
Abstract
Chanodichthys erythropterus is a fierce carnivorous fish widely found in East Asian waters. It is not only a popular food fish in China, it is also a representative victim of overfishing. Genetic breeding programs launched to meet market demands urgently require high-quality genomes to facilitate genomic selection and genetic research. In this study, we constructed a chromosome-level reference genome of C. erythropterus by taking advantage of long-read single-molecule sequencing and de novo assembly by Oxford Nanopore Technology (ONT) and Hi-C. The 1.085 Gb C. erythropterus genome was assembled from 132 Gb of Nanopore sequence. The assembled genome represents 98.5% completeness (BUSCO) with a contig N50 length of 23.29 Mb. The contigs were clustered and ordered onto 24 chromosomes covering roughly 99.49% of the genome assembly with Hi-C data. Additionally, 33,041 (98.0%) genes were functionally annotated from a total of 33,706 predicted protein-coding sequences by combining transcriptome data from seven tissues. This high-quality assembled genome will be a precious resource for future molecular breeding and functional genomics research of C. erythropterus.
Measurement(s) | whole genome sequencing |
Technology Type(s) | Oxford Nanopore Sequencing |
Sample Characteristic - Organism | Chanodichthys erythropterus |
Similar content being viewed by others

Background & Summary
Chanodichthys erythropterus (Basilewsky, 1855), which belongs to the family Cyprinidae, is widely spread in East Asia, inhabiting lakes or slow-moving rivers with rich vegetation1. Its juvenile fish feed on zooplankton, such as copepods, while adults mainly feed on small fish, a small and fierce carnivorous fish2. The C. erythropterus is highly adaptable to its natural environment and is not obviously affected even when living in alkaline lakes like Hulun Lake3,4.
Due to its delicious and delicate flesh, the C. erythropterus is so popular with consumers in the market and has a high commercial value5. Over the last decade, interest in the aquaculture of C. erythropterus has increased to meet market demand as wild stock is under threat due to overfishing and water pollution. Whole-genome sequencing of a given species is an important and essential tool to address important questions in both biological research and aquaculture. Former research on C. erythropterus has mostly focused on reproduction, age and growth6,7, feeding habits2, muscle composition8, and population genetics9. To date, no genomic resources are available for C. erythropterus, however, severely hampering research into its phylogeny, evolution and biology. Both genomic data and resources can provide a basis for our subsequent studies on the species diversity and population dynamics of C. erythropterus, and can provide a solid support for the proposal of logical conservation measures.
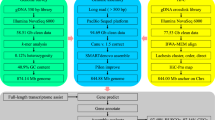
In the current study, the chromosome-level genome of Chanodichthys erythropterus was constructed using Nanopore sequencing and Hi-C technology. We have obtained a scaffold N50 of 42.39 Mb for the final genome assembly, which is approximately 1,085.51 Mb. Using Hi-C data, we identified that 99.49% of the assembled bases were associated with the 24 chromosomes. A valued resource for the conservation and breeding management of C. erythropterus, this genome could serve as the genetic basis for future research into its evolution and biology.
Methods
Sampling and sequencing
The C. erythropterus sample that was obtained in the Hulun Lake (Inner Mongolia, China) was used for genome sequencing and assembly. The muscle tissue was stored at −80 °C and used for DNA extraction, genomic DNA sequencing, and Hi-C library construction. We used a standard SDS extraction method to obtain high-molecular weight DNA.
Following the manufacturer’s recommendations, sequencing libraries were generated using the Truseq Nano DNA HT Sample Preparation Kit (Illumina, USA) and an index code was added to attribute sequences to each sample. These libraries constructed above were sequenced by the Illumina NovaSeq 6000 platform and yielded 150 bp paired-end reads with an insert size of approximately 350 bp. We obtained 41 Gb of raw genomic data for C. erythropterus as a result of Illumina sequencing.
Sequencing was performed on flow cells on the PromethION sequencer according to the manufacturer’s instructions. The Nanopore technology yielded 132 Gb of high-quality data from the long-read library, which covered 117.86-fold of the genome assembly.
In order to obtain chromosome-level assembly of the genome, a high-throughput chromatin conformation capture (Hi-C) library was built for sequencing10. We built the Hi-C library, which used original samples as input. Following grinding with liquid nitrogen, crosslinking was carried out with a 4% formaldehyde solution under vacuum for 30 minutes at room temperature. Add 2.5 M glycine to quench the cross-linking reaction for 5 minutes. Nuclei were digested with 100 units of MboI, tagged with biotin-14-dCTP and subsequently ligated with T4 DNA Ligase. The following incubation overnight to reverse cross-linking, the ligated DNA was segments sheared into 200 to 600 bp fragments. Blunt-end repair and A-tailing of DNA fragments followed by purification through biotin-streptavidin-mediated pulldown. The Hi-C libraries were eventually quantified and sequenced on Illumina PE150.
