
Abstract
Hyperaccumulators are a group of plant species that accumulate high concentrations of one or more metal(loid)s in their above-ground tissues without showing any signs of toxicity. Several hyperaccumulating species belong to the Brassicaceae family, among them the Cd and Zn hyperaccumulator Noccaea praecox. In this paper, we present de novo transcriptome assembled from two naturally occurring N. praecox populations growing in (i) metal-enriched soil and (ii) soil non-contaminated with metals (control site). Total RNA was extracted from the leaves of both populations. We obtained 801,935,101 reads, which were successfully assembled and annotated. The resulting assembly contains 135,323 transcripts, with 103,396 transcripts (76.4%) annotated with at least one function and encoding 53,142 putative proteins. Due to its close relationship with the hyperaccumulating model species N. cearulescens, it will be possible to derive protein functions from sequence comparisons with this species. Comparisons will highlight common and differing pathways of metal acquisition, storage, and detoxification which will allow us to expand our knowledge of these processes.
Similar content being viewed by others

Background & Summary
Hyperaccumulators are defined as plant species that can accumulate extraordinarily high concentrations of one or more metal(loid)s in the above-ground biomass (especially leaves) without apparent toxicity symptoms1,2. Concentrations of metal(loid)s in the leaves of a hyperaccumulating species could be up to 1,000-fold higher compared to non-hyperaccumulators2,3. To date, approximately 500 plant taxa (0.2% of all angiosperms) are acknowledged to hyperaccumulate metal(loid)s, with several belonging to the Brassicaceae family4,5. Although most hyperaccumulators are defined as nickel (Ni) hyperaccumulators, they accumulate other metal(loid)s as well, including arsenic (As), cadmium (Cd), cobalt (Co), chromium (Cr), copper (Cu), manganese (Mn), lead (Pb), antimony (Sb), selenium (Se), thallium (Tl) and zinc (Zn)6,7,8. Metal hyperaccumulation is of interest for several reasons, which include the biofortification of staple crops9, phytoremediation10,11, and food protection against toxic metal(loid)s12.
Some hyperaccumulating species in Brassicaceae were identified in the genus Noccaea, which includes a well-known hyperaccumulating model species, Noccaea caerulescens. The last hyperaccumulating representative of this genus, identified up to date, was Noccaea praecox, a hyperaccumulator of Cd and Zn13. In N. praecox leaves, Zn is primarily stored in the epidermis, whereas most of the Cd is distributed within the mesophyll14. Both metals were also found in the seeds and were preferentially localized in the epidermis of cotyledons15. Even though it is known Brassicaceae do not form mycorrhizal associations, it was demonstrated N. praecox forms symbiosis with arbuscular mycorrhizal fungi, which improved the plant’s nutrient uptake13,16.
Although N. praecox is a well-characterized hyperaccumulating species, in contrast to its closely related N. caerulescens and N. goesingense no studies were performed on the transcriptome or genome of N. praecox17,18. Despite extensive genomic data acquisition in recent years, current knowledge of gene networks in hyperaccumulators providing physiological responses to environmental changes remains incomplete. As such, RNA-seq of a new hyperaccumulating Noccaea species and validation of metabolic pathways and regulation cascades observed in the model species N. caerulescens could facilitate physiological and molecular studies of these species.
Here we provide the transcriptome of N. praecox. To capture the expression of genes relevant to metal homeostasis under high and low metal load, we analyzed samples from two localities (metal-enriched and non-polluted soils). Detailed accumulation data is available for these sites17. A transcriptome comparison between the two populations and analysis of differentially expressed genes with subsequent models on potential detoxification pathways will be the object of future studies.
Methods
Sample collection
Samples representing the whole flowering plant, including the rhizosphere and bulk soil, were collected in Spring 2022 in Lokovec (N 46° 2′ 39.2706″, E 13° 46′ 8.9934″) and Žerjav (N 46° 28′ 26.1258″, E 14° 51′ 56.0118″) and transferred to the lab. Soil from Lokovec is not contaminated soil, whereas Žerjav is metal-contaminated due to the past mining and smelting activities in the region. Leaves of four plants of N. praecox from each site were sampled, flush-frozen in liquid nitrogen, and stored at −80 °C until further analysis.
