
Abstract
Background
The marine dinoflagellate, Symbiodinium, is a well-known photosynthetic partner for coral and other diverse, non-photosynthetic hosts in subtropical and tropical shallows, where it comprises an essential component of marine ecosystems. Using molecular phylogenetics, the genus Symbiodinium has been classified into nine major clades, A-I, and one of the reported differences among phenotypes is their capacity to synthesize mycosporine-like amino acids (MAAs), which absorb UV radiation. However, the genetic basis for this difference in synthetic capacity is unknown. To understand genetics underlying Symbiodinium diversity, we report two draft genomes, one from clade A, presumed to have been the earliest branching clade, and the other from clade C, in the terminal branch.
Results
The nuclear genome of Symbiodinium clade A (SymA) has more gene families than that of clade C, with larger numbers of organelle-related genes, including mitochondrial transcription terminal factor (mTERF) and Rubisco. While clade C (SymC) has fewer gene families, it displays specific expansions of repeat domain-containing genes, such as leucine-rich repeats (LRRs) and retrovirus-related dUTPases. Interestingly, the SymA genome encodes a gene cluster for MAA biosynthesis, potentially transferred from an endosymbiotic red alga (probably of bacterial origin), while SymC has completely lost these genes.
Conclusions
Our analysis demonstrates that SymC appears to have evolved by losing gene families, such as the MAA biosynthesis gene cluster. In contrast to the conservation of genes related to photosynthetic ability, the terminal clade has suffered more gene family losses than other clades, suggesting a possible adaptation to symbiosis. Overall, this study implies that Symbiodinium ecology drives acquisition and loss of gene families.
Similar content being viewed by others


Background
Dinoflagellates are one of the major groups in the supergroup Alveolata, with an estimated ~ 2500 species [1]. They inhabit aquatic environments and nearly half are phototrophic [1]. Dinoflagellate evolution has been resolved using morphological and molecular phylogenetic analyses [2, 3]. Comparative analyses suggest that horizontal gene transfers are linked to major transitions in dinoflagellate evolution [4, 5].
Recent studies of dinoflagellates focused on their life styles in relation to marine environments [1, 6]. Dinoflagellates are the major group of red tide-producing microorganisms [7], specialized for toxin biosynthesis [8]. However, dinoflagellates of the genus Symbiodinium are renowned for their symbiotic relationships with reef-building corals [9, 10], which are foundational to marine ecosystem biodiversity [11,12,13].
The extensive diversification of Symbiodinium has been well described [11,12,13,14,15,16]. Molecular phylogenetics has classified these dinoflagellates into nine major groups, A to I [17]. Symbiodinium strains are hosted by ciliates, foraminiferans, sponges, cnidarians, molluscs, and acoelomorphs [12, 18]. It is thought that clade A diverged first (the oldest) and that lineages C and H in the crown clade are the most recent (the youngest) (Fig. 1a). Clade A Symbiodinium may also form parasitic as well as mutualistic symbioses with other organisms [19,20,21]. The diversity and dominance of clade C in association with reef invertebrates has been reported in the Great Barrier Reef (GBR), Australia, and at Zamami Island, Okinawa, Japan [22].

Phenotypic differences in the production of mycosporine-like amino acids of two divergent Symbiodinium species. a. Phylogenetic positions of the Symbiodinium species analyzed, SymA (green) and SymC (red). A phylogenetic tree was constructed using the Maximum-Likelihood method, based on 28S rDNA sequences [12]. The scale bar shows 0.2 changes per site. b. Zoospore of SymA. Scale bar, 5 μm. c. Zoospore of SymC. A short, transverse flagellum originating from the cingulum and a long longitudinal flagellum originating from the sulcus, are observed in zoospores (arrows). d. High-performance liquid chromatography (HPLC) comparison of aqueous extracts prepared from SymA (black) and SymC (pink) detected at 330 nm. The largest difference between SymA and SymC is seen in peaks with a retention time of ~ 4.5 min (arrow). The large peak in SymA is not detected in SymC. e. High-resolution mass spectrum of isolated Porphyra-334 (MH+ 347.1456, C14H23N2O8, Δ 0.74 mmu), showing the production of mycosporine-like amino acid (MAAs) by SymA (arrow). Inset shows the chemical formula of Porphyra-334
Physiological work on Symbiodinium diversity has been reported using cultured Symbiodinium strains [23,24,25] and recent work clarifies differences in metabolite production among Symbiodinium clades [26]. One of the pioneering studies reported differences in mycosporine-like amino acid (MAA) production [23], but the genetic basis of this chemistry remains unknown in Symbiodinium. MAAs function as anti-oxidants and UV-absorbing molecules [27]. In a cultured dinoflagellate, Gymnodinium sanguineum, MAAs function as specific UV blockers to protect the dinoflagellate photosynthetic machinery [28]. Recently, four biosynthetic enzymes in a cyanobacterium were characterized using heterologous gene expression and an MAA biosynthetic gene cluster encoding those enzymes was characterized [29]. Further reports suggested that three enzymes involved in MAA biosynthesis, dimethyl 4-deoxygadusol (DDG), O-methyltransferase (O-MT), and ATP-grasp, are conserved in bacteria [30, 31] and the enzyme for the fourth biosynthetic step from the cluster is either a non-ribosomal peptide synthetase (NRPS) homolog or D-alanine (D-Ala) D-Ala ligase-like [30, 32]. Although these genes have also been found in eukaryotic genomes [33,34,35,36,37], the gene cluster has been identified only in bacterial [38] and red algal genomes [39]. One hypothesis is that the capacity for MAA production in Symbiodinium may be essential for symbioses involving hosts that cannot produce MAAs [27, 40].
Draft genomes have been published for three Symbiodinium taxa [4, 41, 42]. The most recent report [42] focused on diversification of transmembrane transporter genes. Comparative analysis also described the importance of duplicated genes as an evolutionary mechanism, underscoring the importance of lineage-specific expansions for symbiotic lifestyles, especially for genes encoding ion transporters. Comparative transcriptomic analyses have identified possible lineage- or clade-specific gene families [43]. While genome evolution of parasitic apicomplexans has been extensively studied, genomes of symbiotic dinoflagellates are still comparatively little known. Therefore, comparative genomic studies of diverse Symbiodinium species are essential to better understand Symbiodinium diversity. To clarify the genetic basis for different physiological phenotypes, we decoded the genomes of culturable clade A and clade C Symbiodinium and performed comparative analyses.