RNA was also extracted from seven tissues of the C. erythropterus, including intestine, liver, muscle, spleen, heart, gallbladder and kidney, transcriptome sequencing was performed on the Illumina NovaSeq 6000 platform and the resulting reads were used for gene prediction.
Genome size estimation and contig assembly
The Illumina data were analysed for k-mer depth frequency distribution to estimate the genome size, heterozygosity and the amount of repetitive sequences in C. erythropterus. The genome size (G) was estimated according to the following formula: G = k-mer number/k-mer depth, in which the k-mer number and k-mer depth are the total number and average depth of the 17 mers, respectively11. Using 41 Gb of clean Illumina data, the k-mer depth frequency distribution analysis was used for the genome of C. erythropterus (Fig. 1). On the basis of a total of 30,891,679,507 17-mer and a peak 17-mer depth of 27, the estimated genome size was 1120.68 Mb, the heterozygosity was 0.31%, and the amounts of repetitive sequences and guanine-cytosine were roughly 57.05% and 37.95%, respectively (Table 1).

17-mer frequency distribution in C. erythropterus genome. The X-axis is the k-mer depth, and Y-axis represents the frequency of the k-mer for a given depth.
Using all Nanopore sequencing data, a preliminary assembly of the C. erythropterus genome was performed using NextDenovo assembler (v2.3.1) (https://github.com/Nextomics/NextDenovo) with the following parameters: “read_ cutoff = 1k, pa_correction = 20, sort_options = -m 20 g -t 10, correction_options = -p 10”. Finally, the contigs sequences were corrected by NextPolish (v1.3.1)12 using Illumina raw data as well as Nanopore sequencing data. Assembly of these data was then performed with NextDenovo, yielding a genome assembly of 1,085.49 Mb with a contig N50 of 23.28 Mb (Table 2). For this assembly, the length is the same as the genome size estimated by k-mer analysis.
Chromosomal-level genome assembly using Hi-C data
Through the use of the Hi-C scaffolding method13, the contigs in the initial assembly are anchored and oriented to the chromosomal scale of the assembly. The Hi-C library generated 86 Gb clean data. After the Hi-C corrected contigs were placed in the ALLhic pipeline14 for segmentation, orientation and sequencing, the final 99.49% of the assembled sequences were anchored to 24 pseudochromosomes with chromosome lengths that ranged from 31.72 Mb to 73.07 Mb (Table 3). This result is in agreement with the karyotype results which are based on cytological observations15, as many cyprinid fish such as Ctenopharyngodon idellus16, Ancherythroculter nigrocauda17, Hypophthalmichthys molitrix and Hypophthalmichthys nobilis18 with chromosome numbers of 2n = 48. Further we manually curated the Hi-C scaffolding from the chromatin contact matrix in Juicebox (Fig. 2). The 24 pseudochromosomes are easily distinguishable on the basis of the heatmap, and the strength of the interaction signal around the diagonal is fairly strong, indicating the high quality of this genome assembly. Following Hi-C correction, the final assembled genome was 1,085.51 Mb while the scaffold N50 was 42.39 Mb (Table 2). The genome size of C. erythropterus was similar to those of some cyprinid fishes such as the Ctenopharyngodon idellus (1.07 Gb), Megalobrama amblycephala (1.09 Gb)19, Culter alburnus (1.02 Gb)19, and Ancherythroculter nigrocauda (1.04 Gb), but much lower than that of the Cyprinus carpio (1.69 Gb)20.

Hi-C chromosome contact map.
Assessment of the genome assemblies
For evaluating the accuracy and completeness of the genome assembly, we first compared Illumina reads to the assembly of C. erythropterus with the BWA (v0.7.8)21 in which 98.71% of the reads were able to be mapped to contigs. Additionally, we have assessed the integrity of the genome assembly with Benchmarking Universal Single-Copy Orthologs (BUSCO v5.2.1)22 with the vertebrata_odb10 database and CEGMA (v2.5)23. The final results of both showed that the assembly contained 98.5% of complete genes and 0.4% of fragmentarily conserved single-copy orthologs (Table 4), as well as 97.98% of the 248 core eukaryotic genes. All in all, the results of these assessments indicate to us that the C. erythropterus genome assembly is complete and of high quality.