Total RNA extraction
Total RNA from plant leaves of N. praecox from both sites was extracted according to the protocol for RNA extraction from plant tissues19. Frozen leaves were ground and homogenized in 400 μL of Z6-buffer containing 8 M guanidinium-HCl, 20 mM MES, and 20 mM EDTA (pH = 7). After the addition of 400 μL of phenol:chloroform:isoamyl alcohol (25:24:1), samples were vortexed and centrifuged for phase separation for 10 minutes at 20,000 g. The upper aqueous phase was transferred to a new microcentrifugation tube, and 0.05 volumes of 1 N acetic acid and 0.7 volumes of 96% ethanol were added. After overnight precipitation at −20 °C, samples were centrifuged for 20 min at 4 °C (20,000 g). The pellet was washed with 200 μL sodium acetate (pH = 5.2) and 70% ethanol. After drying, RNA was dissolved in 30 μL of ultrapure water. The removal of the DNA in the RNA samples was carried out using RNAse free DNAse according to the protocol of RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific). Samples were then stored at −80 °C until further analysis.
Quality control of total RNA, library preparation, and sequencing
RNA quality checks, library preparation, and sequencing were performed by Macrogen company. RNA Integrity Number (RIN) was calculated using Agilent Technologies 2100 Bioanalyzer. Three samples per population (six altogether) were used for further analysis. The cDNA libraries of six samples of N. praecox from both locations were constructed following the manufacturer’s instructions using the TruSeq Stranded mRNA Sample Preparation Kit (Illumina, San Diego, USA). All cDNA libraries were sequenced on the Illumina NovaSeq. 6000 platform using 2 × 150 PE (paired-end sequencing with 150 nt reads). Corresponding read depths are presented in Table 1.
De novo transcriptome assembly
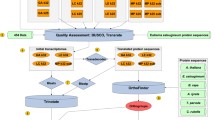
The overall bioinformatic workflow of transcriptome assembly and annotation is summarized in Fig. 1. We used six biological samples, three from contaminated soil and three from non-contaminated soil for the assembly. Raw reads were processed with RCorrector v1.0.520 installed through Anaconda. Uncorrectable reads were removed using FilterUncorrectabledPEfastq.py python script [https://github.com/harvardinformatics/TranscriptomeAssemblyTools]. Cleaned reads were further processed for adapter removal and quality trimming using TrimGalore v0.6.2 installed through Anaconda with default parameters and–length 50 -q 5–stringency 1 -e 0.1. Ribosomal RNAs potentially still present after polyA capture were removed through alignment against the SILVA Ribosomal database (Release 138) with Bowtie2 v2.5.121. Read quality was assessed before and after the processing of reads with FastQC v0.11.822. Retained reads were assembled with Trinity v2.13.223 using default options and --SS_lib_type RF and --min_contig_length 300 (minimum length of contigs 300 nt). Assembly retained the sample information and allows differential expression analysis using native Trinity scripts and deposited raw reads (see Data Records). Finally, we used CD-HIT-EST v4.8.124 to reduce transcript redundancy with the following options: -c 0.90 -n 9 -d 0 -M 0 -T 30 -s 0.9 -aS 0.9. The resulting unique genes (unigenes) were used for the quality check of the assembly and annotations. To find contigs originating outside of the N. praecox transcriptome, we used the NCBI Foreign Contamination Screen (FCS) caller (https://github.com/ncbi/fcs), which flagged 39,641 sequences for removal.

Flowchart of the bioinformatic analysis pipeline.
The quality of the assembly was first analyzed with TrinityStats.pl, and the final transcriptome completeness was estimated using the Benchmarking Universal Single-Copy Orthologs (BUSCO) v525 against the conserved single-copy Viridiplantae genes database on the server gVolante [https://gvolante.riken.jp] Finally, filtered reads were mapped back to the transcriptome to evaluate individual mapping rate with Bowtie2 and ExN50 was generated by Trinity accessory scripts.
Differentially expressed genes
The original sequence reads were mapped to the assembly using the Kallisto pseudoaligner26 and differentially expressed genes (DEGs) were defined as genes having a false discovery rate (FDR) ≤0.05 and an absolute log2 fold change value ≥1 in R using DESeq2 v1.40.2 library27.
Transcriptome annotation
Transcriptome assembly annotation was performed using Trinotate v.4.0.0 pipeline [http://trinotate.github.io]. First, contigs were scanned with Transdecoder v.5.7.0 to predict Open Reading Frames (ORFs). Then unigenes were queries against the SwissProt database (release 2023_02) using blast28, Pfam database (release 35.0) using HMMER29, and Rfam (release 14.9) using infernal v1.1.430. The annotations were associated with Gene Ontology (GO) terms from SwissProt and Pfam databases. In addition, Trinotate was used to predict transmembrane regions (tmHMM v2.0c4331) and signal peptide cleavage sites (signalP v632). The results of these analyses were loaded into a local SQLite database and merged using Trinotate.