Results
Genome assembly and physiological characters in divergent Symbiodinium taxa
To obtain Symbiodinium genome sequences from early- and late-branching lineages, two strains (clade A, Y106 and clade C, Y103) were selected for genome sequencing (Fig. 1b and c). A molecular phylogenetic tree (Fig. 1a) from the recent analysis by Pochon et al. [12] and ITS sequences showed that Y106 belongs to clade A3 (SymA). In contrast, the subclade of Y103 was not clear from our phylogenetic analysis (SymC), although its ITS suggested that it is similar to clade C92 [44].
Physiological characterization by mass spectrometry confirmed that SymA3 produces an MAA, Porphyra-334 (Fig. 1d and e), which has an m/z of 347.1456 Da. On the other hand, MAAs were not detected in SymC (Fig. 1d) or in S. minutum (shown here as SymB) of clade B1 (data not shown). These results are comparable to previously reported differences at the clade level [23].
Using genomic DNA and mRNA from cloned cells, we obtained sequence data using Illumina GAIIx and Hiseq sequencers (Additional file 1: Tables S1 and S2). Approximately ~ 200 Gbp of genomic sequences were used for each assembly (Additional file 1: Table S1). In our previous sequencing of the S. minutum (SymB) genome, sequence data amounted to ~ 56 Gbp from Roche 454 and Illumina GAIIx platforms [4]. Thus, the amount of raw data in this study was much greater than that of the previous study. The assembly was performed as described previously, with several modifications [4]. We produced two assembled genomes suitable for gene analyses (Additional file 1: Table S3), as in our previous report [4]. Scaffolds totaled ~ 767 Mb and ~ 705 Mb for SymA and SymC, respectively (Additional file 1: Table S3). RNAseq data were derived from Symbiodinium cultured under standard conditions, as described previously [4], or under dark conditions (Additional file 1: Table S2). Data were assembled into 76,628 unique cDNAs in SymA and 68,876 in SymC (Additional file 1: Table S4). Gene predictions yielded 69,018 and 65,832 protein-coding models, respectively (Additional file 1: Table S3). The following genome browser provides access to the assembled data and predicted genes: http://marinegenomics.oist.jp/gallery/ [45]. Using TopHat with default parameters [46], RNAseq reads were mapped by library (Additional file 1: Table S2) onto the draft genomes and information for read counts is available on the browser (Additional file 2: Figure S1). For SymA, 67.5% of the gene models were supported by RNAseq data and 62.5% for SymC. A characteristic feature of gene structures in SymC was a higher frequency of genes lacking introns (~ 19.7%) (Additional file 1: Table S3). The GC contents of the assembled SymA and SymC genomes were 50 and 43%, respectively (Additional file 1: Table S3). Unidirectional arrangements of genes and three major types (GT/GC/GA) of the first two nucleotides of introns [4] were found in the genomes of SymA and SymC (Additional file 1: Table S3).
Gene content of each Symbiodinium genome
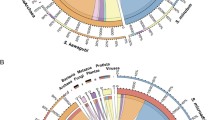
Both genomes were predicted to contain more than 65,000 genes (69,018 for SymA and 65,832 for SymC) (Additional file 1: Table S3). These numbers are larger than those of previously reported Symbiodinium genomes (41,925 for S. minutum, 36,850 for S. kawagutii, and 49,109 for S. microadriaticum) [4, 41, 42], although they fall within the estimated range of 38,000–87,000 [47]. To clarify which gene families are conserved or expanded in each lineage, we annotated predicted proteins using a pfam domain search (http://marinegenomics.oist.jp/gallery/) and compared the proteins with genes of S. minutum. We found 4435 domain classes for 26,261 SymA genes, 4169 for 21,107 SymB genes, and 4122 for 23,808 from SymC (Fig. 2a).

Comparisons and expansions of gene families in Symbiodinium lineages. a. Venn diagram comparing Pfam domains among three divergent Symbiodinium genomes. Total numbers of domain types found in each genome are shown outside the circle. Percentages indicate the ratio of the inside number to the total number. b-d. Molecular phylogenies of dUTPase (b), mitochondrial transcription termination (mTERF) proteins (c), and LRR domain-containing proteins (d), respectively. Scale bars show 0.5 changes per site. The arcs show possible lineage specific expansions in SymC (red) and SymA (green), respectively. A, SymA. B, symbB1. C, SymC. Adi, Acropora digitifera. Ath, Arabidopsis thalina. Cci, Cephus cinctus. Des, Desulfatitalea sp. Epa, Exaiptasia pallida. Hhe, Human herpesvirus. Hsa, Homo sapiens. Lma, Leishmania major. Mhe, Macacine herpesvirus. Nve, Nematostella vectensis. Pma, Perkinsus marinus. Ral, Rhizobium alamii. Tbr, Trypanosoma brucei. Vbr, Vitrella brassicaformis
Next, we compared gene numbers within gene families in each genome. Lineage-specific gene family expansions were defined as Pfam domain groups with multiple copies in Symbiodinium, in which gene numbers were significantly greater in one genome compared to the other two. The 30 most expanded gene families are summarized for SymA (Additional file 1: Table S5) and SymC (Additional file 1: Table S6), respectively. Our analyses indicate that the majority of the top 30 Pfam domains in SymA correspond to those reported previously [42]. These include reverse transcriptase (RVT), regulator of chromosome condensation (RCC1), and endonuclease (Additional file 1: Table S5). In the SymC genome, gene families for RVT, DNA methylase, integrase, and zf-CCHC are expanded. Thus, comparisons of gene numbers with Pfam domains showed many copies of reverse transcriptase in SymA and SymC. Special expansions in the genome of the late-branching group were predicted in gene families with DNA methylase or zf-CCHC domains. Similar observations have been reported in the Symbiodinium kawagutii genome [41]. It is possible that DNA methylation is related to endogenous retroviral expression [48]. zf-CCHC domains have been found in retrovirus GAG proteins [49]. These larger gene numbers in SymA and SymC (Additional file 1: Table S3) seem to be partly related to the richness of enzyme genes in many retroviruses.
