
Abstract
Background
Loose silky bentgrass (Apera spica-venti) is an important weed in Europe with a recent increase in herbicide resistance cases. The lack of genetic information about this noxious weed limits its biological understanding such as growth, reproduction, genetic variation, molecular ecology and metabolic herbicide resistance. This study produced a reference transcriptome for A. spica-venti from different tissues (leaf, root, stem) and various growth stages (seed at phenological stages 05, 07, 08, 09). The de novo assembly was performed on individual and combined dataset followed by functional annotations. Individual transcripts and gene families involved in metabolic based herbicide resistance were identified.
Results
Eight separate transcriptome assemblies were performed and compared. The combined transcriptome assembly consists of 83,349 contigs with an N50 and average contig length of 762 and 658 bp, respectively. This dataset contains 74,724 transcripts consisting of total 54,846,111 bp. Among them 94% had a homologue to UniProtKB, 73% retrieved a GO mapping, and 50% were functionally annotated. Compared with other grass species, A. spica-venti has 26% proteins in common to Brachypodium distachyon, and 41% to Lolium spp. Glycosyltransferases had the highest number of transcripts in each tissue followed by the cytochrome P450s. The GSTF1 and CYP89A2 transcripts were recovered from the majority of tissues and aligned at a maximum of 66 and 30% to proven herbicide resistant allele from Alopecurus myosuroides and Lolium rigidum, respectively.
Conclusions
De novo transcriptome assembly enabled the generation of the first reference transcriptome of A. spica-venti. This can serve as stepping stone for understanding the metabolic herbicide resistance as well as the general biology of this problematic weed. Furthermore, this large-scale sequence data is a valuable scientific resource for comparative transcriptome analysis for Poaceae grasses.
Similar content being viewed by others

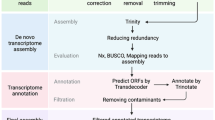
Background
Loose silky bentgrass (Apera spica-venti (L.) Beauv. Poaceae: Pooidea) occurs in Europe, Central Asia, North-West Africa, Caucasus, Turkey, and North-West Iran [1]. It is the most serious agricultural weed in Eastern and Central Europe, infesting many crops particularly winter wheat and winter barley [2–4]. It is an annual, dioecious, outcrossing species with bisexual flowers. Its life cycle consists of seed germination in autumn, then overwintering at the 2-3 leaf stage. It resumes growth in early spring becoming taller than most crop species. It flowers in mid-summer and finally sheds seeds around July [5]. One plant can produce up to 16,000 seeds which enables it to infest whole fields within a few generations [2]. At a density of 200 plants per m2, A. spica-venti decreases the yield of winter wheat up to 30% while another study [5] estimates the reduction in crop yield to be proportional to the quantity of the weed [2]. The European populations have shown a high degree of morphological and genetic variation [6] allowing it to adapt quickly to changing agricultural landscapes.
Control of Apera spica-venti has been, and still is, heavily relying upon herbicides, specifically acetolactate synthase (ALS) and acetyl-CoA (ACCase) inhibitors [7]. However, the continuous use of herbicides favored the evolution of resistance in A. spica-venti populations. Resistance to ALS herbicides was reported for the first time in Switzerland in 1994, and has since been found in 9 other European countries [7]. The study of resistance mechanisms is dependent on previous knowledge of genetic information such as gene identification, abundance, and nucleotide sequences. Information on other genes not related to herbicide resistance, as well as genetic information from susceptible genotypes, are, however, necessary for proper investigations [8].
Transcriptome data have been used in different weed species to study the origin of polyploidy events [9, 10], to understand genetics and biology of weeds [11–14], and to investigate herbicide resistance [15–22]. The majority of transcriptomic studies have focused on dictoyledonous weed species and only four grass weeds have been investigated at the transcriptomic level: Lolium rigidum [22, 23], Eleusine indica [24], Poa annua [15, 25], and Echinocloa cruss-galli [16]. The investigation into evolutionary and molecular processes in these weeds is therefore facilitated by the availability of molecular data. Publicly available genetic information on the diploid species A. spica-venti is very limited: only 12 nucleotide sequences from five genes (ALS, ACCase, rbcL, trnL and matK) are available in the NCBI database [26]. Eight of these come from phylogenetic studies while the four remaining sequences are from herbicide resistance studies [27–29].
The aim of this study was to establish a de novo assembled comprehensive reference transcriptome of A. spica-venti from different tissues (leaf, root, and stem) and early phenological growth stages BBCH05, 07, 08, and 09. We used multiple individuals from various herbicide susceptible populations in order to represent a precise and accurate reference transcriptome for this species which would contain the genetic variation within this outcrossing species. Each dataset, as well as the combined dataset, were de novo assembled and compared among themselves and to similar dataset from the grass weed Lolium rigidum and the model organism Brachypodium distachyon. Known herbicide resistance gene families, cytochrome P450 monooxygenase (P450), glutathione S-transferase (GST), ATP-binding cassette transporters (ABC), elongation initiation factor (EIF), transcription factor (TF) and glycosyltransferase (GT), were identified and transcript abundance was quantified in each tissue.
Results
Transcriptome assembly
A de novo assembly approach was used in order to obtain a comprehensive reference transcriptome of A. spica-venti. The Illumina sequencing resulted in eight libraries having between 54 to 80 × 106 reads each with an average read length of 145 bp for a total dataset of 80,885 Mbp. On average, >80% of the raw reads passed the quality control and normalization process with an average percentage of bases quality score >30 (%Q30) of 88%, an average phred score of 37 with mean base call accuracy of 99.99%. After trimming, quality control and normalization, 13 to 18 × 106 high quality paired-end reads remained in the different libraries. These were assembled into contigs ranging from 32,001 to 83,349 (Table 1). A de novo assembly using Trinity software resulted in a contig number between 117,629 and 319,916 (Table 1). These were passed to the redundancy reduction steps (longest isoform selection, 90% sequence similarity merging and translation into protein sequence using longest ORF: see Methods). The combined assembly show transcript lengths between 99 and 200 bp, while 657 transcripts are longer than 1,000 bp (Additional file 1). Finally, between 28,000 and 43,000 unigenes were identified in the eight individual non-redundant assemblies. Of these, >95% were annotated to UniprotKB and to Brachypodium distachyon coding sequences (Table 1). Some assembly statistics are lower for the combined assembly (N50, average contig lenght) compared to the individual assemblies, some statistics are higher or similar (GC%, protein coding transcripts, max contig lenght).