Repeat annotation
Aiming to annotate repetitive elements in the C. erythropterus genome, methods combining homologous comparison and ab initio prediction were used. For ab initio repeat annotation, in which a de novo repetitive element database is constructed using LTR_FINDER (v1.0.7)24, RepeatScout (v1.0.5)25 and RepeatModeler (v1.0.8)26, the RepeatMasker (v4.0.5)26 was used to annotate the repeat elements in the database. The RepeatMasker and RepeatProteinMask (v4.0.5) were then used for known repeat element types via a search of the Repbase database27. Furthermore, TRF (v4.07b)28 can be used to annotate the tandem repeat. Ultimately, we identified 557 Mb of repetitive sequences, accounting for 51.34% of the assembled genome. These figures are higher than in Ctenopharyngodon idellus genome (38.06%) and Megalobrama amblycephala genome (38.68%), but slightly lower than that in Danio rerio genome (52.2%). Within this, we identified 469 Mb of LTR which dominated the assembled genome (43.23%) (Table 5).
Gene prediction and annotation
We detected protein-coding genes in the C. erythropterus genome assembly by a combination of three methods: Ab initio prediction, homology-based prediction and RNA-Seq prediction. As for ab initio prediction, Augustus (v3.2.3)29, GlimmerHMM (v3.04)30, SNAP (2013-11-29)31, Geneid (v1.4)32, and Genescan (v1.0)33 were used in our automated gene prediction pipeline. As for homology-based predictions, we downloaded the protein sequences of Ancherythroculter nigrocauda (GWHAAZV00000000), Cyprinus carpio (GCF_000951615.1), Danio rerio (GCF_000002035.6), Sinocyclocheilus anshuiensis (GCF_001515605.1), Sinocyclocheilus grahami (GCF_001515645.1), Sinocyclocheilus rhinocerous (GCF_001515625.1) from the NCBI database and used TblastN (v2.2.26)34 to match with the C. erythropterus genome with an e-value cutoff of 1E-5, and then the matched proteins were accurately spliced against the homologous genomic sequences using GeneWise (v2.4.1)35 software. As for RNA-Seq prediction, RNA-Seq data from seven tissues (including intestine, liver, muscle, spleen, heart, gallbladder and kidney) were aligned with genomic fasta using TopHat (v2.0.11)36 and gene structures were predicted using Cufflinks (v2.2.1)37. The non-redundant reference gene set was generated by combining genes predicted from three methods using EvidenceModeler (EVM, v1.1.1), using PASA (Program to Assemble Spliced Alignment) terminal exon support38, as well as including masked transposable elements as input to the gene predictions. Overall, a total of 33,706 protein-coding genes were predicted and annotated, with an average exon number per gene of 7.77 and an average CDS length of 1,363.50 bp (Table 6). In the final analysis, we compared the distribution of gene number, gene length, coding DNA sequence (CDS) length, exon length and intron length with that of other stiff bony fishes (Table 7 and Fig. 3).

Comparisons of the prediction gene models in C. erythropterus genome to other species. (a) CDS length distribution and comparison with other species. (b) Exon length distribution and comparison with other species. (c) Exon number distribution and comparison with other species. (d) Gene length distribution and comparison with other species. (e) Intron length distribution and comparison with other species.
The predicted genes of C. erythropterus were functionally annotated by using BLAST39 against SwissProt40, Nr from NCBI, KEGG41, InterPro42, GO43, and Pfam44 databases with an e-value cutoff of 1E-5. The InterproScan (v4.8)45 tool is used to predict protein function based on conserved protein structural domains using the InterPro database. The result was that 33,041 genes were successfully annotated for C. erythropterus, representing 98.0% of all predicted genes (Table 8 and Fig. 4).

Venn diagram of the number of genes with functional annotation using multiple public databases.
Eventually, miRNAs and snRNAs were identified via a search of the Rfam database using the default parameters of INFERNAL46. We chose the human rRNA sequences as a reference and used BLAST39 to predict the rRNA sequences of C. erythropterus. The tRNAs were predicted using the program tRNASCAN-SE47. As a result, we annotated 1,609 miRNA, 8,135 tRNA, 1,251 rRNA and 1,060 snRNA genes (Table 9).
Data Records
The genomic Illumina sequencing data were deposited in the Sequence Read Archive at NCBI SRR1869180448-SRR1869180549.
The genomic Nanopore sequencing data were deposited in the Sequence Read Archive at NCBI SRR1882894250.
The transcriptome Illumina sequencing data were deposited in the Sequence Read Archive at NCBI SRR1869729251-SRR18697298.