To confirm the relationship between lineage-specific expansions and potential retrogenes, we constructed a molecular phylogenetic tree of UTPase proteins from Symbiodinium genomes (Fig. 2b). dUTPases prevent the misincorporation of uracil into DNA, and these enzymes have recently been suggested to regulate host interactions [50]. The two Symbiodinium genomes (SymA and SymB) encode one or two eukaryotic dUTPases per genome (Fig. 2b). On the other hand, the dUTPases in SymC are expanded and some of them had RVT domains. In addition, many of them are intronless, suggesting that gene expansion in SymC is due to integration of processed cDNAs [51].
Genes of endosymbiotic origin are conserved in the early-branching genome
When proteins containing transposable elements or retrovirus-related Pfam domains were removed from the calculation, it became apparent that organelle-related genes (mitochondrial or plastid proteins) have been expanded in SymA (Additional file 1: Table S5). In particular, the gene expansion for mitochondrial transcription termination factor family protein (mTERF) was found in the early-diverging genome (Fig. 2c). mTERF genes have been identified as putative organellar transcription factors [52, 53]. Land plants have the highest number of mTERF genes (~ 30 members), which are targeted to plastids and mitochondria [54]. The mammalian mTERF family (four genes) is important in mitochondrial gene expression. In addition, gene numbers for Peridinin-chlorophyll A binding protein (PCP), chlorophyll A-B binding proteins, and Rubiscos were more numerous than those of other Symbiodinium. Expansion of chlorophyll a-binding proteins has also been reported in Symbiodinium minutum [55]. Several genes for Rubisco are tandemly aligned in the SymA genome, consistent with a previous report in the dinoflagellate, Prorocentrum minimum [56]. Differences in plastid physiological responses to heat stress were analyzed in SymA and SymB [57] and may be due to the expanded plastid-related proteins. In a future study, the relationship between stress and expansion of organelle-related genes will be determined, although gene functions in organelle genomes might also be important to understand differences in sensitivity to heat and light stress [58, 59].
Expansions of repeat domain-containing genes in the late-branching genome
There were fewer gene families in the SymC genome than in SymA or SymB. On the other hand, genes for repeated domains are expanded, including leucine-rich repeats (LRR), FNIP (initial “FNIP” amino acids) repeats, and tetratrico peptide repeats (TPR) (Additional file 1: Table S6). Those domains are involved in protein-protein interactions [60, 61]. Therefore, these expansions may be similar to those of apicomplexans [62]. To characterize expanded LRR-containing proteins, we performed molecular phylogenetic analyses. Most of the expansion in SymC pertained to one subfamily similar to FNIP repeats, which has also been expanded in the Dictyostelium discoideum genome [63]. Other proteins with expanded LRRs were similar to those of sds22 and PfLRR1, which relate to cell cycle regulation [64]. Molecular phylogeny showed that lineage-specific expansions occurred in both SymB and SymC (Fig. 2d), suggesting that numbers of genes for protein-protein interactions are expanded in the late-branching genome.
Gene cluster for mycosporine-like amino acid (MAA) biosynthesis in Symbiodinium
To determine the genetic basis for the difference in MAA production (Fig. 1d and e), we surveyed the decoded Symbiodinium genomes. MAA biosynthetic genes have not been found in the genome of Symbiodinium kawagutii [41], but they have been identified in the host genomes of cnidarian anthozoans [35, 36]. We found genes for MAA biosynthesis [29, 30] in the SymA genome. In addition, preliminary RNAseq analysis indicated that expression levels of those genes were similar between light and dark conditions (Additional file 2: Figure S1). Unexpectedly, the gene cluster corresponds to that of bacteria, although the gene arrangement of D-Ala D-Ala ligase differs from the bacterial arrangement (Fig. 3). We constructed four phylogenetic trees incorporating the genes in this cluster (Fig. 3a, Additional file 2: Figures S2-S4). Dinoflagellate DDG synthases clustered with those of anthozoans (Fig. 3a), while O-methyltransferases and D-Ala D-Ala ligases are shared with those of bacteria (Additional file 2: Figures S2 and S4). The phylogenetic relationship of ATP-grasp is unclear (Additional file 2: Figures S3). This complicated result suggests that those genes have been lost in eukaryotes or have been transferred several times. It has been suggested that the fusion gene (3-dehydroquinate synthase+O-methyltransferase) came from cyanobacteria [33] or via secondary endosymbiosis [65]. Our analysis implies that these genes were likely acquired in gene transfers via secondary endosymbiosis (Fig. 3a).

The nuclear genome of the earliest-branching Symbiodinium (SymA) harbors a gene cluster to produce MAAs. a. Molecular phylogenetic analysis of dimethyl 4-deoxygadusol (DDG) synthase by maximum likelihood. Numbers denote bootstrap values (WAG+G model and RAxML) and posterior probabilities (MrBayes). nd: “not determined” by a different topology. DDG genes of Symbiodinium are clustered with other dinoflagellate proteins from Karlodinium veneficum and Heterocapsa triquetra. In addition, dinoflagellate proteins are phylogenetically close to those of anthozoans. b. Potential evolutionary relationships between MAA gene clusters in cyanobacteria and those of eukaryotes with DDG synthase are shown. Deep evolutionary conservation of the bacterial-like gene cluster is suggested in SymA. On the other hand, genes within the cluster have been lost in the crown lineage (red box). The topology of the tree is based on phylogenetic analysis of DDG synthase
Discussion
Our comparative analysis identified genomic characters of SymA and SymC, both of which were originally isolated from bivalve molluscs. The higher GC content of the SymA genome was similar to that reported in S. microadriaticum [42], suggesting that this may be an attribute of the earliest-branching lineage. Comparisons of gene families suggest that the late-branching lineage has lost more gene families than early-branching lineages, or that the early-branching lineages have acquired more gene families than the late-branching lineage. In other words, in SymC, there are fewer gene families, even though total gene numbers are expanded in late-branching Symbiodinium.
Finally, we found that the genome of SymA in the early-branching clade encoded a gene cluster for MAA biosynthesis. As this gene cluster is conserved, the transfer of large DNA segments probably occurred at an early stage of endosymbiosis. However, we cannot exclude the possibility that the cluster formed in the Symbiodinium lineage. Our survey shows that the three genes for MAA biosynthesis are found in S. microadriaticum and their genomic locations are dispersed on three scaffolds, 22, 397 and 882 [66] (http://smic.reefgenomics.org). Differences between the two genomes of clade A Symbiodinium also support reports of diversity in this early diverging lineage [20, 21]. Although it is suggested that adaptation to shallow-water environments may have been maintained in clade A Symbiodinium [67], previous reports for 54 species of symbiotic cnidarians have shown that highly variable MAA concentrations are not depth-dependent [68]. On the other hand, genes in this cluster were not found in the SymC genome. Although we surveyed raw data from publicly available Symbiodinium genomes by BLAST search, orthologs of the fusion gene (3-dehydroquinate synthase+O-methyltransferase) were not found in SymB, S. kawagutii or SymC (data not shown). Since SymB and S. kawagutii (clade F) genomes also lack these genes [4, 41], it is likely that gene losses occurred in the common ancestor of the crown lineages.