Transcriptome assembly quality
The eight assemblies show a high percentage of completeness (Table 2), both when comparing them to all plants using Benchmarking Universal Single-Copy Orthologs (BUSCO) and Reads Mapped Back to the Transcriptome (RMBT) where paired-end reads are aligned to their respective transcriptome assembly. For example, the combined assembly is 83% complete and 90% of its reads mapped back to its own transcripts. This indicates that these assemblies are properly assembled and are of good quality. The percentage of reads from each library that are properly paired when mapped to the combined assembly (RMBT to Full in Table 2) is greater than the percentage properly paired when mapped to the assembly from that library (RMBT to themselves in Table 2) indicating that our combined assembly represent well the individual assemblies. Intermediate levels of duplication within assemblies (18-24%) were found in the BUSCO analysis (Table 2).
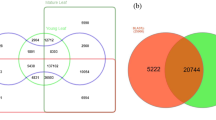
This observed level of redundancy led us to create a zero-redundancy protein ID list for each assembly in order to compare the similarity between the different assemblies without the redundancy. We wanted to ensure that the level of sequence duplication was not introducing a bias in the comparative analysis of the assemblies. The comparison of zero-redundant protein IDs with their respective original assemblies reveals a 24, 27, 25, 25, 22, 23 and 18% of redundancy in seed BBCH05, 07, 08, 09, leaves, root and stem, respectively. These are similar levels of redundancy as was observed with BUSCO. The zero-redundant protein lists were further used to compare the number of shared proteins between tissues and growth stages (Fig. 1). The four germinated seed stages share 25% of proteins, while the three tissues share 27% of proteins (Fig. 1). The percentage of shared proteins between the 3 seed stages are lower (3%) indicating that each growth stage is genetically distinct. The assemblies for roots and stems are more similar than to the leaves assembly. Each tissue/stage has a large number of unique proteins. Root assembly has the highest proportion of unique proteins with 12,171 sequences representing 36% of all its non-redundant expressed proteins recovered.

Venn diagram of non-redundant proteins. Left, between the four seed growth stages BBCH05, 07, 08, and 09. Right, between the three tissues leaves, roots, and stems
Functional annotation
The lack of functional molecular data regarding A. spica-venti prompted us to perform a functional annotation on all eight assemblies. Similar to the individual assemblies, the combined dataset has 10,163 sequences that have an enzyme code representing 12% of the total transcript number. The e-values distribution of the combined assembly shows that 78% of the transcripts were annotated with an e-value of <1e−10 (Additional file 2) indicating high confidence in the annotation performed. The combined assembly has 56,716 sequences belonging to the gene ontology (GO) category of biological process, 63,685 to metabolic function, and 30,063 to cellular component for a total of 150,464 GO annotations (Additional file 3). The “organic substance metabolic process” was the most represented within the category Biological Process (BP) with 15,792 transcripts, then “ion binding” for the Metabolic Function (MF) category with 18,318, and “intracellular function” for the cellular component (CC) with 9,168 transcripts. The comparison of functional annotations between individual assemblies shows that these are rather similar in their GO annotations (Fig. 2). There does not appear to be any disproportionally represented GO categories (BP, MF, CC) between the different tissues and seed growth stages (Fig. 2).

Comparison of Gene Ontology terms found in the seven individual and combined assemblies. BP; biological processes, MF; metabolic function, CC; cellular component
Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis can be used to identify potential pathways most represented in each tissue/growth stages of A. spica-venti. The top ten KEGG pathways, based on sequences number, were compared between each assembly. Purine metabolism was the pathway with the most sequences in each individual assembly with 1,206, 1,400, 1,348, 1,324, 1,061, 1,309, 1,031 and 1,932 sequences in seeds BBCH05, 07, 08, 09, leaves, stem, root and combined assembly, respectively. Carbon fixation in photosynthetic organisms was also observed in the top ten for seeds at BBCH07, 08, 09, and in leaves with 116, 136, 118, and 108 sequences, respectively. All assemblies have similar pathways in their top ten such as important metabolisms pathways (pyruvate, pyrimidine, methane) and sugar pathways (starch and sucrose biosynthesis, amino and nucleotide sugar biosynthesis, and glycolysis and gluconeogenesis). These most represented pathways are important basic pathways necessary for cell life and therefore found in high amounts in all plants. In the top ten KEGG pathways, biosynthesis of antibiotics was recovered in all assemblies while the plant secondary metabolite pathway was not recovered in any assembly. In order to assess for fungal or bacterial contamination in our assemblies, every homologue species recovered from the annotation was verified and confirmed as either algae or green plant organisms. Secondly, the annotation of twenty random sequences that were categorized as part of the biosynthesis of antibiotics was verified. The twenty sequences were all annotated to grass plants with a similarity percentage greater than 70%. We believe that many plant secondary metabolites were most likely defined as antibiotics (such as phytoalexins) in the KEGG database. Our results indicate that plant antibiotics are produced to a large extent in all tissues of A. spica-venti.
The GO enrichment analysis revealed no significantly enriched GO category between individual assemblies and the combined assembly. This result is consistent with Figs. 1 and 2, indicating that the individual assemblies are well represented within the combined and that the individual assemblies are similar to each other.