Statistics
R v4.3.0 with the library TrinotateR (https://github.com/cstubben/trinotateR) was used for summarisation and visualizations of the obtained transcriptome assembly. For better clarity of the results, GO terms in the figures were filtered with cut-off of 1000 genes (terms with less than 1,000 genes are not included).
Data Records
The filtered and cleaned original RNA sequencing data have been deposited at the NCBI Sequence Read Archive under the SRA study accession SRX20705925-SRX2070593033. This Transcriptome Shotgun Assembly project has been deposited at DDBJ/EMBL/GenBank under the accession GKNA0000000034. The version described in this paper is the first version, GKNA01000000. A full functional annotation of the Trinity transcriptome assembly file of the assembly, including the contaminants (39,641 sequences) flagged by NCBI (https://github.com/ncbi/fcs) as not belonging to N. praecox are available as a supplementary.tsv file at Zenodo as well as the list of genes, their counts and transcripts per million (TMP) values (https://doi.org/10.5281/zenodo.10148119)35.
Technical Validation
Quality of the raw reads and assembly validation
Over 800 million 150 bp preprocessed reads were obtained from six biological samples of N. praecox. After trimming, filtering, and error correction, approximately 689 million (86% of raw reads) of high-quality paired reads were retained and used for de novo assembly. The initial Trinity assembly yielded 210,927 transcripts with an N50 of 1,343 bp. BUSCO score for the initial assembly against orthologs from Viridiplantae showed 95.1% complete, 4.2% fragmented, and 0.7% missing genes. Reducing the redundancy of the initial assembly resulted in an assembly of 177,907 transcripts with an N50 of 1,154 bp, an average sequence length of 834 bp, and a GC content of 44.0%. The assembly showed a reads mapped back to the transcriptome (RMBT) value of 62.3%, whereas transcriptome BUSCO completeness scores for the final assembly showed that the final assembly was 95.0% complete and 4.2% fragmented (Fig. 2a). The final assembly exhibited low levels of missing single-copy orthologs (0.8% missing), indicating good coverage and quality of the assembly. On the other hand, the BUSCO completeness score for protein-coding genes showed the final assembly was 88.0% complete and 9.9% fragmented (Fig. 2b). The percentage of missing single-copy orthologs was higher compared to the BUSCO score for all transcripts (2.1%). Additionally, ExN50 for all transcripts was calculated as it has been suggested to be more informative than the contig N50 and, therefore, a more reliable measure of transcriptome assembly quality36. Our assembly showed a peak saturation point at 90% of the normalized expression data.

BUSCO transcriptome assessment of (a) all transcripts and (b) protein-coding genes.
After reducing redundancy, the length distribution of unigenes was assessed. Most unigenes were 400–600 nucleotides long, and their number decreased with the increasing length. An increased number of unigenes is then detected at the length of >3000 nucleotides (Fig. 3). Differential expression analysis yielded 11,128 differentially expressed genes: between plants grown on the mental-enriched site Žerjav, and the control location in Lokovec. 5,074 genes were down-regulated and 6,054 up-regulated at the metal-enriched site in Žerjav. If contigs were filtered by annotation and only those an annotation for plant taxa were included in the analysis, 3,288 differentially expressed genes were observed. Of those 1,440 were up-regulated in plants at Žerjav and 1,848 were down-regulated at Žerjav, the metal-enriched site.

Length distribution for unigenes after reduced redundancy in the assembly.
Quality control of annotation
The quality of functional annotation depends on the read quality and on the reference data used in the analysis. Therefore, it is crucial to choose appropriate data source to achieve appropriate annotation quality. Search against the SwissProt database yielded results for 80,717 (59.65% of all) unigenes (Table 2), whereas the search for protein sequences found 53,142 (39.27% of all) and 51,479 (38.04% of all) matches for SwissProt and Pfam databases, respectively. Furthermore, 77,738 (57.45%) of unigenes were identified to possess trans-membrane regions and 3,639 (2.69%) were flagged for signal peptides. A search against Rfam identified 893 (0.61%) transcripts as belonging to non-mRNA families.
The highest number of searches for proteins against SwissProt showed affinity to Viridiplantae (Fig. 4a) with 44,264 transcripts (83.3% of all hits), followed by Metazoa and Fungi. At the genus level, the highest number of hits against the database was assigned to Arabidopsis, with 42,462 transcripts (54.6% of all hits) (Fig. 4b).

BLASTp taxonomic results against SwissProt database: (a) at higher taxonomic levels and (b) at the genus level.