Loss of the capacity to produce UV-absorbing molecules may have been compensated by expansion of other genes for UV stress. We surveyed possible enzymes for repairing UV-damaged DNA [69] because cryptochromes/photolyases in dinoflagellates have not been surveyed in detail. Molecular phylogenetic analyses revealed no large differences in such gene families. Genomes of Symbiodinium encode three groups of cryptochromes/photolyases [69] (Additional file 2: Figure S5). Therefore, it is difficult to conclude that there is any relationship between acquisitions of repair genes and losses of MAA biosynthetic genes. On the other hand, diverse MAAs have been detected in coral tissues [27, 70] and in shallow-water bivalves [40], so adaptation to UV radiation may depend largely on symbioses with MAA-producing or -using hosts [40, 71]. For example, a report about Symbiodinium evolution and bivalve symbiosis suggests that the Symbiodinium in the bivalve, Fragum, might be a shade-loving alga [72]. SymC, which was originally isolated from Fragum, had no MAA biosynthetic gene cluster, so our analysis supports that suggestion [72].
Conclusions
Gene expansions in Symbiodinium have occurred both by tandem duplication and integration of processed cDNA, possibly transposon-mediated. Comparative analyses indicate that expanded genes in the early-branching lineage include organelle-related genes. The crown lineage retains fewer gene families, but has acquired repeat-domain genes for protein-protein interactions, resembling massive gene losses and extracellular protein expansions in apicomplexans [62]. Finally, our decoded genomes show that the MAA gene cluster of secondary endosymbiotic origin, which is present in some dinoflagellate genomes, has been lost in the crown lineage of Symbiodinum. Taken together, these studies suggest that gene losses and expansions of genes transferred via secondary endosymbiosis drive Symbiodinium evolution.
Methods
Biological materials
Two dinoflagellates, Symbiodinium spp. clade A (SymA) and clade C (SymC) were cultured to produce genomic DNA and mRNA for sequencing. SymA and SymC are harbored by the cardiid clams, Tridacna crocea and Fragum sp., respectively, obtained in Okinawa, Japan. In regard to host habitats, T. crocea is epifaunal and Fragum is infaunal [72]. In the 1980s, isolations of Symbiodinium cells were performed by Prof. Terufumi Yamasu at the University of the Ryukyus using sterilized seawater and micropipettes [73]. The cultured Symbiodinium have been maintained since then in the laboratory of Prof. Michio Hidaka, at the University of the Ryukyus. SymA and SymC were designated as strains “Y106” and “Y103,” respectively. By manually isolating single cells under a microscope using a glass micropipette, each isoclonal line was established at the Marine Genomics Unit of Okinawa Institute of Science and Technology Graduate University in 2009. Repetitive subculture in 250-mL flasks has continued for 8 years, as previously described [4]. Using an incubator (CLE-303, TOMY), all cultures were maintained at 25 °C on a 12 h-light/12-dark cycle at about 20 μmol.m− 2 s− 1 illumination with white fluorescent lamps. The culture solution was artificial seawater containing 1× Guillard’s (F/2) marine-water enrichment solution (Sigma-Aldrich), plus three antibiotics, ampicillin (100 μg/mL), kanamycin (50 μg/mL), and streptomycin (50 μg/mL). Although culturing difficulties for some clade C Symbiodinium have been reported [74], the same culturing conditions have resulted in similar growth rates for SymA, SymC, and S. minutum (SymB).
Genome sequencing and assembly
DNA obtained from clonal cultures (25 °C) of SymA and SymC was used for Illumina library construction (Additional file 1: Table S1), as described previously. Libraries were sequenced using the Illumina Genome Analyzer IIx (GAIIx) and Hiseq (Additional file 1: Table S1). Paired-end reads were assembled de novo with IDBA_UD (ver. 1.1.0) [75], and subsequent scaffolding was performed with SSPACE (ver. 3.0) [76] using Illumina mate-pair information. Gaps inside scaffolds were closed with Illumina paired-end data using Gapcloser [77]. As described previously [4], sequences that aligned to another sequence by more than 70% using BLASTN (1e− 100) were removed from the assembly. Scaffolds > 1 kb were added in version 1.0 of the genome assembly.
Transcriptome sequencing and assembly
Cells cultured at 25 °C were aliquoted and freshly cultured under three types of conditions, 25 °C on 12 h-light/12-dark (Control), 31 °C on 12-light/12-dark (heat-stressed), and 25 °C under 24-dark (dark condition) (Additional file 1: Table S2). After 48 h, cells were collected and frozen for RNA extraction, as done previously [4]. RNAseq library preparation followed manufacturer protocols. RNA sequencing was performed using the GAIIx platform. De novo transcriptome assembly was performed using Trinity software [78].
Gene prediction and annotation
A set of gene model predictions (Gene Model ver. 1) was generated mainly with AUGUSTUS [4], and a genome browser has been established using the Generic Genome Browser (JBrowser) [79]. Annotation and identification of Symbiodinium genes were performed using three methods or combinations of methods: reciprocal BLAST analyses, screening of gene models against the Pfam database [80] at an E-value cutoff of 0.001, and phylogenetic analyses. Gene annotations are available at the genome browser site (http://marinegenomics.oist.jp/gallery/). Scaffold 1 of both SymA and SymC manifested similarities to a bacterial genome, which was identified by genome sequencing of Symbiodinium minutum [4], but which was not included for gene annotation. Expansions of gene families were predicted by chi-square values from comparisons of gene numbers with Pfam domains.
