
Abstract
Decapterus maruadsi is one of the representative offshore fish in the Western Pacific. Since the last century, it has become a commercially valuable marine fishery species in the Western Pacific region. Despite its high economic value, there is still a lack of high-quality reference genome of D. maruadsi in germplasm resource evaluation research. Here we report a chromosome-level reference genome of D. maruadsi based on Nanopore sequencing and Hi-C technologies. The whole genome was assembled through 169 contigs with a total length of 723.69 Mb and a contig N50 length of 24.67 Mb. By chromosome scaffolding, 23 chromosomes with a total length of 713.58 Mb were constructed. In addition, a total of 199.49 Mb repetitive elements, 33,515 protein-coding genes, and 6,431 ncRNAs were annotated in the reference genome. This reference genome of D. maruadsi will provide a solid theoretical basis not only for the subsequent development of genomic resources of D. maruadsi but also for the formulation of policies related to the protection of D. maruadsi.
Similar content being viewed by others

Background & Summary
Decapterus maruadsi, a pelagic fish in the family Carangidae, lives widely in distributed warm offshore waters of East and Southeast Asia1. And it is especially abundant along the coasts of the South China Sea2. From last century, D. maruadsi has become one of the most commercially valuable marine fishery species in Chinese aquaculture. It is also one of the main species captured by pelagic trawls and light-luring fishing vessels3. A short lifespan and fast growth and reproduction rates are the most notable features of D. maruadsi4. Meanwhile, as an r-selection strategy species, it is vulnerable to the environmental deterioration and fishing intensity including those unregulated fishing methods and advanced technologies5,6. In recent decades, under the multiple stresses of continuous high-intensity fishing, increasing temperature and feed structure changes caused by global climate change, the population of D. maruadsi has been subjected to strong selection pressure, which gradually showing adaptive evolution phenomena such as miniaturization, sex precocity, and the population size has also been decreasing year by year7,8. To deal with this dilemma, artificial cultivation of juvenile fish of D. maruadsi has been gradually realized in the offshore area of Dongshan island at present.
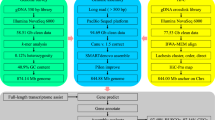
To ensure the preservation of economically significant species, it is crucial to safeguard their germplasm resources and prevent any potential decline9. Genomic data are essential tools for investigating species germplasm resources and assessing population genetic structure and diversity. These resources are of great significance for managing fishery resources and promoting their sustainable use. High-quality reference genomes are essential basic genetic data, and their application value to the aquaculture is also very important. Furthermore, the value of long Nanopore reads which includes low cost, high-throughput sequencing, and high-quality assembly of genomes has been reported by many researches10,11. By combining third-generation sequencing and high-through chromosome conformation capture (Hi-C)12 technologies, we can assemble the chromosome-level genome rapidly, efficiently and accurately. On this basis, the annotation of D. maruadsi genome can be completed. In this report, we provided a high-quality genome assembly of D. maruadsi using Illumina short-reads sequencing, Nanopore sequencing, and Hi-C technologies. We obtained a total of 47.61 Gb clean reads by Illumina platform, and through K-mer frequency distribution analysis, the genome size of D. maruadsi was about 720.70 Mb. For Nanopore genome sequencing, we assembled a total genome length of 723.69 Mb, which includes a total of 169 contigs. In addition, N50 and N90 lengths of filtered reads were respectively 24.67 Mb and 2.78 Mb, and contigs with a length of 2Kb accounted for 100%. We generated 76.37 Gb of Hi-C filtered data, after chromosome-level scaffolding, there are 23 chromosomes with a total length of 713.58 Mb, resulting in a scaffold N50 of 32.35 Mb. The reference genome of D. maruadsi can assist in subsequent population genomics and adaptive genome microevolution studies13.
Methods
Ethics statement
The D. maruadsi in our experiments were collected from Dongshan, Zhangzhou City, Fujian Province, China. Furthermore, the methods used in this work are strictly in accordance with the Guidelines for The Care and Use of Laboratory Animals and followed by the Laboratory Animal Laboratory Committee School of Ocean and Earth Sciences, Xiamen University.