Comparison to other grass species
Comparative approaches are effective for finding differences and analogies between molecular dataset. Sequence conservation of the A. spica-venti assembly to phylogenetically related grasses and weeds was thus performed. Following annotation to UniprotKB, the highest numbers of homologues for the combined assembly were found in Aegilops tauschii with 10,266 transcripts representing 12% of the species distribution, followed by Triticum aestivum with 8,990 transcripts for 11% of transcripts (Additional file 4). Overall, the top twenty species account for 77% of homologues in the combined assembly. The individual assemblies showed a similar species distribution (data not shown). A comparison of non-redundant protein ID was used (similar to above) between A. spica-venti, Lolium spp. and B. distachyon. The Lolium spp. sequences used come from Schliesky et al. 2012 [30] and originate from a concatenation of three species (L. perenne, L. multiflorum, and L. rigidum). The annotation of the Lolium spp. transcriptome yielded 86,417 annotated protein sequences of which 48,251 (56%) were non-redundant. Brachypodium distachyon yielded 32,086 protein transcripts of which 25,048 (78%) were non-redundant. Apera spica-venti had 53,005 (58%) non-redundant protein annotations. These three species share 21% proteins (Fig. 3). Apera has a higher percentage of protein similarity to Lolium (41%) than to B. distachyon (26%). Apera also has a similar annotation redundancy percentage to Lolium spp with 42% and 44%, respectively. Twentytwo percentage of annotations in B. distachyon was redundant. This lower number is expected considering that this species is a model organism with better annotated data and also because the sequences used here are ab initio predicted from cDNA extracted from genomic studies.

Venn diagram of non redundant proteins between A. spica-venti, Lolium, and B. distachyon
Herbicide resistance gene family
Genes involved in herbicide resistance mechanisms were identified in varying number in the different tissues of A. spica venti. Glycosyltransferase is the most abundant (Fig. 4) in each tissue and growth stage. Seed growth stages had the highest number of GT (excluding the combined assembly) with an average of 250 sequences versus 170 sequences for the three tissues (Fig. 4). The majority of GT sequences found were homologues of barley (Hordeum vulgare var. distichum). The GST transcripts were found in low amount in A. spica-venti overall (Fig. 4). Five sequences identified as GSTF1 in Apera were identified from seeds BBCH05, BBCH07, BBCH08, root and stem. Sequences identified as GSTF1 were aligned with the AmGSTF1 identified in blackgrass (Alopecurus myosuroides; Additional file 5) as this specific sequence was shown to endow herbicide resistance [31]. These had a 38-66% similarity to the A. myosuroides AmGSTF1 sequence. This large range of percentage of similarity is attributable to length differences as no SNPs were identified between the different Apera GSTF1 sequences. However, a three amino acids insertion was found in the blackgrass sequence. The three amino acids are Asparagine, Glutamine and Valine (DEV) at position 104 to 106 bp (Additional File 5). Both alleles are functional. These amino acids were not present in the Apera neither in the Triticum uratu sequence. It is possible that this insertion in A. myosuroides is actually a deletion in A. spica-venti and T. uratu. A GST1 identified in E. crus-galli also responsible for herbicide resistance [32] was also identified in Apera.

Transcript abundance for major herbicide resistance gene families in the seven different tissues and growth stages from A. spica-venti. P450; cytochrome P450, GST; glutathione S-transferase, GT; glycosyltransferase, ABC; ABC transporters, EIF; elongation initiation factor, TF; transcription factor
The cytochrome P450s transcripts were found in high numbers across A. spica-venti tissues and growth stages (Fig. 4). We identified a total of 85, 107, 100, 133, 61, 102 and 67 cytochrome P450 sequences in seeds BBCH 05, 07, 08, 09, leaves, root and stem respectively (Fig. 4). A total of 49 different cytochrome P450s were identified throughout the seven individuals assemblies. Many sequences were identified as P450s but did not have a subfamily identification and therefore were not counted in this number. The most abundant P450 was from subfamily CYP71 (CYP71D7) with 28 sequences identified as such in the combined assembly (Fig. 5). This specific P450 was also the most abundant of the P450s in the seed stages and in leaves with 15, 16, 10, 17 and 6 sequences respectively. The second most abundant P450 is CYP89A2 (Fig. 5). Sequences identified as CYP89A2 in A. spica-venti were aligned with the CYP89A2 identified in L. rigidum (Additional file 6) shown to confer herbicide resistance [22]. Multiple CYP89A2 sequences (mostly from T. uratu and Aegilops tauschii) were identified in each tissue, with the highest number (13) found in seeds at BBCH 05. Overall, 58 sequences of P450 CYP89A2 were found. A large variation in length was observed with the shortest being 99 amino acid long and the longest 488 amino acids (average: 208 aa). The alignment of all CYP89A2 sequences show a 14-40% sequence similarity to Lolium spp, with the highest score belonging to sequences that were annotated to the gene TRIUR3_23510 (Uniprot ORF number) from T. uratu. This demonstrates high sequence polymorphism among the four grass species for CYP89A2. The P450 family was further compared based on protein identification between the seven tissues and growth stages. Overall, the four seed growth stages express 18% of the same P450 in optimal growing conditions (Fig. 6). Seeds at the BBCH 09 had the highest number of uniquely expressed P450s (Fig. 6). The three tissues have almost 20% of identical P450s expressed while root have the highest number of uniquely expressed P450s.

Top 10 most abundant cytochrome P450s in A. spica-venti

Venn diagram of shared P450 proteins. Left, between germinated seeds at BBCH stage 05, 07, 08, and 09. Right, between leaf, root, and stem
Discussion
The aim of this study was to enhance genetic resources for A. spica-venti and through the provision of a reference transcriptome creating a platform to decipher, among others, the development of herbicide resistance. In this study, through a de novo assembly and redundancy reduction strategy, 74,724 unigenes were found in the combined assembly of A. spica-venti which showed a high level of completeness, as well as in the seven individual assemblies. The combined assembly is expected to have assembly statistics differences with the individual assemblies since it contains much more reads and has a 7-fold duplication of many transcripts.