The Hi-C sequencing data were were deposited in the Sequence Read Archive at NCBI SRR1869693552.
The final chromosome assembly were deposited in the GenBank at NCBI JALPSW00000000053.
The annotation results of repeated sequences, gene structure and functional prediction were deposited in the Figshare database54.
Technical Validation
The concentration of DNA was determined using Qubit Fluorometer and agarose gel electrophoresis, and the absorbance was approximately 1.8 at 260/280.
For the SNP discovery, Samtools (v0.1.19)55 was applied, resulting in the identification of 950,346 SNPs, including 947,721 heterozygous SNPs and 2,625 homozygous SNPs. The proportion of homozygous SNPs was extremely low, indicating the high accuracy of this assembly.
Code availability
No specific code or script was used in this work. The commands used in the processing were all executed according to the manuals and protocols of the corresponding bioinformatics software.
References
Chen, L., Li, B., Zhou, L. & Zhao, G. The complete mitochondrial genome sequence of Predatory carp Chanodichthys erythropterus (Cypriniformes: Cyprinidae). Mitochondrial DNA Part A. 27, 1119–1120 (2016).
Li, Y. & Zhang, M. Ontogenetic changes in isotopic signatures of an omnivorous fish Cultrichthys erythropterus in East Lake Taihu, China. Journal of Oceanology and Limnology. 33, 725–731 (2015).
Mao, Z., Gu, Z. & Zeng, Q. The structure of fish community and changes of fishery resources in Lake Hulun. Journal of Lake Sciences. 28, 387–394 (2016).
Wang, J., Feng, W. & Zhang, L. Monitoring and Evaluation on Water Quality and Biology Resource Quantity in Hulun Lake. Journal of Hydroecology. 32, 64–68 (2011).
Kindong, R., Prithiviraj, N., Apraku, A., Larbi Ayisi, C. & Dai, X. Biochemical composition of Predatory carp (Chanodichthys erythropterus) from Lake Dianshan, Shanghai, China. Egyptian Journal of Basic and Applied Sciences. 4, 297–302 (2019).
Ma, B., Li, L. & Wu, S. Length-weight relationships of five fishes from the middle Heilongjiang River, China. Journal of Applied Ichthyology. 32, 156–157 (2016).
Wang, T., Wang, H., Sun, G., Huang, D. & Shen, J. Length-weight and length-length relationships for some Yangtze River fishes in Tian-e-zhou Oxbow, China. Journal of Applied Ichthyology. 28, 660–662 (2012).
Li, H., Xia, C., Li, S., Gao, Q. & Zhou, Q. The nutrient contents in the muscle of Culter erythropterus and its nutritional evaluation. Acta Nutrimenta Sinica. 31, 285–288 (2009).
Wang, C., Yu, X. & Tong, J. Microsatellite diversity and population genetic structure of redfin culter (Culter erythropterus) in fragmented lakes of the Yangtze River. Hydrobiologia. 586, 321–329 (2007).
Belton, J.-M. et al. Hi-C: A comprehensive technique to capture the conformation of genomes. Methods. 58, 268–276 (2012).
Marcais, G. & Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 27, 764–770 (2011).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics 36, 2253–2255 (2020).
Wingett, S. et al. HiCUP: pipeline for mapping and processing Hi-C data. F1000Research. 4, 1310–1310 (2015).
Zhang, X., Zhang, S., Zhao, Q., Ming, R. & Tang, H. Assembly of allele-aware, chromosomal-scale autopolyploid genomes based on Hi-C data. Nature Plants 5, 833–845 (2019).
Arai, R. Fish karyotypes: a check list. (Springer Science & Business Media, 2011).
Wang, Y. et al. The draft genome of the grass carp (Ctenopharyngodon idellus) provides insights into its evolution and vegetarian adaptation. Nature genetics 47, 625–631 (2015).
Zhang, H. H. et al. High‐quality genome assembly and transcriptome of Ancherythroculter nigrocauda, an endemic Chinese cyprinid species. Molecular ecology resources 20, 882–891 (2020).
Jian, J. et al. Whole genome sequencing of silver carp (Hypophthalmichthys molitrix) and bighead carp (Hypophthalmichthys nobilis) provide novel insights into their evolution and speciation. Molecular Ecology Resources 21, 912–923 (2021).
Ren, L. et al. The subgenomes show asymmetric expression of alleles in hybrid lineages of Megalobrama amblycephala× Culter alburnus. Genome research 29, 1805–1815 (2019).