Sample collection and nucleic acid preparation
A healthy alive female D. maruadsi was collected from the Dongshan Pacific Ocean Observation and Experiment Station, Xiamen University. Ten fresh tissue samples, including muscle, eye, skin, gill, kidney, liver, intestine, spleen, heart and stomach, were frozen in liquid nitrogen immediately and then stored in −80 °C. Following the standard protocol of QIAGEN DNeasy Blood & Tissue Kit (Qiagen, Shanghai, China), genomic DNA (gDNA) of muscle was extracted. Total RNA was extracted from ten tissues by a TRIzoL kit (Invitrogen, Shanghai, China) and mixed equally for RNA-seq. The quality of nucleic acid was detected by 1.0% agarose gel electrophoresis and quantified by a Qubit 4.0 fluorometer (Thermo Fisher Scientific, Waltham, MA).
Library construction and sequencing
For Illumina data, a pair-end sequencing library with 350 bp insert size was constructed using the Illumina TruSeq Nano DNA Library Prep Kit (Illumina, San Diego, CA, USA) and sequenced on the Illumina HiSeq X Ten platform with the 2 × 150 bp read strategy at Novogene company (Tianjin, China). A total of 47.85 Gb raw data were obtained and 47.61 Gb clean data were retained after quality filtering by fastp (V.0.23.1)14 software (Table 1). For the Nanopore sequencing, the frozen muscle sample was lysed in SDS digestion buffer, and then the lysate was purified with AMPure XP microbeads (Beckman Coulter, High Wycombe, UK) to obtain High-Molecular-Weight(HMW) gDNA. DNA fragment sizes were selected with the BluePippin system (Sage Science, Beverly, MA, USA) and fragments larger than 20 kb were used for subsequent Nanopore sequencing. The Nanopore libraries were prepared using the Ligation Sequencing Kit (SQK-LSK109, Oxford Nanopore Technologies, Oxford, UK) according to the manufacturer’s instructions and sequenced on the flow cells of the PromethION sequencer at Novogene company (Tianjin, China). Finally, we obtained 86.39 Gb Nanopore data, which average and N50 read length were 22.07Kb and 26.23Kb, respectively. The Nanopore data were further screened before assembly to remove reads less than 1500 bp in length. For Hi-C sequencing, DNA fixed with formaldehyde was digested with the restriction enzyme (DpnII), and after being repaired by 5’overhangs biotinylated and blunt-end ligation, these fragments are connected in situ, the DNA is cross-linked and purified15. In the end, the Hi-C sequencing library was performed on the Illumina HiSeq X Ten platform with a strategy of 2 × 150 bp and generated 76.37 Gb raw reads overall. The RNA-seq library was constructed using Illumina standard protocol (San Diego, CA, USA) and sequenced on the Illumina HiSeq X Ten platform. Finally, we obtained 33.65 Gb paired-end raw reads and 32.61 Gb paired-end clean reads for the following gene prediction (Table 1).
De novo genome assembly
The Illumina clean reads were used for further assembly and estimation of genome size using 17-kmer analysis. With K-mer numbers of 434,759,917,857 and a dominant peak depth of 47.24, the genome size was approximately 720.70 Mb, which was similar to the species in Genus Decapterus and the heterozygosity and repetitive sequence content were about 0.69 and 32.6%, respectively16 (Supplementary Table 1 & Supplementary Fig. 1).
NextDenovo was used for genome assembly based on the overlap layout-consensus algorithm with default parameters. To obtain the contig-level genome, we utilized Racon17 for three iterations of polish using the three-generation Nanopore data. Nextpolish18 was then employed to correct the genome based on the Illumina data. Lastly, we utilized Purge_Dups19 (v.1.25) to de-redundant the genome, resulting in the final contig-level genome. The assembled genome size was 723.69 Mb, including 169 contigs in total, with a contig N50 of 24.67 Mb (Table 2). The assembled genome size almost matched the estimated results of genome survey, which reflected the high assembly integrity.
Hi-C sequencing data was used for chromosome assembly of D. maruadsi. Firstly, we filtered out Hi-C raw reads(low-quality and duplicated reads) using HiC-Pro20. Juicer21 was used to map Hi-C clean reads to the reference genome. Subsequently, we used the genomic proximity signal in the Hi-C data sets to get chromosome-level scaffolds. Then, the 3D-DNA pipeline22 was used to scaffold the D. maruadsi genome. Afterwards, scaffolds were fine-tuned to correct the misassemblies by Juicebox23 assembly tools. Finally, we generated a chromosome-level genome assembly of 724.05 Mb and scaffold N50 is up to 32.35 Mb (Table 2). The genome assembly contained 23 chromosomes, with a total length of 713.58 Mb (98.6% of the total length of all contigs). Chromosome sizes ranged from 21.36 to 45.1 Mb, with an average chromosome length of 31.03 Mb (Fig. 1A,B & Table 3).