We then classified the transcripts based on their annotated GO terms (Fig. 5). In the Biological processes category (45,254, 32.2% of all transcripts with GO term annotation), the three top GO terms are ‘protein phosphorylation’ (2,666, 1.7%), ‘regulation of DNA-templated transcription’ (1,912, 1.2%) and ‘defense response’ (1,632, 1.1%). Cellular Component category has 46,400 (34.1%) transcripts with GO term annotation, among which ‘nucleus’ (13,850, 11.5%), ‘cytoplasm’ (9,273, 7,7%), ‘cytosol’ (7,994, 6.7%), and ‘plasma membrane’ (7,904, 6.6%) are the most abundant. There are 44,547 (32.7%) transcripts identified within the Molecular Function category, with ‘ATP binding’ and ‘metal ion binding’ having the largest number of matched transcripts, with 8,898 (7.6%) and 6,526 (5.6%), respectively.

Distribution of Gene Ontology (GO) classified into three functional categories: biological process, cellular component, and molecular function. GO categories with less than 1,000 unigenes were removed for this representation.
In the end, we examined the annotations from the KEGG database for the A. thaliana transcripts (Fig. 6). The largest number of transcripts was annotated within ‘Metabolic pathways’ (9,176, 44.5% of all transcripts with KEGG annotation) and ‘Biosynthesis of secondary metabolites’ (4,947, 24.0% of all transcripts with KEGG annotation).

Annotations against the KEGG database for A. thaliana genes. The top ten KEGG pathways with the highest number of unigenes are represented.
Code availability
The specific codes for analyses of RNA-seq data are available at https://github.com/matevzl533/Noccaea_praecox_transcriptome.
References
Brooks, R. R., Lee, J., Reeves, R. D. & Jaffre, T. Detection of nickeliferous rocks by analysis of herbarium specimens of indicator plants. J. Geochem. Explor. 7, 49–57 (1977).
Rascio, N. Metal accumulation by some plants growing on zinc-mine deposits. Oikos 29, 250–253 (1977).
Reeves, R. D. in Phytoremediation of Metal-Contaminated Soils Vol. 68 (eds. Morel, J. L., Echevarria, G. & Goncharova, N.) Ch. 2 (Springer, 2006).
van der Ent, A., Baker, A. J. M., Reeves, R. D., Pollard, A. J. & Schat, H. Hyperaccumulators of metal and metalloid trace elements: facts and fiction. Plant Soil 362, 319–334 (2013).
Reeves, R. D. & Baker, A. J. M. in Phytoremediation of Toxic Metals: Using Plants to Clean Up the Environment (eds. Raskin, I. & Finsley, B. D.) Ch. 12 (Wiley, 2000).
Baker, A. J. M. & Brooks, R. R. Terrestrial higher plants which hyperaccumulate metallic elements - a review of their distribution, ecology and phytochemistry. Biorecovery 1, 81–126 (1989).
Baker, A. J. M., McGrath, S. P., Reeves, R. D. & Smith, J. A. C. in Phytoremediation of Contaminated Soil and Water (eds. Terry, N. & Banuelos, G. S.) Ch. 5 (CRC Press, 2000).
Reeves, R. D. et al. A global database for plants that hyperaccumulate metal and metalloid trace elements. New Phytol. 218, 407–411 (2018).
Clemens, S. How metal hyperaccumulating plants can advance Zn biofortification. Plant Soil 411, 111–120 (2017).
Raskin, L., Smith, R. D. & Salt, D. E. Phytoremediation of metals: using plants to remove pollutants from the environment. Curr. Opin. Biotechnol. 8, 221–226 (1997).
Marques, A. P. G. C., Rangel, A. O. S. S. & Castro, P. M. L. Remediation of heavy metal contaminated soils: phytoremediation as a potentially promising clean-up technology. Crit. Rev. Environ. Sci. Technol. 39, 622–654 (2009).
Hu, R. et al. Intercropping with hyperaccumulator plants decreases the cadmium accumulation in grape seedlings. Acta Agric. Scand. – B Soil Plant Sci. 69, 304–310 (2019).
Vogel-Mikuš, K., Drobne, D. & Regvar, M. Zn, Cd and Pb accumulation and arbuscular mycorrhizal colonisation of pennycress Thlaspi praecox Wulf. (Brassicaceae) from the vicinity of a lead mine and smelter in Slovenia. Environ. Pollut. 133, 233–242 (2005).
Vogel-Mikuš, K. et al. Comparison of essential and non-essential element distribution in leaves of the Cd/Zn hyperaccumulator Thlaspi praecox as revealed by micro-PIXE. Plant Cell Environ. 31, 1484–1496 (2008).