The assembly and homology search statistics are all congruent with previous transcriptome studies in Poaceae. The observed N50 for A. spica-venti are similar to the values from Lolium spp de novo transcriptome (771 bp; [23]), L. rigidum (1,150: [22]), Leymus chinensis (813: [33]) and Phragmites australis (1,187 bp; [34]), but are lower when compared to Zea mays (1,612 bp :[30]), and Arundo donax (2,229 bp ; [35]), Poa annua (1,602 bp: [25]), L. perenne (1,500 bp: [36]), and Eleusine indica (2,095 bp: [24]). The number of transcript after redundancy reduction is slightly lower than the number found after filtering in Lolium spp. [23]. The percentage of functionally annotated protein sequence is also in accordance with Lolium spp. results (57%: [25]). The observed top GO terms in each category (BP, CC, MF) are also found in top positions in E. indica [24], L. chinensis [33], and E. crus-galli [16]. The percentage of properly paired reads to their respective assembly (RMBT) is in line with similar results from A. donax [34]. Overall, the similarity found between the A. spica-venti transcriptome and other grass transcriptomes indicate a properly assembled and annotated dataset presented here.
Although a strict redundancy reduction strategy was used during the assembly, the A. spica-venti transcriptome still shows an intermediate level of transcript redundancy. This indicates that many reconstructed transcript were similar enough to annotate to the same description, yet different enough not to be combined during the assembly. Several biological and technical biases could explain this. Biologically, different gene copy number, alternative splicing, heterozygosity and transcripts from large gene families could explain intermediate levels of redundancy. Alternatively, library preparation such as the amplification step and RNA fragmentation steps, as well as fragmented contigs could also be an explanation. This redundancy can be seen in the alignment of CYP89A2 (see below), where there is a large variation in sequence similarity and length. Both A. spica-venti and Lolium spp showed similar levels of redundancy. This high level of sequence heterogeneity most likely stems from the heterogeneous genetic background of multiple individuals from different populations used in this study to create the transcriptome representing natural variation within A. spica-venti. Transcript redundancy is a biological artifact that complicates de novo transcriptome assembly when using heterogeneous data [23]. Experimentally, contig redundancy could be minimized by performing several generational crosses between individuals from the same population. The level of redundancy in A. spica-venti is consistent across tissues and growth stages and can be identified by multiple methods presented here (BUSCO, non-redundant protein ID) and most importantly, does not affect the levels of similarity between A. spica-venti assemblies. With and without the redundancy, the A. spica-venti transcriptome has high levels of similarity with phylogenetically related grass species such as B. distachyon and Lolium spp. Our study shows that a reference transcriptome can be successfully created from multiple genetically heterogeneous individuals. Using extra steps after the initial de novo assembly can reduce redundancy drastically.
The absence of GO enrichment between the different tissues and growth stages is unsurprising considering the large sample size being compared and the fact that the assemblies were constructed from material under the same biotic and abiotic conditions. This lack of enrichment indicates also that the combined assembly represents well the variation within each tissue and growth stages. The fact that the A. spica-venti transcriptome is more similar to Lolium spp. than to B. distachyon, despite B. distachyon being a much better annotated reference, is most likely caused by the close phylogenetic relationship or could indicate a genetic similarity between grass weeds. The genera Apera, Lolium, Alopecurus and Poa are all part of the large monophyletic core Pooideae while Brachypodium is part of the Brachypodieae [37].
Many populations of A. spica-venti are resistant to herbicides and some have been identified as metabolic resistance cases, with a strong suspicion on the cytochrome P450s family, but never confirmed [38]. We find a higher number of total sequences corresponding to some key herbicide resistance gene families in A. spica-venti than compared to Lolium rigidum reference, which contained 56 GST and 7 GT sequences [22]. The total number of different P450s recovered in this study (49) is in line with the 57 recovered in the transcriptome of Lolium rigidum [22]. However, this number is far below the number of P450s identified in other Poaceae species such as rice (228) and sorghum (326), respectively [39]. Transcriptome studies do not recover the total number of genes present in an organism. This tells us that herbicide susceptible populations of A. spica-venti in Denmark express a low number of P450s in optimal growing conditions. This is consistent with the knowledge of P450s as being involved in stress and xenobioticide defense in plants. The fact that the roots presented the largest number of unique P450s could indicate they were under some undetermined stress and more likely to indicate higher basal P450 expression. Roots in Zea mays and in Arabidopsis are known to express P450s [40, 41] and some P450s are expressed predominantly in roots [42], but there is no evidence that this is a general trend for the P450 family. The most abundant P450, CYP71D7, has been shown to be implicated in glycoalkaloid and ginsenosides biosynthesis, stress-induced secondary metabolites, as well as green tissue regeneration, especially in shoot development stages, in many crops [43–46]. Interestingly, CYP71D7 is also highly upregulated in blight-resistant potato [47]. This hints at a potential role in herbicide resistance in A. spica-venti, but remains to be investigated.
The large sequence variation in CYP89A2 between Lolium, Apera, and Triticum, could indicate that these sequences are not highly conserved as they are implicated in environmental adaptation. The A. spica-venti and A. myosuroides GSTF1 sequences are similar indicating a potential for similar metabolic activity in the presence of herbicide as was demonstrated in Cummins et al [31]. These two known resistance genes (CYP89A2 and GSTF1) were recovered in the majority of tissues of this reference transcriptome of A. spica-venti indicate that these are constitutively expressed at many early stages and overall in the plant. GST are known to be expressed at every stage of plant development in every tissue [48]. Many of the known herbicide resistant P450s, GSTs, and EIFs identified in other weeds species [22, 23, 49–53] were not recovered.
Conclusion
In the present study, we have described the construction and functional annotation of a de novo reference transcriptome assembly for Apera spica-venti with emphasis on gene families involved in herbicide detoxification. These transcriptomes represents a considerable addition to the molecular knowledge available for this important weed species. Moreover, GO and KEGG analysis were conducted and all unigenes were classified into functional categories with the aim to understand their role and metabolic pathways. This data can be used to develop oligo-nucleotide microarray to study gene expression studies at large scale. We have characterized the major gene families responsible for herbicide resistance and shown strong potential of two specific genes to have a role in herbicide resistance in A. spica-venti. Finally, this study can serve as a reference to unravel genes and pathways involved in metabolic herbicide resistance mechanisms, weed adaption, grass genetics and trait evolution in the future.