Characteristics of Decapterus maruadsi genome assembly. (A) Contact map of chromosomal interactions in the D. maruadsi genome using Hi-C data. (B) A circos plot of 23 chromosomes in D. maruadsi genome, the tracks from outside to inside are: a. Lines represent D. maruadsi chromosomes; b. GC content; c. Gene density; d. Repeat element density. (C) Circos diagram showing synteny relations between D. maruadsi and O. latipes. Each coloured line represents a 1 Kb fragment match between two species. We reordered the chromosome numbers of D. maruadsi for better illustration.
Anotation of repeat sequences
Both homology-based and de novo methods were used to annotate repeat sequences in the D. maruadsi genome. RepeatModeler24 (v.2.0.1) and LTR_Finder25 (v.1.07) were utilized to detect repetitive sequences in the D. maruadsi genome and generate a de novo repeat library. Combined with Repbase26, the final repeat library was constructed. RepeatMasker27 (v.4.1.0) was used to search and classify repeats based on this library. Unclassified repeats were further annotated using TEclass28 (v.2.1.3). Transposable Elements (TEs) annotation results were summarized by adopting the buildSummary.pl of RepeatMasker. Moreover, calcDivergenceFromalign.pl was used to calculate the Kimura divergence value of TEs and createRepeatLandscape.pl was used to draw TEs landscapes29. To estimate the insertion age, we compared the nucleotide distances between all copies of each TE using the Kimura two-parameter method29. We identified tandem repeats using the Tandem Repeats Finder30 (v.4.0.9) and soft-masked all repetitive regions except for tandem repeats in the process of protein-coding gene annotation. Finally, a total of 199.49 Mb (27.57% in genome) of consistent and non-redundant repeat sequences were obtained by combining novel, known and tandem repeats. The most abundant repetitive elements were DNA transposons, which spanned more than 102.57 Mb, accounting for 14.17% of the genome of D. maruadsi. Besides, the repetitive sequences were also composed of long interspersed elements (LINE) in 37.62 Mb (5.20% in genome), short interspersed nuclear elements (SINEs) in 2.82 Mb (0.39% in genome) and long terminal repeats (LTRs) in 40.99 Mb (5.66% in genome) (Fig. 2A & Table 4).

Gene and repeat annotations of the D. maruadsi genome. (A) Distribution of divergence rate for each type of TEs in the D. maruadsi genome. (B) Venn diagram of the functionally annotated protein-coding genes based on diferent databases.
Prediction and functional annotation of protein-coding genes
For non-coding RNA (ncRNA) annotation, the programs tRNAScan-SE31 (v.1.3.1) and RNAmmer32 (v.1.2) were used to predict tRNA and rRNA respectively. The other ncRNAs were predicted by searching the Rfam database33 (http://eggnogdb.embl.de/). As a result, we annotated four types of non-coding RNAs, including 1,285 miRNAs, 3,820 tRNAs, 1,592 rRNAs and 762 snRNAs (Table 4).
For gene structure prediction, ab-initio strategies, homologous searching and transcriptome-assisted approaches were used to predict protein-coding genes in the D. maruadsi genome after soft-masking all repeat sequences. In homology-based prediction, the genetically proximal coding sequences of related species, containing Oryzias latipes, Seriola lalandi, Seriola dumerili, Oreochromis niloticus, and Trachinotus ovatus were downloaded from European Nucleotide Archive and provided to GenomeTreader34 (v.1.7.0) (Supplementary Table 4). Additionally, the RNA-seq data was subjected to the assembly using Trinity35 (v.2.10.0). The ab-initio gene prediction was performed using the transcripts assembled from RNA-seq and known genes of O. latipes, S. lalandi, S. dumerili, O. niloticus, and T. ovatus by Braker236. After two rounds of model training, the optimal parameters are determined. Another gene prediction method involved aligning RNA-seq data to the D. maruadsi genome to assemble the transcriptome using Hisat237 and StringTie38 (v.2.1.4). Then, the open reading frame (ORF) regions were predicted using TransDecoder (v.5.5.0). Ultimately, EvidenceModeler was utilized to create a thorough gene set, which was then further annotated for protein-coding gene structure via PASA39 (v.2.4.1). For functional annotation of predicted gene, Diamond40 (v.2.0.6) was applied to align protein-coding genes to the Swiss-Prot (http://www.uniprot.org/), InterPro(https://www.ebi.ac.uk/interpro/) and NR protein databases with E-values < 1*10−5. Additionally, GO and KEGG pathway annotations were performed by InterProScan41 (v.4.8) (https://www.ebi.ac.uk/interpro/) and KEGG Automatic Annotation Server (KAAS42, https://www.genome.jp/tools/kaas/) (Table 5).