Xu, P. et al. Genome sequence and genetic diversity of the common carp, Cyprinus carpio. Nature genetics 46, 1212–1219 (2014).
Li, H. & Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 (2009).
Simao, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212 (2015).
Parra, G., Bradnam, K. & Korf, I. CEGMA: a pipeline to accurately annotate core genes in eukaryotic genomes. Bioinformatics 23, 1061–1067 (2007).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research 35, W265–W268 (2007).
Price, A. L., Jones, N. C. & Pevzner, P. A. De novo identification of repeat families in large genomes. Bioinformatics 21, i351–i358 (2005).
Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Current protocols in bioinformatics 4, 4–10 (2004).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mobile DNA 6, 1–6 (2015).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Research 27, 573–580 (1999).
Stanke, M. & Morgenstern, B. AUGUSTUS: a web server for gene prediction in eukaryotes that allows user-defined constraints. Nucleic Acids Research 33, W465–W467 (2005).
Majoros, W. H., Pertea, M. & Salzberg, S. L. TigrScan and GlimmerHMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20, 2878–2879 (2004).
Korf, I. Gene finding in novel genomes. BMC Bioinformatics 5, 1–9 (2004).
Parra, G., Blanco, E. & Guigo, R. GeneID in Drosophila. Genome research 10, 511–515 (2000).
Burge, C. & Karlin, S. Prediction of complete gene structures in human genomic DNA. Journal of molecular biology 268, 78–94 (1997).
Gertz, E. M., Yu, Y., Agarwala, R., Schaffer, A. A. & Altschul, S. F. Composition-based statistics and translated nucleotide searches: Improving the TBLASTN module of BLAST. BMC Biology 4, 1–14 (2006).
Birney, E., Clamp, M. & Durbin, R. GeneWise and Genomewise. Genome research 14, 988–995 (2004).
Trapnell, C., Pachter, L. & Salzberg, S. L. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111 (2009).
Ghosh, S. & Chan, C.-K. K. Analysis of RNA-Seq Data Using TopHat and Cufflinks. Methods in molecular biology. 1374, 339–361 (2016).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the program to assemble spliced alignments. Genome Biology 9, 1–22 (2008).
Altschul, S. F., Gish, W., Miller, W., Myers, E. W. & Lipman, D. J. Basic local alignment search tool. Journal of molecular biology. 215, 403–410 (1990).
Bairoch, A. & Apweiler, R. The SWISS-PROT protein sequence database and its supplement TrEMBL in 2000. Nucleic Acids Research 28, 45–48 (2000).
Kanehisa, M. & Goto, S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Research 28, 27–30 (2000).
Finn, R. D. et al. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Research 45, D190–D199 (2017).
Ashburner, M. et al. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nature Genetics 25, 25–29 (2000).
Mistry, J. et al. Pfam: The protein families database in 2021. Nucleic Acids Research 49, D412–D419 (2021).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848 (2001).
Griffiths-Jones, S. et al. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Research 33, D121–D124 (2005).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research 25, 955–964 (1997).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR18691804 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR18691805 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR18828942 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR18697292 (2022).
NCBI Sequence Read Archive https://identifiers.org/ncbi/insdc.sra:SRR18696935 (2022).
Zhao, S. Chanodichthys erythropterus isolate Z2021, whole genome shotgun sequencing project, GenBank https://identifiers.org/ncbi/bioproject:PRJNA827856 (2022).
Zhao, S. Whole genome sequencing of the redfin culter (Chanodichthys erythropterus). figshare https://doi.org/10.6084/m9.figshare.20337048.v1 (2022).
Li, H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27, 2987–2993 (2011).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 31872242; No. 32070405; No. 31900311; No. 32000291; No. 32170530). We appreciate the help from Hulunbuir Academy of Inland Lakes in Northern Cold & Arid Areas who provided the C. erythropterus samples.
Author information
Authors and Affiliations
Contributions
Zhao S.H., Yang X.F. and Zhang H.H. designed the study; Pang B., Zhang L., Wang Q. and Dou H.S. collected the samples and extracted the genomic DNA; Zhao S.H., Yang X.F, Pang B., Zhang L., Wang Q. and He S.B. performed data analysis; Zhao S.H. and Yang X.F. wrote the paper. All authors have read, revised, and approved the final manuscript for submission.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Zhao, S., Yang, X., Pang, B. et al. A chromosome-level genome assembly of the redfin culter (Chanodichthys erythropterus). Sci Data 9, 535 (2022). https://doi.org/10.1038/s41597-022-01648-0
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-022-01648-0
- Springer Nature Limited