Molecular phylogenetic analysis
Maximum likelihood (ML) phylogenetic trees were constructed using MEGA 5.2, as previously described [81]. ML phylogenetic analysis of the DDG synthase family was also carried out using RaxML with 1000 bootstraps and using the GAMMA and Le-Gasquel model of rate heterogeneity [82]. Bayesian inference was conducted with MrBayes v.3.2 [83] using the same replacement model and run for 4 million generations and four chains until the posterior probability approached 0.01. Statistics and trees were summarized using a burn-in of 25% of the data. Phylogenetic trees were visualized using Figtree (http://tree.bio.ed.ac.uk/software/figtree/) and edited with Treegraph 2 [84].
MAA extraction from Symbiodinium
Symbiodinium cells were cultured at 25 °C for 1 mo on a 12 h-light/12-dark cycle at about 20 μmol.m− 2 s− 1 illumination, as described in the section, Biological materials, and cells were not exposed to UV. Biomass was collected by centrifugation and extracted with methanol (10 mL × 2) at room temperature. Methanol (10 mL) was added to the biomass (0.4–0.6 g, wet weight) followed by vortexing (1 min), sonication (10 min), and centrifugation (7000 g, 10 min, 10 °C) to yield a methanol extract. The resulting clear solution was transferred to a new tube and stored at − 30 °C. Additional methanol was added to the residue, vortexed, and kept overnight at room temperature. After centrifugation, the second methanol extract was decanted, pooled with the first extract, and dried in a vacuum concentrator (40 °C), and the crude extract was stored at − 30 °C before HPLC analysis and purification. The dried methanol extract was suspended in TFA (0.2%, 1 mL) followed by vortexing (1 min), sonication (10 min), and centrifugation (7000 g, 10 min, 10 °C) to give a clear aqueous solution, which was collected and analyzed by HPLC and LC-MS.
MAA analysis by high performance liquid chromatography (HPLC)
HPLC was run on a Nexera (LC-10 AD, Shimadzu) equipped with an autosampler (SIL-30 AC), a column oven (CTO-20 AC), and diode-array detector (SPD-M20A). An ODS column (150 × 2.1 mm, 5 μm, Hypersil Gold, Thermo) was used for MAA analysis and an ODS column (250 × 4.6 mm, 5 μm, Cosmosil) was used for purification. A 16-min gradient was used (A/B 100/0 for 0.0–5.0 min, 100/0 to 85/15 for 5.0–10.0 min, followed by washing 5/95 for 10.0–13.0 min and equilibration 100/0 for 13.0–16.0 min. Solvents (A) Milli Q water and (B) acetonitrile, both containing 0.1% TFA) were used for separation of compounds. A 15 μL sample was injected into the column using the auto-sampler and MAAs were detected at λ330 nm. A constant flow rate 300 μL/min was used and the column was kept at 25 °C.
MAA crude extracts purification by HPLC
The aqueous MAA extract from Symbiodinium was dried and redissolved in 0.2% TFA (300 μL) and injected into the preparative ODS column (250 × 4.6 mm, 5 μm, Cosmosil) using the above HPLC and the target peak (retention time, 8.0 min) was collected. The purified component showed homogeneity in HPLC analysis and was identified as porphyrin-334 by high-resolution mass spectrometry.
Identification of MAAs from Symbiodinium by NanoLC-mass spectrometry (NanoLC-MS)
As described previously [8], a Thermo Scientific hybrid (LTQ Orbitrap) mass spectrometer was used for MS data collection. The mass spectrometer was equipped with an HPLC (Paradigm MS4, Michrom Bioresources Inc.), an auto-sampler (HTC PAL, CTC Analytics), and a nanoelectrospray ion source (NSI). The high-resolution MS spectrum was acquired at 60,000 resolution in FTMS mode (Orbitrap), full mass range m/z 150–500 Da, with capillary temperature (200 °C) and spray voltage (1.9 kV), in positive ion mode. Crude extracts and purified MAA were separated on a capillary ODS column (50 × 0.15 mm, 3-μm, C18, Vydac). A 15-min gradient was employed (100% A for 0.0–10.0 min, 100 to 50% A from 10.1 to 12.0 min, hold at 50% A until 15.0 min, equilibration at 100% A for 15.0 to 18.0 min, where solvent A was water-acetonitrile 98:2 and solvent B was water-acetonitrile 2:98, both containing 0.1% formic acid. The flow rate was 2.0 μL/min and a 2.0 μL sample loop was used for MAA separation.
References
Horiguchi T. Diversity and phylogeny of marine parasitic dinoflagellates. In: Marine Protists. Tokyo: Springer; 2015. p. 397–419.
Wisecaver JH, Hackett JD. Dinoflagellate genome evolution. Annu Rev Microbiol. 2011;65:369–87.
Lin S. Genomic understanding of dinoflagellates. Res Microbiol. 2011;162:551–69.
Shoguchi E, Shinzato C, Kawashima T, Gyoja F, Mungpakdee S, Koyanagi R, et al. Draft assembly of the Symbiodinium minutum nuclear genome reveals dinoflagellate gene structure. Curr Biol. 2013;23:1399–408.
Janouškovec J, Gavelis GS, Burki F, Dinh D, Bachvaroff TR, Gornik SG, et al. Major transitions in dinoflagellate evolution unveiled by phylotranscriptomics. Proc Natl Acad Sci U S A. 2017;114:E171–80.
Wham DC, LaJeunesse TC. Symbiodinium population genetics: testing for species boundaries and analysing samples with mixed genotypes. Mol Ecol. 2016;25:2699–712.
Wang D-Z. Neurotoxins from marine dinoflagellates: a brief review. Mar Drugs. 2008;6:349–71.
Beedessee G, Hisata K, Roy MC, Satoh N, Shoguchi E. Multifunctional polyketide synthase genes identified by genomic survey of the symbiotic dinoflagellate, Symbiodinium minutum. BMC Genomics. 2015;16:941.
Meyer E, Weis VM. Study of cnidarian-algal Symbiosis in the “omics” age. Biol Bull. 2012;223:44–65.
Shinzato C, Mungpakdee S, Satoh N, Shoguchi E. A genomic approach to coral-dinoflagellate symbiosis: studies of Acropora digitifera and Symbiodinium minutum. Front Microbiol. 2014;5:336.
Baker AC. Flexibility and specificity in coral-algal Symbiosis: diversity, ecology, and biogeography of Symbiodinium. Annu Rev Ecol Evol Syst Annual Reviews. 2003;34:661–89.
Pochon X, Putnam HM, Gates RD. Multi-gene analysis of Symbiodinium dinoflagellates: a perspective on rarity, symbiosis, and evolution. PeerJ. 2014;2:e394.