In this study, a high-quality reference genome of D. maruadsi was generated, which could provide a solid foundation for species diversity and population genetic studies in the future. Nowadays, genomics is gradually being applied in every stage of large-scale aquaculture production and domestication. As an important aquatic economic fish, D. maruadsi is necessary to identify genetic diversity under phenotypic traits by a high-precision chromosome-scale genome to improve the economic benefits of aquaculture species. In addition, high-quality genome-wide maps are important as essential basic genetic data for industrial and scientific research applications, providing a genetic basis and more accurate genetic evaluation tools for the management and sustainable use of D. maruadsi fisheries resources. Finally, as a potential cultured fish, the genome of D. maruadsi will help to the breeding program for selecting excellent growth-related traits.
Data Records
The raw sequencing reads of all libraries have been deposited into NCBI SRA database via the accession number of SRP40850543. The assembled genome has been deposited at Genbank under the accession number GCA_030347415.244. Moreover, data of the assembled genome and sequence annotations are available at Figshare45.
Technical Validation
Genome assembly and annotation completeness evaluation
To ensure the accuracy and integrity of the assembly, we assessed the completeness of the final genome assembly using Benchmarking Universal Single-Copy Orthologues (BUSCO)46 with the Actinopterygii_odb10 lineage database. Out of 3,640 single-copy orthologues, approximately 97.8% were completely identified in the D. maruadsi genome (Supplementary Table 3). Besides, the Illumina short reads were aligned to the genome using the BWA MEM algorithm. Subsequently, employing samtools on the generated BAM files, we calculated the sequencing depth across the genome. The non-zero sequencing depth positions were tallied and summed, then compared to the total base positions for the final coverage percentage. This yielded a mapping ratio of 99.76% and a genome coverage of 98.80% (Supplementary Table 2). Moreover, a total of 33,515 protein-coding genes were successfully obtained by combining ab-initio strategies, homologous searching and transcriptome-assisted approaches. A total of 25,933 genes were successfully functionally annotated in at least one of these databases (Fig. 2B & Table 5). The high integration efficiency, mapping ratio, recognition rate of single-copy orthologues and gene number collectively suggest that the assembled D. maruadsi genome was of superior quality.
Genome assembly accuracy evaluation
To validate the precise arrangement of the D. maruadsi genome, we aligned the assembly to the O. latipes genome using minimap247 with a unit of 1 Kbp (Fig. 1C). Additionally, we performed the same alignment method with Trachurus trachurus, a closely related species in the Carangidae family. The 23 chromosomes identified in the D. maruadsi genome showed a significant level of collinearity with the other two species, indicating the high genomic continuity of our assembly (Supplementary Fig. 2 & Supplementary Fig. 3).
Notably, Chromosome 2 of D. maruadsi aligns with both chromosome 2 and chromosome 4 of O. latipes and T. trachurus. To confirm the accuracy of the chromosome number, we performed a nucmer48 alignment of chromosome 2 of D. maruadsi with chromosome 2 and chromosome 4 of O. latipes and T. trachurus. The results revealed that chromosomes 4 and 2 of O. latipes aligned to the regions 0.34 M - 31.19 M and 31.82 M - 44.96 M, respectively, on chromosome 2 of D. maruadsi. Similarly, chromosomes 4 and 2 of T. trachurus aligned to the regions 0.09 M - 31.62 M and 31.66 M - 45.06 M, respectively, on chromosome 2 of D. maruadsi (Supplementary Fig. 4B). These comparative analyses collectively indicate the presence of a distinct alignment gap within the 31 M - 32 M region of the reported chromosome 2 of D. maruadsi. This alignment gap suggests the possibility of a structural alteration and connection region between the two chromosomes in this location.