Methods
Tissue collection
Seeds from six herbicide susceptible populations of Apera spica-venti originating at six different locations in Denmark, were mixed together to create a meta-population encompassing a range of genetic variations possible within the species. Each of these six populations were previously tested between 2007 and 2010 with ALS and ACCase inhibitor herbicides and found to be susceptible (Mathiassen, pers. comm). Seeds were sown in pots and grown under greenhouse conditions on a table with an automatic watering system. Leaf, stem, and root material from individual plant were harvested once the plants were at BBCH stage 34 to 36 (stem elongation). Tissues were cut in small pieces and immediately put in liquid nitrogen and then stored at -80 °C. These tissues were kept separated by individual plant. Seeds from this meta-population were sown in 9 cm petri dishes with four filter papers and 5 mL of deionized water and grown in a climate cabinet (Termaks Series KB8000L) at night/day temperatures of 10 °C/17 °C for 10 h/14 h. Germinated seeds were harvested at stage BBCH 05 (radicle emerges from seed), 07 (coleoptile emerges from caryopsis), 08 (coleoptile elongation), and 09 (Emergence: coleoptile breaks through soil surface). Germinated seeds were put in RNAlater RNA stabilization reagent (Qiagen GmbH, Hilden, Germany) and then stored at 4 °C.
RNA extraction and cDNA sequencing
A total of 0.5 g of frozen tissue from leaf, stem, and root was grounded separately using a 2010 Geno/Grinder (SPEX Sample prep, Stanmore, UK) for 45 s at 1,500 Hz and then soaked in liquid nitrogen. A total of 0.7 g of germinated seeds were grounded using the same method but for 2 cycles of 45 s at 1,500 Hz and were not soaked in liquid nitrogen. The RNA mini plant kit (Qiagen, Hilsen, Germany) was used for RNA extraction following the manufacturer protocol, three wash steps were performed. RNA quality was verified using a spectrophotometer (Nanodrop 3300, Wilmington, USA) and bioanalyzer (Agilent 2100, Santa Clara, USA). Samples that showed a RIN value of 6.0 and more were treated with DNAse 1 according to the protocol (Qiagen, Hilsen, Germany) and suspended in a final volume of 14 μL. RNA quality was verified again with spectrophotometer (NanoDrop-1000 v.3.1.0) and bioanalyzer using the RNA 6000 Plant Nano program (Agilent 2100). Samples with a RIN value higher than 6 were selected for cDNA library construction. The lower than recommended (>8) RIN value threshold selected here is because of difficulties in extracting high quality RNA from germinated seed samples. High quality RNA from minimum three individuals was pooled for each tissue and seed growth stages. Nine libraries were created; seven for each of the individual tissues or growth stage, and libraries for leaf and root, respectively. Samples of mRNA were selected, fragmented and transformed to 150-400 bp short insert, strand specific cDNA libraries for sequencing on Illumina HiSeq 2500 (Eurofins MGW, Germany).
De novo transcriptome assembly of Apera spica-venti
The raw reads were quality-clipped before performing the assembly using the software Trimmomatic [54]. Using a sliding window approach, raw reads with a Phred score below 20, containing only “N”, and/or length below 80 bp were removed. These were normalized using kmer size of 20 [55]. Transcript reconstruction is computationally challenging, especially for non-model plant species [30]. The de Bruijn’s graph strategy has shown to be the best performing for Illumina reads [56]. The Trinity assembler was selected as it has been shown to be more accurate than Velvet to reconstruct full length transcriptome [57]. A combined dataset was created by merging all reads from individual libraries. The assembly method was a composite of different methods in order to reduce transcripts redundancy. First the de novo Trinity pipeline (version 2.1.1; [58]) was used with a minimum contig length of 200 bp and a minimum kmer covariance of 2. The longest isoform for each gene was selected (perl script by Brian Haas on trinityrnaseq-users google group), then contigs were merged according to a similarity criterion of 90% in CD-HIT-EST (version 4.6.3, [59]). The contigs were then translated to coding protein sequences using Transdecoder (version 2.0.1; [56]) following identification of the longest ORFs. This pipeline was performed for each of the seven tissue- and growth stage- specific libraries. Non-redundant assemblies were used in downstream functional annotation KEGG, and GO enrichment analysis. Raw reads, assembled and annotated data set are available at NCBI Gene Expression Omnibus; submission GSE86989.
Assembly quality
The quality of the assemblies was assessed using three methods. First, peptide sequences from the longest ORF from each library were blasted (blastp) against Brachypodium distachyon peptide sequences (Ensembl) as this species is the closest model plant species to A. spica-venti. Second, the assemblies were evaluated against a database of single copy orthologue genes for plants as implemented in BUSCO (version 1.161; [60]). Thirdly, the number of reads that mapped back to the combined transcriptome assembly, and to their own respective assembly, was assessed (RMBT; [61]). Comparing the number of unique proteins IDs recovered (no redundancy) to the original protein list allowed us to calculate a redundancy percentage.
Functional annotation, KEGG and GO enrichment
Individual libraries and the combined, were each aligned (blastp v.3.2.2) to the non-redundant plant protein database UniprotKB (viridiplantae 75_2015-11) with an e-value filter of 1e−1 and only the best homologue was reported. GO mapping was performed against the Gene Ontology database implemented in B2G (version 3.3.5; [62]). Annotation was performed with an e-value hit filter set to 1e−1, annotation cutoff of 55, and evidence code set to 0.8 for the different categories as implemented in B2G. The KEGG pathways was performed in B2G based on enzyme code in each assembly. GO enrichment analysis was performed using a two sided Fisher Exact Test with the multiple testing corrections of Benjamini and Hochberg (FDR) of 0.05. The combined assembly was used as the reference while each annotated assemblies were tested individually against it.