Yamashita H, Koike K. Biology of symbiotic dinoflagellates (Symbiodinium) in corals. In: Marine Protists. Tokyo: Springer; 2015. p. 421–39.
Carlos AA, Baillie BK, Kawachi M, Maruyama T. Phylogenetic position of Symbiodinium (Dinophyceae) isolates from tridacnids (Bivalvia), cardiids (Bivalvia), a sponge (Porifera), a soft coral (Anthozoa), and a free-living strain. J Phycol. Wiley Online Library. 1999;35:1054–62.
Coffroth MA, Santos SR. Genetic diversity of symbiotic dinoflagellates in the genus Symbiodinium. Protist. 2005;156:19–34.
Hirose M, Reimer JD, Hidaka M, Suda S. Phylogenetic analyses of potentially free-living Symbiodinium spp. isolated from coral reef sand in Okinawa, Japan. Mar biol. Springer-Verlag. 2008;155:105–12.
Pochon X, Gates RD. A new Symbiodinium clade (Dinophyceae) from soritid foraminifera in Hawai’i. Mol Phylogenet Evol. 2010;56:492–7.
Hikosaka-Katayama T, Koike K, Yamashita H, Hikosaka A, Koike K. Mechanisms of maternal inheritance of dinoflagellate symbionts in the acoelomorph worm Waminoa litus. Zool Sci. 2012;29:559–67.
Stat M, Morris E, Gates RD. Functional diversity in coral-dinoflagellate symbiosis. Proc Natl Acad Sci U S A. 2008;105:9256–61.
LaJeunesse TC, Lee SY, Gil-Agudelo DL, Knowlton N, Jeong HJ. Symbiodinium necroappetens sp. nov.(Dinophyceae): an opportunist “zooxanthella” found in bleached and diseased tissues of Caribbean reef corals. Eur J Phycol. Taylor & Francis. 2015;50:223–38.
Lee SY, Jeong HJ, Kang NS, Jang TY, Jang SH, Lajeunesse TC. Symbiodinium tridacnidorum sp. nov., a dinoflagellate common to indo-Pacific giant clams, and a revised morphological description of Symbiodinium microadriaticum Freudenthal, emended trench & blank. Eur J Phycol. Taylor & Francis. 2015;50:155–72.
LaJeunesse TC, Bhagooli R, Hidaka M, deVantier L, Done T, Schmidt GW, et al. Closely related Symbiodinium spp. differ in relative dominance in coral reef host communities across environmental, latitudinal and biogeographic gradients. Mar Ecol Prog Ser. Inter-Research Science Center. 2004;284:147–61.
Banaszak AT, LaJeunesse TC, Trench RK. The synthesis of mycosporine-like amino acids (MAAs) by cultured, symbiotic dinoflagellates. J Exp Mar Bio Ecol. 2000;249:219–33.
Takahashi S, Whitney SM, Badger MR. Different thermal sensitivity of the repair of photodamaged photosynthetic machinery in cultured Symbiodinium species. Proc Natl Acad Sci U S A. 2009;106:3237–42.
Díaz-Almeyda EM, Prada C, Ohdera AH, Moran H, Civitello DJ, Iglesias-Prieto R, et al. Intraspecific and interspecific variation in thermotolerance and photoacclimation in Symbiodinium dinoflagellates. Proc Biol Sci. 2017;284:20171767. Available from: https://doi.org/10.1098/rspb.2017.1767
Klueter A, Crandall JB, Archer FI, Teece MA, Coffroth MA. Taxonomic and environmental variation of metabolite profiles in marine dinoflagellates of the genus Symbiodinium. Meta. 2015;5:74–99.
Shick JM, Dunlap WC. Mycosporine-like amino acids and related Gadusols: biosynthesis, accumulation, and UV-protective functions in aquatic organisms. Annu Rev Physiol. Annual Reviews. 2002;64:223–62.
Neale PJ, Banaszak AT, Jarriel CR. Ultraviolet sunscreens in Gymnodinium sanguineum (Dinophyceae): Mycosporine-like amino acids protect against inhibition of photosynthesis. J Phycol. 1998;34:928–38.
Balskus EP, Walsh CT. The genetic and molecular basis for sunscreen biosynthesis in cyanobacteria. Science. 2010;329:1653–6.
Miyamoto KT, Komatsu M, Ikeda H. Discovery of gene cluster for mycosporine-like amino acid biosynthesis from Actinomycetales microorganisms and production of a novel mycosporine-like amino acid by heterologous expression. Appl Environ Microbiol. 2014;80:5028–36.
Shimura Y, Hirose Y, Misawa N, Osana Y, Katoh H, Yamaguchi H, et al. Comparison of the terrestrial cyanobacterium Leptolyngbya sp. NIES-2104 and the freshwater Leptolyngbya boryana PCC 6306 genomes. DNA Res. 2015;22:403–12.
Gao Q, Garcia-Pichel F. Microbial ultraviolet sunscreens. Nat Rev Microbiol. 2011;9:791–802.
Waller RF, Slamovits CH, Keeling PJ. Lateral gene transfer of a multigene region from cyanobacteria to dinoflagellates resulting in a novel plastid-targeted fusion protein. Mol Biol Evol. 2006;23:1437–43.
Starcevic A, Akthar S, Dunlap WC, Shick JM, Hranueli D, Cullum J, Long PF. Enzymes of the shikimic acid pathway encoded in the genome of a basal metazoan, Nematostella vectensis, have microbial origins. Proc Natl Acad Sci U S A. 2008;105(7):2533–7.
Shinzato C, Shoguchi E, Kawashima T, Hamada M, Hisata K, Tanaka M, et al. Using the Acropora digitifera genome to understand coral responses to environmental change. Nature. 2011;476:320–3.
Baumgarten S, Simakov O, Esherick LY, Liew YJ, Lehnert EM, Michell CT, et al. The genome of Aiptasia, a sea anemone model for coral symbiosis. Proc Natl Acad Sci U S A. 2015;112:11893–8.
Bhattacharya D, Agrawal S, Aranda M, Baumgarten S, Belcaid M, Drake JL, Erwin D, Foret S, Gates RD, Gruber DF, et al. Comparative genomics explains the evolutionary success of reef-forming corals. Elife. 2016;5:e13288.
