%) (Table 2). The NR annotation showed that most sequences were aligned to those of Symbiodinium microadriaticum (28,544, 65.94%), and 9,041 (20.89%) sequences were still not annotated (Fig. 3). With regard to functional annotations, a total of 37,256 sequences in GO database were annotated to three classes of GO terms including “biological process”, “cellular component” and “molecular function” (Fig. 4). In GOG, eggNOG, KOG database (Fig. 5), transcripts were annotated to 26 function classes, respectively.

Homologous species distribution by NR annotation.

GO classification of the P. shikokuense full-length transcripts. The x-axis represents GO categories, the left y-axis represents the percentage of transcripts number, and the right y-axis represents transcripts number. The figure shows transcript GO classification on second level.

KOG function classification of P. shikokuense full-length transcripts. The x-axis represents different KOG categories (represented by the legend on the right), and y-axis represents the number of the transcripts.
A stepwise screening method was used to predict LncRNAs, and 11,917 candidates LncRNAs were finally predicted by Pfam database. (Fig. 6a). Besides, a total of 78,265 ORFs were identified, of them 15,501 were the protein coding ones. Length distribution of protein coded by predicted CDS is shown in the Fig. 6b. In addition, a total of 437 putative transcription factor (TF) members from 17 TF families were predicted, with the largest number being the C3H family, followed by the CSD family (Fig. 6c).

The analysis of LncRNAs, CDS, TF and SSRs. (a) The long non-coding RNA prediction by cnci, cpc, cpat and pfam datebase. (b) Length distribution of protein coded by predicted CDS. (c) The transcription factors prediction. (d) The structure of simple sequence repeats. The enlarged image in the red box is shown by the arrow.
Structural analysis of full-length transcripts
A total of 34,723 transcripts were identified to contain more than one SSR marker, of which most SSR type was tri-nucleotide (three bases) 54.49%, followed by compound SSR (hybrid microsatellite, distance of two SSRs less than 100 bp) 24.52%, di-nucleotide (two bases) 9.45%, mono-nucleotide (single base) 8.90%, and the least was penta-nucleotide (five nucleotides) 0.1%. The density distribution of different SSR types was summarized in Fig. 6d.
Code availability
(1) SMRTLink: version10.1, default parameters.
(2) ccs: version 6.2.0, main parameters: --min-rq 0.9 --min-passes 3 -j 6 --min-length 200.
(3) Lima: version 2.1.0, main parameters: --isoseq --num-threads 6.
(4) Isoseq3: version 3.4.0, main parameters: refine --require-polya, cluster --num-threads 6 --verbose --use-qvs.
(5) CD-HIT: version 4.6.1, main parameters: -c 0.99 -M 0 (cd-hit-est).
(6) BUSCO: version 3.0.2, main parameters: -m tran -c 4 -f.
(7) BLAST: version 2.2.31, main parameters: -outfmt 5 (Alternative splicing).
(8) IsoSeq_AS_de_novo: version 1.0, default parameters.
(9) MISA: version 1.0, default parameters.
(10) TransDecoder: version5.0.0, main parameters: -m 50 -G universal -S.
(11) CPAT: version 1.2.2, main parameters: -cutoff 0.38.
(12) CPC2: version 0.1, default parameters.
(13) CNCI: version 2, default parameters.
(14) PfamScan: version 1.60, main parameters: -translate orf.
(15) iTAK: version 1.7a, default parameters.
(16) diamond: version 2.0.15, -k 100 -e-evalue 1e-5 -f 5.
(17) InterProScan: version 5.34–73.0, main parameters: -appl Pfam -goterms -iprlookup -pa -f xml -dp -t p.
(18) Hmmscan: version 3.3.2, main parameters: --noali --cut_nc --acc -notextw.
References
Bujak, J. P. & Williams, G. L. The evolution of dinoflagellates. Can. J. Bot. 59, 2077–2087 (1981).
Gómez, F. A checklist and classification of living Dinoflagellates (Dinoflagellata, Alveolata). CICIMAR Oce. 27, 65–140 (2012).
Rizzo, P. J. & Nooden, L. D. Chromosomal proteins in the dinoflagellate alga Gyrodinium cohnii. Science 176, 796–797 (1972).
Haapala, O. & Soyer, M. O. Structure of dinoflagellate chromosomes. Nat. New Biol. 244, 195–197 (1973).
Bachvaroff, T. R. & Place, A. R. From stop to start: tandem gene arrangement, copy number and trans-splicing sites in the dinoflagellate Amphidinium carterae. PLoS. One 3, e2929 (2008).
Beauchemin, M. et al. Dinoflagellate tandem array gene transcripts are highly conserved and not polycistronic. Proc. Natl. Acad. Sci. USA 109, 15793–15798 (2012).
Erdner, D. L. & Anderson, D. M. Global transcriptional profiling of the toxic dinoflagellate Alexandrium fundyense using massively parallel signature sequencing. BMC Genomics 7, 88 (2006).