Homology to other model grass species
Protein sequences from Lolium spp. and Brachypodium distachyon were aligned by blastp (e-value filter of 1e−1 and only the best homologue was reported) to the same version of UniprotKB database used for A. spica-venti. This was done in order to compare the three species based on exactly the same search parameters and database type and version. B. distachyon is the closest model species, while Lolium is a genus of grass weed with a well-characterized transcriptome [22, 23, 36]. The coding sequences used for Lolium spp. were taken from the recent work of Duhoux et al. [30] which was generated using tissues from the three species of rye-grasses (L. perenne, L. multiflorum, and L. rigidum). Similarly to the A. spica-venti transcriptome generated here which contains different individuals from field conditions, the Lolium spp. transcriptome contains a high level of heterogeneity stemming from different field individuals and from different species within the Lolium genus. The number of identical non-redundant proteins recovered between Apera, Lolium, and Brachypodium was compared, as well as redundancy percentage.
Herbicide resistance gene families
The number of transcripts annotated to known herbicide resistance gene families (P450, GST, GT, TF, ABC and EIF were compared between the seven assemblies, and also to B. distachyon genome and L. rigidum transcriptome. Specific transcripts identified to genes conferring herbicide resistance in previous studies were aligned to assess identity, homology, and potential resistance phenotype in A. spica-venti. Because the majority of resistance genes identified in herbicide resistant weeds are P450s, this gene family was further investigated. The top ten most abundant P450s recovered were identified. The number of P450s common between tissues and growth stages were compared.
Abbreviations
- ABC:
-
ATP-binding cassette transporters
- ACCase:
-
Acetyl-CoA
- ALS:
-
Acetolactate synthase
- BBCH:
-
Bundesanstalt Bundessortenamt und Chemische Industrie
- BP:
-
Biological process
- BUSCO:
-
Benchmarking universal single-copy orthologs
- CC:
-
Cellular component
- EIF:
-
Elongation initiation factor
- GO:
-
Gene ontology
- GST:
-
Glutathione S-tranferase
- GT:
-
Glycosyltransferase
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- MF:
-
Metabolic function
- NTSR:
-
Non-target site resistance
- P450:
-
Cytochrome monooxygenase P450
- RMBT:
-
Reads mapped back to the transcriptome
- TF:
-
Transcription factor
References
Apera spica-venti (L.) P.Beauv. - Checklist View [Internet]. [cited 2016 Aug 15]. Available from: http://www.gbif.org/species/5289642.
Soukup J, Novakova K, Hamouz P, Namestek J. Ecology of silky bent grass (Apera spiva-venti (L.) Beauv.), its importance and control in the Czech Republic. J Plant Dis Prot. 2006;73–80.
Schulz A, Pallutt B, Gerowitt B. Effect of crop rotation and reduced nitrogen fertilisation on Apera spica-venti populations in long-term experiments. Commun Agric Appl Biol Sci. 2011;76:479–83.
Massa D, Gerhards R. Investigation on herbicide resistance in European silky bent grass (Apera spica-venti) populations. J Plant Dis Prot. 2011;118:31–9.
Melander B, Holst N, Jensen PK, Hansen EM, Olesen JE. Apera spica-venti population dynamics and impact on crop yield as affected by tillage, crop rotation, location and herbicide programmes. Weed Res. 2008;48:48–57.
Warwick SI, Thompson BK, Black LD. Genetic Canadian and European populations of the colonizing weed species Apera spica-venti. New Phytol. 1987;106:301–17.
International survey of herbicide resistant weeds - [weedscience.org cited 2016 Aug 15]. Available from: http://www.weedscience.org/.
DéLye C, Gardin JAC, Boucansaud K, Chauvel B, Petit C. Non-target-site-based resistance should be the centre of attention for herbicide resistance research: Alopecurus myosuroides as an illustration: Why we need more research on NTSR. Weed Res. 2011;51:433–7.
Lai Z, Kane NC, Kozik A, Hodgins KA, Dlugosch KM, Barker MS, et al. Genomics of compositae weeds: EST libraries, microarrays, and evidence of introgression. Am J Bot. 2012;99:209–18.
Estep MC, Gowda BS, Huang K, Timko MP, Bennetzen JL. Genomic characterization for parasitic weeds of the genus by sample sequence analysis. Plant Genome J. 2012;5:30.
Lee RM, Tranel PJ. Utilization of DNA microarrays in weed science research. Weed Sci. 2008;56:283–9.
Horvath DP, Chao WS, Suttle JC, Thimmapuram J, Anderson JV. Transcriptome analysis identifies novel responses and potential regulatory genes involved in seasonal dormancy transitions of leafy spurge (Euphorbia esula L.). BMC Genomics. 2008;9:536.
Huang Y-L, Fang X-T, Lu L, Yan Y-B, Chen S-F, Hu L, et al. Transcriptome analysis of an invasive weed Mikania micrantha. Biol Plant. 2012;56:111–6.
Ranjan A, Ichihashi Y, Farhi M, Zumstein K, Townsley B, David-Schwartz R, et al. De novo assembly and characterization of the transcriptome of the parasitic weed dodder identifies genes associated with plant parasitism. Plant Physiol. 2014;166:1186–99.
Chen S, McElroy JS, Dane F, Peatman E. Optimizing transcriptome assemblies for leaf and seedling by combining multiple assemblies from three de novo assemblers. Plant Genome. 2015;8.
Yang X, Yu X-Y, Li Y-F. De novo assembly and characterization of the barnyardgrass (Echinochloa crusgalli) transcriptome using next-generation pyrosequencing. PLoS ONE. 2013;8:e69168.
Riggins CW, Peng Y, Stewart CN, Tranel PJ. Characterization of de novo transcriptome for waterhemp (Amaranthus tuberculatus) using GS-FLX 454 pyrosequencing and its application for studies of herbicide target-site genes. Pest Manag Sci. 2010;66:1042–52.