Shih PM, Wu D, Latifi A, Axen SD, Fewer DP, Talla E, et al. Improving the coverage of the cyanobacterial phylum using diversity-driven genome sequencing. Proc Natl Acad Sci U S A. 2013;110:1053–8.
Brawley SH, Blouin NA, Ficko-Blean E, Wheeler GL, Lohr M, Goodson HV, et al. Insights into the red algae and eukaryotic evolution from the genome of Porphyra umbilicalis (Bangiophyceae, Rhodophyta). Proc Natl Acad Sci U S A. 2017;114:E6361–70.
Ishikura M, Kato C, Maruyama T. UV-absorbing substances in zooxanthellate and azooxanthellate clams. Mar Biol. Springer-Verlag. 1997;128:649–55.
Lin S, Cheng S, Song B, Zhong X, Lin X, Li W, et al. The Symbiodinium kawagutii genome illuminates dinoflagellate gene expression and coral symbiosis. Science. 2015;350:691–4.
Aranda M, Li Y, Liew YJ, Baumgarten S, Simakov O, Wilson MC, et al. Genomes of coral dinoflagellate symbionts highlight evolutionary adaptations conducive to a symbiotic lifestyle. Sci Rep. 2016;6:39734.
González-Pech RA, Ragan MA, Chan CX. Signatures of adaptation and symbiosis in genomes and transcriptomes of Symbiodinium. Sci Rep. 2017;7:15021.
Franklin EC, Stat M, Pochon X, Putnam HM, Gates RD. GeoSymbio: a hybrid, cloud-based web application of global geospatial bioinformatics and ecoinformatics for Symbiodinium--host symbioses. Mol Ecol Resour. Wiley Online Library. 2012;12:369–73.
Koyanagi R, Takeuchi T, Hisata K, Gyoja F, Shoguchi E, Satoh N, et al. MarinegenomicsDB: an integrated genome viewer for community-based annotation of genomes. Zool Sci. 2013;30:797–800.
Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11.
Hou Y, Lin S. Distinct gene number-genome size relationships for eukaryotes and non-eukaryotes: gene content estimation for dinoflagellate genomes. PLoS One. 2009;4:e6978.
Harbers K, Schnieke A, Stuhlmann H, Jähner D, Jaenisch R. DNA methylation and gene expression: endogenous retroviral genome becomes infectious after molecular cloning. Proc Natl Acad Sci U S A. 1981;78:7609–13.
Marchler-Bauer A, Bo Y, Han L, He J, Lanczycki CJ, Lu S, et al. CDD/SPARCLE: functional classification of proteins via subfamily domain architectures. Nucleic Acids Res. 2017;45:D200–3.
Penadés JR, Donderis J, García-Caballer M, Tormo-Más MÁ, Marina A. dUTPases, the unexplored family of signalling molecules. Curr Opin Microbiol. 2013;16:163–70.
Slamovits CH, Keeling PJ. Widespread recycling of processed cDNAs in dinoflagellates. Curr Biol. 2008;18:R550–2.
Linder T, Park CB, Asin-Cayuela J, Pellegrini M, Larsson N-G, Falkenberg M, et al. A family of putative transcription termination factors shared amongst metazoans and plants. Curr Genet. 2005;48:265–9.
Robles P, Micol JL, Quesada V. Unveiling plant mTERF functions. Mol Plant. 2012;5:294–6.
Babiychuk E, Vandepoele K, Wissing J, Garcia-Diaz M, De Rycke R, Akbari H, et al. Plastid gene expression and plant development require a plastidic protein of the mitochondrial transcription termination factor family. Proc Natl Acad Sci U S A. 2011;108:6674–9.
Maruyama S, Shoguchi E, Satoh N, Minagawa J. Diversification of the light-harvesting complex gene family via intra- and intergenic duplications in the coral symbiotic alga Symbiodinium. PLoS One. 2015;10:e0119406.
Zhang H, Lin S. Complex gene structure of the form II rubisco in the dinoflagellate Prorocentrum minimum (Dinophyceae). J Phycol. Blackwell Science Inc. 2003;39:1160–71.
Aihara Y, Takahashi S, Minagawa J. Heat induction of cyclic Electron flow around photosystem I in the symbiotic dinoflagellate Symbiodinium. Plant Physiol. 2016;171:522–9.
Allen JF. Why chloroplasts and mitochondria contain genomes. Comp Funct Genomics. 2003;4:31–6.
Reynolds JM, Bruns BU, Fitt WK, Schmidt GW. Enhanced photoprotection pathways in symbiotic dinoflagellates of shallow-water corals and other cnidarians. Proc Natl Acad Sci U S A. 2008;105:13674–8.
Blatch GL, Lässle M. The tetratricopeptide repeat: a structural motif mediating protein-protein interactions. BioEssays. 1999;21:932–9.
Kobe B, Kajava AV. The leucine-rich repeat as a protein recognition motif. Curr Opin Struct Biol. 2001;11:725–32.
Woo YH, Ansari H, Otto TD, Klinger CM, Kolisko M, Michálek J, et al. Chromerid genomes reveal the evolutionary path from photosynthetic algae to obligate intracellular parasites. elife. 2015;4:e06974.
Song J, Xu Q, Olsen R, Loomis WF, Shaulsky G, Kuspa A, et al. Comparing the Dictyostelium and Entamoeba genomes reveals an ancient split in the Conosa lineage. PLoS Comput Biol. 2005;1:e71.
Daher W, Browaeys E, Pierrot C, Jouin H, Dive D, Meurice E, et al. Regulation of protein phosphatase type 1 and cell cycle progression by PfLRR1, a novel leucine-rich repeat protein of the human malaria parasite Plasmodium falciparum. Mol Microbiol. Wiley Online Library. 2006;60:578–90.
Méheust R, Zelzion E, Bhattacharya D, Lopez P, Bapteste E. Protein networks identify novel symbiogenetic genes resulting from plastid endosymbiosis. Proc Natl Acad Sci U S A. 2016;113:3579–84.
Liew YJ, Aranda M, Voolstra CR. Reefgenomics.Org - a repository for marine genomics data. Database [Internet]. 2016;2016 Available from: https://doi.org/10.1093/database/baw152
Santos RS, LaJeunesse TC. Searchable database of Symbiodinium diversity—geographic and ecological diversity (SD2-GED). Auburn University. 2006. http://www.auburn.edu/~santosr/sd2_ged.htm.