Waller, R. F. & Jackson, C. J. Dinoflagellate mitochondrial genomes: stretching the rules of molecular biology. Bioessays 31, 237–245 (2009).
Hallegraeff, G. M. et al. Perceived global increase in algal blooms is attributable to intensified monitoring and emerging bloom impacts. Commun. Earth & Environ. 2, 117 (2021).
Takano, Y. & Matsuoka, K. A comparative study between Prorocentrum shikokuense and P. donghaiense (Prorocentrales, Dinophyceae) based on morphology and DNA sequences. Plankton Benthos Res. 6, 179–186 (2011).
Lu, D. D. & Goebel, J. Five red tide species in genus Prorocentrum including the description of Prorocentrum donghaiense Lu sp. nov. from the East China Sea. Chin. J. Oceanol. Limn. 19, 337–344 (2001).
Lu, D. D. et al. Morphological and genetic study of Prorocentrum donghaiense Lu from the East China Sea, and comparison with some related Prorocentrum species. Harmful Algae 4, 493–505 (2005).
Gu, H. F. et al. Emerging harmful algal bloom species over the last four decades in China. Harmful Algae 111, 102059 (2022).
Shin, H. H. et al. Harmful dinoflagellate Prorocentrum donghaiense Lu is widely distributed along the East China Sea and Korean coastal area. Ocean Sci. J. 54, 685–691 (2019).
Madhu, N. V. et al. Phytoplankton characterisation in the Alappuzha mud banks during the pre-/post phases of a red-tide, Prorocentrum shikokuense Hada. Reg. Stud. Mar. Sci. 40, 101486 (2020).
Marampouti, C., Buma, A. G. J. & de Boer, M. K. Mediterranean alien harmful algal blooms: origins and impacts. Environ. Sci. Pollut. Res. 28, 3837–3851 (2021).
Su, M. & Koike, K. A red tide off the Myanmar coast: morphological and genetic identification of the dinoflagellate composition. Harmful Algae 27, 149–158 (2013).
Li, H. M., Tang, H. J., Shi, X. Y., Zhang, C. S. & Wang, X. L. Increased nutrient loads from the Changjiang (Yangtze) river have led to increased harmful algal blooms. Harmful Algae 39, 92–101 (2014).
Huang, X. Z., Huang, B. Q., Chen, J. X. & Liu, X. Cellular responses of the dinoflagellate Prorocentrum donghaiense Lu to phosphate limitation and chronological ageing. J. Plankton Res. 38, 83–93 (2016).
Zhou, Z. X., Yu, R. C. & Zhou, M. J. Seasonal succession of microalgal blooms from diatoms to dinoflagellates in the East China Sea: a numerical simulation study. Ecol. Model. 360, 150–162 (2017).
Zhou, Z. X., Yu, R. C. & Zhou, M. J. Resolving the complex relationship between harmful algal blooms and environmental factors in the coastal waters adjacent to the Changjiang River estuary. Harmful Algae 62, 60–72 (2017).
Li, Y., Lu, S. H., Jiang, T. J., Xiao, Y. P. & You, S. P. Environmental factors and seasonal dynamics of Prorocentrum populations in Nanji Islands National Nature Reserve, East China Sea. Harmful Algae 10, 426–432 (2011).
Hadjadji, I., Frehi, H., Ayada, L., Abadie, E. & Collos, Y. A comparative analysis of Alexandrium catenella/tamarense blooms in Annaba Bay (Algeria) and Thau lagoon (France); phosphorus limitation as a trigger. C.R. Biol. 337, 117–122 (2014).
Lin, X., Zhang, H., Huang, B. & Lin, S. Alkaline phosphatase gene sequence characteristics and transcriptional regulation by phosphate limitation in Karenia brevis (Dinophyceae). Harmful Algae 17, 14–24 (2012).
Paytan, A. & McLaughlin, K. The oceanic phosphorus cycle. Chem. Rev. 107, 563–576 (2007).
Takeda, E. et al. A novel function of phosphate-mediated intracellular signal transduction pathways. Adv. Enzyme. Regul. 46, 154–161 (2006).
Lin, S. J., Litaker, R. W. & Sunda, W. G. Phosphorus physiological ecology and molecular mechanisms in marine phytoplankton. J. Phycol. 52, 10–36 (2016).
Zhang, S. F., Yuan, C. J., Chen, Y., Lin, L. & Wang, D. Z. Transcriptomic response to changing ambient phosphorus in the marine dinoflagellate Prorocentrum donghaiense. Sci. Total Environ. 692, 1037–1047 (2019).
Yu, L. et al. Comparative metatranscriptomic profiling and microRNA sequencing to reveal active metabolic pathways associated with a dinoflagellate bloom. Sci. Total Environ. 699, 134323 (2020).
Shi, X. et al. Transcriptomic and microRNAomic profiling reveals multi-faceted mechanisms to cope with phosphate stress in a dinoflagellate. ISME J. 11, 2209–2218 (2017).