Leslie T, Baucom RS. De novo assembly and annotation of the transcriptome of the agricultural weed Ipomoea purpurea uncovers gene expression changes associated with herbicide resistance. Genes Genomes Genet. 2014;4:2035–47.
Peng Y, Lai Z, Lane T, Rao M, Okada M, Jasieniuk M, et al. De novo genome assembly of the economically-important weed Conyza canadensis using integrated data from multiple sequencing platforms. Plant Physiol. 2014;166:1241–54.
Yuan JS, Abercrombie LLG, Cao Y, Halfhill MD, Zhou X, Peng Y, et al. Functional genomics analysis of horseweed (Conyza canadensis) with special reference to the evolution of non–target-site glyphosate resistance. Weed Sci. 2010;58:109–17.
Peng Y, Abercrombie LL, Yuan JS, Riggins CW, Sammons RD, Tranel PJ, et al. Characterization of the horseweed (Conyza canadensis) transcriptome using GS-FLX 454 pyrosequencing and its application for expression analysis of candidate non-target herbicide resistance genes. Pest Manag Sci. 2010;66:1053–62.
Gaines TA, Lorentz L, Figge A, Herrmann J, Maiwald F, Ott M-C, et al. RNA-Seq transcriptome analysis to identify genes involved in metabolism-based diclofop resistance in Lolium rigidum. Plant J. 2014;78:865–76.
Duhoux A, Carrère S, Gouzy J, Bonin L, Délye C. RNA-Seq analysis of rye-grass transcriptomic response to an herbicide inhibiting acetolactate-synthase identifies transcripts linked to non-target-site-based resistance. Plant Mol Biol. 2015;87:473–87.
An J, Shen X, Ma Q, Yang C, Liu S, Chen Y. Transcriptome profiling to discover putative genes associated with paraquat resistance in goosegrass (Eleusine indica L.). PLoS ONE. 2014;9:e99940.
Chen S, McElroy JS, Dane F, Goertzen LR. Transcriptome assembly and comparison of an allotetraploid weed species, annual bluegrass, with its two diploid progenitor species. Plant Genome. 2016;9:1.
Apera - GQuery: Global Cross-database NCBI search - [NCBI cited 2016 Aug 15]. Available from: http://www.ncbi.nlm.nih.gov/gquery/?term=Apera.
Délye C, Michel S. “Universal”primers for PCR-sequencing of grass chloroplastic acetyl-CoA carboxylase domains involved in resistance to herbicides. Weed Res. 2005;45:323–30.
Hamouzová K, Soukup J, JursíK M, Hamouz P, Venclová V, TůMová P. Cross-resistance to three frequently used sulfonylurea herbicides in populations of Apera spica-venti from the Czech Republic: ALS-resistant Apera spica-venti. Weed Res. 2011;51:113–22.
Massa D, Krenz B, Gerhards R. Target-site resistance to ALS-inhibiting herbicides in Apera spica-venti populations is conferred by documented and previously unknown mutations: Target-site resistance in Apera spica-venti populations. Weed Res. 2011;51:294–303.
Schliesky S, Gowik U, Weber APM, Bräutigam A. RNA-Seq assembly – Are we there yet? Front Plant Sci. 2012;3:220.
Cummins I, Wortley DJ, Sabbadin F, He Z, Coxon CR, Straker HE, et al. Key role for a glutathione transferase in multiple-herbicide resistance in grass weeds. Proc Natl Acad Sci. 2013;110:5812–7.
Li G, Wu SG, Yu RX, Cang T, Chen LP, Zhao XP, et al. Identification and expression pattern of a glutathione S-transferase in Echinochloa crus-galli. Zwerger P, editor. Weed Res. 2013;53:314–21.
Chen S, Huang X, Yan X, Liang Y, Wang Y, Li X, et al. Transcriptome analysis in sheepgrass (Leymus chinensis): A dominant perennial grass of the Eurasian steppe. PLoS ONE. 2013;8:e67974.
He R, Kim M-J, Nelson W, Balbuena TS, Kim R, Kramer R, et al. Next-generation sequencing-based transcriptomic and proteomic analysis of the common reed, Phragmites australis (Poaceae), reveals genes involved in invasiveness and rhizome specificity. Am J Bot. 2012;99:232–47.
Sablok G, Fu Y, Bobbio V, Laura M, Rotino GL, Bagnaresi P, et al. Fuelling genetic and metabolic exploration of C 3 bioenergy crops through the first reference transcriptome of Arundo donax L. Plant Biotechnol J. 2014;12:554–67.
Farrell JD, Byrne S, Paina C, Asp T. De novo assembly of the perennial ryegrass transcriptome using an RNA-Seq strategy. PLoS ONE. 2014;9:e103567.
Döring E, Schneider J, Hilu KW, Röser M. Phylogenetic relationships in the Aveneae/Poeae complex (Pooideae, Poaceae). Kew Bull. 2007;62:407–424.
Hamouzová K, Košnarová P, Salava J, Soukup J, Hamouz P. Mechanisms of resistance to acetolactate synthase-inhibiting herbicides in populations of Apera spica-venti from the Czech Republic: Mechanisms of herbicide resistance in Apera spica-venti. Pest Manag Sci. 2014;70:541–8.
Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, et al. The Sorghum bicolor genome and the diversification of grasses. Nature. 2009;457:551–6.
Requejo R, Tena M. Proteome analysis of maize roots reveals that oxidative stress is a main contributing factor to plant arsenic toxicity. Phytochemistry. 2005;66:1519–28.
Ljung K. Sites and Regulation of auxin biosynthesis in Arabidopsis roots. Plant Cell. 2005;17:1090–104.
Werck-Reichhart D, Bak S, Paquette S. Cytochromes P450. Arabidopsis Book. 2002;1:e0028.
Wu Q, Song J, Sun Y, Suo F, Li C, Luo H, et al. Transcript profiles of Panax quinquefolius from flower, leaf and root bring new insights into genes related to ginsenosides biosynthesis and transcriptional regulation. Physiol Plant. 2010;138:134–49.