Vogel-Mikuš, K. et al. Localisation and quantification of elements within seeds of Cd/Zn hyperaccumulator Thlaspi praecox by micro-PIXE. Environ. Pollut. 147, 50–59 (2007).
Pongrac, P. et al. Changes in elemental uptake and arbuscular mycorrhizal colonisation during the life cycle of Thlaspi praecox Wulfen. Chemosphere 69, 1602–1609 (2007).
Likar, M., Pongrac, P., Vogel-Mikuš, K. & Regvar, M. Molecular diversity and metal accumulation of different Thlaspi praecox populations from Slovenia. Plant Soil 330, 195–205 (2010).
Assunção, A. G. L., Schat, H. & Aarts, M. G. M. Thlaspi caerulescens, an attractive model species to study heavy metal hyperaccumulation in plants. New Phytol. 159, 351–360 (2003).
Longemann, J., Schell, J. & Willmitzer, L. Improved method for the isolation of RNA from plant tissues. Anal. Biochem. 163, 16–20 (1987).
Song, L. & Florea, L. Rcorrector: efficient and accurate error correction for Illumina RNA-seq reads. GigaScience 4, 1–8 (2015).
Langmead, B. & Salzberg, S. L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012).
Wingett, S. W. & Andrews, S. FastQ screen: a tool for multi-genome mapping and quality control. F1000Res. 7, 1338 (2018).
Simon, A. et al. Replicated anthropogenic hybridisations reveal parallel patterns of admixture in marine mussels. Evol. Appl. 13, 575–599 (2020).
Fu, L., Niu, B., Zhu, Z., Wu, S. & Li, W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152 (2012).
Manni, M., Berkeley, M. R., Seppey, M. & Zdobnov, E. M. BUSCO: assessing genomic data quality and beyond. Curr. Protoc. 1, 1–41 (2021).
Bray, N. L., Pimentel, H., Melsted, P. & Pachter, L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 34, 525–527 (2016).
Love, M. I., Huber, W. & Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol. 15, 1–21 (2014).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. J. Mol. Biol. 215, 403–410 (1990).
Eddy, S. R. A new generation of homology search tools based on probabilistic inference. Genome Inform. 23, 205–211 (2009).
Nawrocki, E. P. & Eddy, S. R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 29, 2933–2935 (2013).
Krogh, A., Larsson, B., Von Heijne, G. & Sonnhammer, E. L. L. Predicting transmembrane protein topology with a hidden Markov model: application to complete genomes. J. Mol. Biol. 305, 567–580 (2001).
Teufel, F. et al. SignalP 6.0 predicts all five types of signal peptides using protein language models. Nat. Biotechnol. 40, 1023–1025 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRP444323 (2023).
Likar, M. & Bočaj, V. TSA: Thlaspi praecox, transcriptome shotgun assembly, GenBank, https://identifiers.org/ncbi/insdc:GKNA00000000.1 (2023).
Likar, M., Bočaj, V., Fischer, S. & Pongrac, P. De novo transcriptome assembly of hyperaccumulating Noccaea praecox. Zenodo https://doi.org/10.5281/zenodo.10148119 (2023).
Dolmatov, I. Y., Afanasyev, S. V. & Boyko, A. V. Molecular mechanisms of fission in echinoderms: transcriptome analysis. PLoS One 13, e0195836 (2018).
Acknowledgements
The authors acknowledge the financial support from the Slovenian Research Agency (research core funding No. P1-0212), project funding (Lessons from nutrient-use-efficient plants to benefit dietary mineral intake; J4-3091), and Young Researcher Scholarship to V.B. Part of this work was performed under the financial support of Short-term scientific mission of V.B. to stay with S.F. by COST Action 19116: Trace metal metabolism in plants (PLANTMETALS). Authors would like to thank the Reviewers for taking their time and effort to improve the quality of the manuscript.
Author information
Authors and Affiliations
Contributions
Valentina Bočaj collected plant material, performed the lab work and some bioinformatic analyses, and wrote most of the manuscript. Paula Pongrac organized the molecular part of this study and helped with the manuscript. Sina Fischer helped with the lab work and with the manuscript. Matevž Likar conceived and designed the study, coordinated its implementation and performed most of the bioinformatic analyses.
Corresponding author
Ethics declarations
Competing interests
All authors declare the research was conducted in the absence of any competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Bočaj, V., Pongrac, P., Fischer, S. et al. De novo transcriptome assembly of hyperaccumulating Noccaea praecox for gene discovery. Sci Data 10, 856 (2023). https://doi.org/10.1038/s41597-023-02776-x
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-023-02776-x
- Springer Nature Limited