Moreover, based on the Hi-C assisted assembly data, we have identified that this connection region is completely covered by the precisely assembled contig (Supplementary Fig. 4). Additionally, we selected the genomic range from 31 M to 32 M and utilized the minimap2 tool to align Illumina and Nanopore reads to this region. Subsequently, we implemented a sliding window approach with a window size of 50 kb to calculate the average depth at each genomic position. In this important region, both Illumina and Nanopore data consistently exhibited stable depth profiles, with no significant decrease in depth observed (Supplementary Fig. 5). These observations further emphasize the integrity and continuity of our assembly results.
Code availability
Genome annotation:
(1) RepeatMasker: parameters: -e ncbi -a -nolow -no_is -norna.
(2) TE-class: parameters: all parameters were set as default.
(3) Braker2: parameters: all parameters were set as default.
(4) PASA: --ALIGNERS blat.
(5) EvidenceModeler: parameters: all parameters were set as default.
Genome assembly:
(1) NextDenovo: parameters: all parameters were set as default.
Gene family identifcation and phylogenetic analysis:
(1) RAxML: parameters: -f a -m PROTGAMMAAUTO.
(2) MCMCTREE: parameters: all parameters were set as default. Other analysis modules that were not mentioned parameters were used default parameters. The other custom codes used in this analysis were mentioned in methods sections.
References
Jamaludin, N. A. et al. Phylogeography of the Japanese scad, Decapterus maruadsi (Teleostei; Carangidae) across the Central Indo-West Pacific: evidence of strong regional structure and cryptic diversity. Mitochondrial DNA A DNA Mapp. Seq. Anal. 31, 298–310 (2020).
Chen, G. & Li, Y. Distribution of the Carangidae fishes in the continental shelf waters of northern South China Sea. J. Shanghai Ocean Univ. 12, 146–151 (2003).
Zheng, Y., Li, J., Zhang, Q. & Hong, W. Research progresses of resource biology of important marine pelagic food fishes in China. J. Fish. China. 38, 149–160 (2014).
Ohshimo, S., Yoda, M., Itasaka, N., Morinaga, N. & Ichimaru, T. Age, growth and reproductive characteristics of round scad Decapterus maruadsi in the waters off west Kyushu, the East China Sea. Fish. Sci. 72, 855–859 (2006).
Niu, S., Su, Y., Wang, J. & Zhang, L. Population genetic structure analysis of Decapterus maruadsi from Fujian coastal waters. J. Xiamen Univ. Nat. Sci. 51, 759–766 (2012).
Yu, J., Liu, Z., Chen, P. & Yao, L. Environmental factors affecting the spatiotemporal distribution of Decapterus maruadsi in the western Guangdong waters, China. Appl. Ecol. Environ. Res. 17, 8485–8499 (2019).
Marty, L., Dieckmann, U. & Ernande, B. Fisheries‐induced neutral and adaptive evolution in exploited fish populations and consequences for their adaptive potential. Evol. Appl. 8, 47–63 (2015).
Enberg, K. et al. Fishing‐induced evolution of growth: Concepts, mechanisms and the empirical evidence. Mar. Ecol. 33, 1–25 (2012).
Gong, D. et al. Protection and utilization status of Parabramis and Megalobrama germplasm resources. Reprod. Breed. 3, 26–34 (2023).
Deamer, D., Akeson, M. & Branton, D. Three decades of nanopore sequencing. Nat. Biotechnol. 34, 518–524 (2016).
Branton, D. et al. The potential and challenges of nanopore sequencing. Nat. Biotechnol. 26, 1146–1153 (2008).
Belton, J. M. et al. Hi–C: a comprehensive technique to capture the conformation of genomes. Methods. 58, 268–276 (2012).
Nielsen, E. E., Hansen, J. H., Larsen, P. F. & Bekkevold, D. Population genomics of marine fishes: identifying adaptive variation in space and time. Mol. Ecol. 18, 3128–3150 (2009).
Chen, S., Zhou, Y., Chen, Y. & Gu, J. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics. 34, i884–i890 (2018).
Rao, S. S. et al. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 159, 1665–1680 (2014).
Mirsky, A. & Ris, H. The desoxyribonucleic acid content of animal cells and its evolutionary significance. J. Gen. Physiol. 34, 451 (1951).
Vaser, R., Sović, I., Nagarajan, N. & Šikić, M. Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res. 27, 737–746 (2017).
Hu, J., Fan, J., Sun, Z. & Liu, S. NextPolish: a fast and efficient genome polishing tool for long-read assembly. Bioinformatics. 36, 2253–2255 (2020).