Hutvágner G, Barta E, Bánfalvi Z. Isolation and sequence analysis of a cDNA and a related gene for cytochrome P450 proteins from Solanum chacoense. Gene. 1997;188:247–52.
Tyagi N, Dahleen LS, Bregitzer P. Candidate genes within tissue culture regeneration QTL revisited with a linkage map based on transcript-derived markers. Crop Sci. 2010;50:1697.
Mekapogu M, Sohn H-B, Kim S-J, Lee Y-Y, Park H-M, Jin Y-I, et al. Effect of light quality on the expression of glycoalkaloid biosynthetic genes contributing to steroidal glycoalkaloid accumulation in potato. Am J Potato Res. 2016;93:264–77.
Yogendra KN, Kushalappa AC. Integrated transcriptomics and metabolomics reveal induction of hierarchies of resistance genes in potato against late blight. Funct Plant Biol. 2016;43:766.
McGonigle B, Keeler SJ, Lau S-MC, Koeppe MK, O’Keefe DP. A genomics approach to the comprehensive analysis of the glutathione S-transferase gene family in soybean and maize. Plant Physiol. 2000;124:1105–20.
Iwakami S, Uchino A, Watanabe H, Yamasue Y, Inamura T. Isolation and expression of genes for acetolactate synthase and acetyl-CoA carboxylase in Echinochloa phyllopogon, a polyploid weed species. Pest Manag Sci. 2012;68:1098–106.
Werck-Reichhart D, Hehn A, Didierjean L. Cytochromes P450 for engineering herbicide tolerance. Trends Plant Sci. 2000;5:116–23.
Didierjean L. Engineering Herbicide Metabolism in Tobacco and Arabidopsis with CYP76B1, a Cytochrome P450 Enzyme from Jerusalem Artichoke. Plant Physiol. 2002;130:179–89.
Saika H, Horita J, Taguchi-Shiobara F, Nonaka S, Nishizawa-Yokoi A, Iwakami S, et al. A novel rice cytochrome P450 gene, CYP72A31, confers tolerance to acetolactate synthase-inhibiting herbicides in rice and Arabidopsis. Plant Physiol. 2014;166:1232–40.
Hu T, Qv X, Xiao G, Huang X. Enhanced tolerance to herbicide of rice plants by over-expression of a glutathione S-transferase. Mol Breed. 2009;24:409–18.
Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30:2114–20.
Brown CT, Howe A, Zhang Q, Pyrkosz AB, Brom TH. A reference-free algorithm for computational normalization of shotgun sequencing data. 2012. arXiv:1203.4802 [q-bio.GN].
Pevzner PA, Tang H, Waterman MS. An Eulerian path approach to DNA fragment assembly. Proc Natl Acad Sci. 2001;98:9748–53.
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, et al. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 2011;29:644–52.
Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8:1494–512.
Fu L, Niu B, Zhu Z, Wu S, Li W. CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics. 2012;28:3150–2.
Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015;31:3210–2.
Zhao Q-Y, Wang Y, Kong Y-M, Luo D, Li X, Hao P. Optimizing de novo transcriptome assembly from short-read RNA-Seq data: a comparative study. BMC Bioinf. 2011;12:S2.
Conesa A, Gotz S, Garcia-Gomez JM, Terol J, Talon M, Robles M. Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics. 2005;21:3674–6.
Acknowledgements
The authors thank Adrian Czaban, Istvan Nagy and Torben Asp for bioinformatics assistance.
Funding
The project was funded by Innovation Fund Denmark EvoPPM; 0603-00516B provided by Bayer Cropsciences and the Danish council for strategic research.
Availability of data and material
The datasets generated during and/or analysed during the current study are available in the NCBI repository, NCBI Bioproject accession: PRJNA342677, NCBI Gene Expression Omnibus GSE86989. Brachypodium distachyon protein retrieved from Ensembl: ftp://ftp.ensemblgenomes.org/pub/plants/release-31/fasta/brachypodium_distachyon/pep sp source, LOLbase: https://lipm-browsers.toulouse.inra.fr/plants/Lolium?download=1. The datasets supporting the conclusions of this article are included within the article and its additional files.
Authors’ contribution
MB participated in the study design, carried out greenhouse and laboratory work, performed bioinformatics analyses and drafted the manuscript. KM provided critical input to the study design and bioinformatics analyses. SKM provided critical input to the study design and supervised the study. MK conceived and supervised the study and revised the manuscript. PK supervised and revised the manuscript. All authors have read and approve the manuscript.
Competing Interests
The authors declare that they have no competing interests. The funding bodies had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
Consent to publish
Not applicable.
Ethics approval and consent to participate
Not applicable.
Author information
Authors and Affiliations
Corresponding author
Additional files
Additional file 1:
Sequence length distribution for combined assembly. (JPG 25 kb)
Additional file 2:
E-value distribution for combined assembly. (PNG 9 kb)
Additional file 3:
Plant Gene Ontology terms associated with the combined assembly of all tissues and growth stage of A. spica-venti. BP; biological processes, MF; metabolic function, CC; cellular component. (PNG 34 kb)
Additional file 4:
Top 20 species identification for the combined dataset. (JPG 74 kb)
Additional file 5:
Alignment of cytochrome P450s CYP89A2 protein sequences from Apera, Lolium, Triticum, and Aegilops. (RTF 1753 kb)
Additional file 6:
Alignment of GSTF1 protein sequences from Apera and Alopecurus. (RTF 159 kb)
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The Creative Commons Public Domain Dedication waiver (http://creativecommons.org/publicdomain/zero/1.0/) applies to the data made available in this article, unless otherwise stated.
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Babineau, M., Mahmood, K., Mathiassen, S.K. et al. De novo transcriptome assembly analysis of weed Apera spica-venti from seven tissues and growth stages. BMC Genomics 18, 128 (2017). https://doi.org/10.1186/s12864-017-3538-4
Received:
Accepted:
Published:
DOI: https://doi.org/10.1186/s12864-017-3538-4