Li, M. Z., Li, L., Shi, X. G., Lin, L. X. & Lin, S. J. Effects of phosphorus deficiency and adenosine 5′-triphosphate (ATP) on growth and cell cycle of the dinoflagellate Prorocentrum donghaiense. Harmful Algae 47, 35–41 (2015).
Zhang, C., Chen, G., Wang, Y., Guo, C. & Zhou, J. Physiological and molecular responses of Prorocentrum donghaiense to dissolved inorganic phosphorus limitation. Mar. Pollut. Bull. 129, 562–572 (2018).
Hu, Z. X., Liu, Y. Y., Deng, Y. Y. & Tang, Y. Z. The notorious harmful algal blooms-forming dinoflagellate Prorocentrum donghaiense produces sexual resting cysts, which widely distribute along the coastal marine sediment of China. Front. Mar. Sci. 9, 826736 (2022).
Gaonkar, C. C. & Campbell, L. De novo transcriptome assembly and gene annotation for the toxic dinoflagellate Dinophysis. Sci. Data 10, 345 (2023).
Dougan, K. E. et al. Multi-omics analysis reveals the molecular response to heat stress in a “red tide” dinoflagellate. Genome Biol. 24, 265 (2023).
Lin, S. J. et al. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science 350, 691–694 (2015).
Liu, H. et al. Symbiodinium genomes reveal adaptive evolution of functions related to coral-dinoflagellate symbiosis. Commun. Biol. 1, 95 (2018).
Aranda, M. et al. Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci. Rep. 6, 39734 (2016).
Shoguchi, E. et al. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr. Biol. 23, 1399–1408 (2013).
Shoguchi, E. et al. Two divergent Symbiodinium genomes reveal conservation of a gene cluster for sunscreen biosynthesis and recently lost genes. BMC Genomics. 19, 458 (2018).
John, U. et al. An aerobic eukaryotic parasite with functional mitochondria that likely lacks a mitochondrial genome. Sci. Adv. 5, eaav1110 (2019).
Wang, L. et al. Comprehensive analysis of full-length transcriptomes of Schizothorax prenanti by single-molecule long-read sequencing. Genomics 114, 456–464 (2022).
Abdel-Ghany, S. et al. A survey of the sorghum transcriptome using single molecule long reads. Nat. Commun. 7, 11706 (2016).
Rhoads, A. & Au, K. F. PacBio sequencing and its applications. Genom. Proteom. Bioinf. 13, 278–289 (2015).
Ardui, S., Ameur, A., Vermeesch, J. R. & Hestand, M. S. Single molecule real-time (SMRT) sequencing comes of age: applications and utilities for medical diagnostics. Nucleic. Acids. Res. 46, 2159–2168 (2018).
Simão, F. A. et al. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Deng, Y. Y. et al. Integrated NR database in protein annotation system and its localization. Comput. Eng. 32, 71–74 (2006).
Apweiler, R. et al. UniProt: the Universal Protein knowledgebase. Nucleic Acids Res. 32, 115–119 (2004).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29 (2000).
Tatusov, R. L., Galperin, M. Y. & Natale, D. A. The COG database: a tool for genome scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36 (2000).
Koonin, E. V. et al. A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 5, R7 (2004).
Finn, R. D. et al. Pfam: the protein families database. Nucleic Acids Res. 42, D222–D230 (2014).
Kanehisa, M. et al. The KEGG resource for deciphering the genome. Nucleic Acids Res. 32, D277–D280 (2004).
Altschul, S. F. et al. Gapped BLAST and PSI BLAST: A new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 (1997).
Kong, L. et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 35, W345–W349 (2007).
Wang, L. G. et al. CPAT: Coding-Potential Assessment Tool using an alignment-free logistic regression model. Nucleic Acids Res. 41, e74 (2013).
Chen, T. et al. The Genome Sequence Archive Family: Toward Explosive Data Growth and Diverse Data Types. Genom. Proteom. Bioinf. 19, 578–583 (2021).
CNCB-NGDC Members and Partners. Database Resources of the National Genomics Data Center, China National Center for Bioinformation in 2022. Nucleic Acids Res. 50, D27–D38 (2022).
Genome Sequence Archive https://ngdc.cncb.ac.cn/gsa/browse/CRA014836 (2024).
Acknowledgements
This work was financially supported by the Science & Technology Innovation Project of Laoshan Laboratory (LSKJ202203205); the Fundamental Research Funds for the Central Universities (202262001); the Key R & D Program of Shandong Province, China (2022LZGC004); National Natural Science Foundation of China (32102757); and National Key R & D Program of China (2022YFF1102300).
Author information
Authors and Affiliations
Contributions
Xiaohui Pan carried out the study and drafted the manuscript; Hang Liu, Leili Feng, Yanan Zong, and Zihao Cao participated the data processing; Li Guo and Guanpin Yang directed the project, discussed and revised the manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Pan, X., Liu, H., Feng, L. et al. Full-length transcriptome analysis of a bloom-forming dinoflagellate Prorocentrum shikokuense (Dinophyceae). Sci Data 11, 430 (2024). https://doi.org/10.1038/s41597-024-03269-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-03269-1
- Springer Nature Limited