Guan, D. et al. Identifying and removing haplotypic duplication in primary genome assemblies. Bioinformatics. 36, 2896–2898 (2020).
Servant, N. et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 1–11 (2015).
Durand, N. C. et al. Juicer provides a one-click system for analyzing loop-resolution Hi-C experiments. Cell Syst. 3, 95–98 (2016).
Dudchenko, O. et al. De novo assembly of the Aedes aegypti genome using Hi-C yields chromosome-length scaffolds. Science. 356, 92–95 (2017).
Durand, N. C. et al. Juicebox provides a visualization system for Hi-C contact maps with unlimited zoom. Cell Syst. 3, 99–101 (2016).
Flynn, J. M. et al. RepeatModeler2 for automated genomic discovery of transposable element families. PNAS. 117, 9451–9457 (2020).
Xu, Z. & Wang, H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Res. 35, W265–W268 (2007).
Bao, W., Kojima, K. K. & Kohany, O. Repbase Update, a database of repetitive elements in eukaryotic genomes. Mob. DNA. 6, 1–6 (2015).
Chen, N. Using Repeat Masker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 5, 4.10. 11–14.10. 14 (2004).
Abrusán, G., Grundmann, N., DeMester, L. & Makalowski, W. TEclass—a tool for automated classification of unknown eukaryotic transposable elements. Bioinformatics. 25, 1329–1330 (2009).
Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 16, 111–120 (1980).
Benson, G. Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580 (1999).
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997).
Lagesen, K. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 35, 3100–3108 (2007).
Kalvari, I. et al. Rfam 14: expanded coverage of metagenomic, viral and microRNA families. Nucleic Acids Res. 49, D192–D200 (2021).
Gremme, G., Brendel, V., Sparks, M. E. & Kurtz, S. Engineering a software tool for gene structure prediction in higher organisms. Inform. Softw. Technol. 47, 965–978 (2005).
Grabherr, M. G. et al. Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644 (2011).
Brůna, T., Hoff, K. J., Lomsadze, A., Stanke, M. & Borodovsky, M. BRAKER2: automatic eukaryotic genome annotation with GeneMark-EP+ and AUGUSTUS supported by a protein database. NAR Genom. Bioinform. 3, lqaa108 (2021).
Kim, D., Paggi, J. M., Park, C., Bennett, C. & Salzberg, S. L. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915 (2019).
Pertea, M. et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295 (2015).
Haas, B. J. et al. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol. 9, 1–22 (2008).
Buchfink, B., Xie, C. & Huson, D. H. Fast and sensitive protein alignment using DIAMOND. Nat. Methods. 12, 59–60 (2015).
Zdobnov, E. M. & Apweiler, R. InterProScan–an integration platform for the signature-recognition methods in InterPro. Bioinformatics. 17, 847–848 (2001).
Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A. C. & Kanehisa, M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 35, W182–W185 (2007).
NCBI Sequence Read Archive. https://identifiers.org/ncbi/insdc.sra:SRP408505 (2023).
NCBI Genbank. https://identifiers.org/ncbi/insdc.gca:GCA_030347415.2 (2023).
Chen, L. The genome of Decapterus maruadsi. Figshare. https://doi.org/10.6084/m9.figshare.22574206.v3 (2023).
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V. & Zdobnov, E. M. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 31, 3210–3212 (2015).
Li, H. Minimap2: pairwise alignment for nucleotide sequences. Bioinformatics. 34, 3094–3100 (2018).
Marçais, G. et al. MUMmer4: A fast and versatile genome alignment system. PLoS Comput. Biol. 14, e1005944 (2018).
Acknowledgements
We acknowledge financial support from the Fundamental Research Funds for the Central Universities (No.20720200119).
Author information
Authors and Affiliations
Contributions
P.X. conceived and supervised the study. Z.X.Z., Y.C.D. and P.X. colledcted the sample. L.Y.C., Z.X.Z. and Z.Y.Z. performed bioinformatics analysis. L.Y.C., Z.X.Z. and J.Y.Y. drafted the manuscript. F.P. helped on manuscript preparation. P.X., T.Z. and Y.L.B. provided review and modification of the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, L., Zhou, Z., Zhou, Z. et al. Chromosome-level assembly and gene annotation of Decapterus maruadsi genome using Nanopore and Hi-C technologies. Sci Data 11, 69 (2024). https://doi.org/10.1038/s41597-024-02912-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41597-024-02912-1
- Springer Nature Limited