Banaszak AT, Barba Santos MG, LaJeunesse TC, Lesser MP. The distribution of mycosporine-like amino acids (MAAs) and the phylogenetic identity of symbiotic dinoflagellates in cnidarian hosts from the Mexican Caribbean. J Exp Mar Bio Ecol. 2006;337:131–46.
Liu Z, Wang L, Zhong D. Dynamics and mechanisms of DNA repair by photolyase. Phys Chem Chem Phys. 2015;17:11933–49.
Yakovleva IM, Hidaka M. Survey of mycosporine-like amino acids in different morphotypes of the coral Galaxea fascicularis from Okinawa, Japan, Galaxea. J Coral Reef Stud. 2009;11:109–18.
Hidaka M. Life History and Stress Response of Scleractinian Corals. In: Kayanne H, editor. Coral Reef Science: Strategy for Ecosystem Symbiosis and Coexistence with Humans under Multiple Stresses. Tokyo: Springer Japan; 2016. p. 1–24.
Ohno T, Katoh T, Yamasu T. The origin of algal-bivalve photo-symbiosis. Palaeontology. London: Palaeontological Association. 1995;38:1–22.
Kobayashi J, Ishibashi M, Nakamura H, Hirata Y, Yamasu T, Sasaki T, et al. Symbioramide, a novel Ca2+-ATPase activator from the cultured dinoflagellate Symbiodinium sp. Experientia. 1988;44:800–2.
Krueger T, Gates RD. Cultivating endosymbionts—host environmental mimics support the survival of Symbiodinium C15 ex hospite. J Exp Mar Bio Ecol. 2012;413:169–76. [Internet]. Available from: https://www.sciencedirect.com/science/article/pii/S0022098111005405
Peng Y, Leung HCM, Yiu SM, Chin FYL. IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics. 2012;28:1420–8.
Boetzer M, Henkel CV, Jansen HJ, Butler D, Pirovano W. Scaffolding pre-assembled contigs using SSPACE. Bioinformatics. 2011;27(4):578–9.
Li R, Fan W, Tian G, Zhu H, He L, Cai J, Huang Q, Cai Q, Li B, Bai Y, et al. The sequence and de novo assembly of the giant panda genome. Nature. 2010;463(7279):311–7.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Skinner ME, Uzilov AV, Stein LD, Mungall CJ, Holmes IH. JBrowse: a next-generation genome browser. Genome Res. 2009;19:1630–8.
Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–22.
Nishitsuji K, Arimoto A, Iwai K, Sudo Y, Hisata K, Fujie M, et al. A draft genome of the brown alga, Cladosiphon okamuranus, S-strain: a platform for future studies of “mozuku” biology. DNA Res. 2016;23:561–70.
Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–90.
Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst Biol. 2012;61:539–42.
Stöver BC, Müller KF. TreeGraph 2: combining and visualizing evidence from different phylogenetic analyses. BMC Bioinf. 2010;11:7.
Acknowledgements
We thank the IT section of OIST for supercomputing support. We also thank Dr. Steven D. Aird for language editing and helpful comments on the manuscript. We acknowledge Ms. Mayuki Fujiwara for isolation of Symbiodinium clones and members of the NIES collection staff for acceptance and maintenance of deposited Symbiodinium strains. We thank members of the Marine Genomics Unit for technical support, especially Dr. Makiko Tanaka and Ms. Mariia Khalturina, for cell culturing and RNAseq library preparation.
Funding
This work was supported in part by Grants-in-Aid from MEXT (no. 25128712 to E.S) and JSPS (no.16 K07454 to E.S, no. 24241071 to N.S.) of Japan, and by generous support from OIST Graduate University to the Marine Genomics Unit.
Availability of data and materials
All sequence data from cultured Symbiodinium are accessible in the DDBJ/EMBL/NCBI database at BioProject IDs, PRJDB3242 for the Y106 strain and PRJDB3243 for the Y103 strain. Accession numbers of raw sequence data are shown in Tables S1 and S2 (Additional file 1). Sequence datasets generated during the current study and raw data (mzXML format) from the mass spectrometer are also available at the genome browser site (http://marinegenomics.oist.jp/gallery/) [45]. Both strains were deposited in the National Institute for Environmental Studies (NIES) collection, at Tsukuba, Japan, and are available as NIES-4076 (Symbiodinium sp. Y-106, clade A3) and NIES-4077 (Symbiodinium sp. Y103, clade C), respectively.
Author information
Authors and Affiliations
Contributions
ES, TK, MH, NS, and CS designed the project. NA and MF extracted DNA samples for genome sequencing and RK purified RNA samples. NA, MF, and RK prepared and sequenced the libraries. ES, GB, IT, KH, TK, TT, MK, and CS analyzed sequence data. ES, GB, IT, KH, NS, and CS interpreted sequence data. IT, KH, and TT performed gene family analysis. ES and MR analyzed MAAs. ES, GB, MR, MK, MH, NS, and CS prepared the manuscript. All authors edited and commented on the manuscript and approved the final manuscript.
Corresponding authors
Ethics declarations
Ethics approval and consent to participate
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Additional files
Additional file 1:
Table S1. Summary of Illumina data used for assembling Symbiodinium genomes. Table S2. Summary of Illumina data used for assembling Symbiodinium transcriptomes. Table S3. Genomic compositions of three genomes of the genus Symbiodinium. Table S4. Summary of assembled transcriptome contigs. Table S5. Expanded genes having Pfam domains in SymA. Table S6. Expanded genes having Pfam domains in SymC. (DOCX 119 kb)
Additional file 2:
Figure S1. A screen shot of scaffold 314 on the SymA genome browser, which is accessible via http://marinegenomics.oist.jp/gallery/, and RNAseq read counts showing the expressions of MAA biosynthetic genes. Figure S2. A molecular phylogenetic tree of O-methyltransferase. Figure S3. A molecular phylogenetic tree of ATP-grasp family proteins. Figure S4. A molecular phylogenetic tree of D-Ala D-Ala ligase family proteins. Figure S5. A molecular phylogenetic tree of cryptochromes/photolyases. (PDF 651 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

Cite this article
Shoguchi, E., Beedessee, G., Tada, I. et al. Two divergent Symbiodinium genomes reveal conservation of a gene cluster for sunscreen biosynthesis and recently lost genes. BMC Genomics 19, 458 (2018). https://doi.org/10.1186/s12864-018-4857-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-018-4857-9