

Abstract
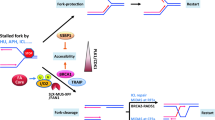
Fanconi anemia (FA) is a devastating hereditary disease characterized by bone marrow failure (BMF) and acute myeloid leukemia (AML). As FA-deficient cells are hypersensitive to DNA interstrand crosslinks (ICLs), ICLs are widely assumed to be the lesions responsible for FA symptoms. Here, we show that FA-mutated cells are hypersensitive to persistent replication stress and that FA proteins play a role in the break-induced-replication (BIR)-like pathway for fork restart. Both the BIR-like pathway and ICL repair share almost identical molecular mechanisms of 53BP1–BRCA1-controlled signaling response, SLX4- and FAN1-mediated fork cleavage and POLD3-dependent DNA synthesis, suggesting that the FA pathway is intrinsically one of the BIR-like pathways. Replication stress not only triggers BMF in FA-deficient mice, but also specifically induces monosomy 7, which is associated with progression to AML in patients with FA, in FA-deficient cells.








Similar content being viewed by others

Data availability
All data supporting the findings of this study are available from the corresponding author upon reasonable request. Source data are provided with this paper.
References
Ceccaldi, R., Sarangi, P. & D’Andrea, A. D. The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol. 17, 337–349 (2016).
Okamoto, Y., Hejna, J. & Takata, M. Regulation of R-loops and genome instability in Fanconi anemia. J. Biochem. 165, 465–470 (2019).
Langevin, F., Crossan, G. P., Rosado, I. V., Arends, M. J. & Patel, K. J. Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475, 53–58 (2011).
Hira, A. et al. Variant ALDH2 is associated with accelerated progression of bone marrow failure in Japanese Fanconi anemia patients. Blood 122, 3206–3209 (2013).
Hubal, E. A., Schlosser, P. M., Conolly, R. B. & Kimbell, J. S. Comparison of inhaled formaldehyde dosimetry predictions with DNA–protein cross-link measurements in the rat nasal passages. Toxicol. Appl. Pharmacol. 143, 47–55 (1997).
Duxin, J. P. & Walter, J. C. What is the DNA repair defect underlying Fanconi anemia? Curr. Opin. Cell Biol. 37, 49–60 (2015).
García-Calderón, C. B. et al. Genotoxicity of tetrahydrofolic acid to hematopoietic stem and progenitor cells. Cell Death Differ. 25, 1967–1979 (2018).
Oberbeck, N. et al. Maternal aldehyde elimination during pregnancy preserves the fetal genome. Mol. Cell 55, 807–817 (2014).
Rosado, I. V., Langevin, F., Crossan, G. P., Takata, M. & Patel, K. J. Formaldehyde catabolism is essential in cells deficient for the Fanconi anemia DNA-repair pathway. Nat. Struct. Mol. Biol. 18, 1432–1434 (2011).
Rochowski, A. et al. Patients with Fanconi anemia and AML have different cytogenetic clones than de novo cases of AML. Pediatr. Blood Cancer 59, 922–924 (2012).
Nagamachi, A. et al. Haploinsufficiency of SAMD9L, an endosome fusion facilitator, causes myeloid malignancies in mice mimicking human diseases with monosomy 7. Cancer Cell 24, 305–317 (2013).
Asou, H. et al. Identification of a common microdeletion cluster in 7q21.3 subband among patients with myeloid leukemia and myelodysplastic syndrome. Biochem. Biophys. Res. Commun. 383, 245–251 (2009).
Mehta, P. A. et al. Numerical chromosomal changes and risk of development of myelodysplastic syndrome–acute myeloid leukemia in patients with Fanconi anemia. Cancer Genet. Cytogen. 203, 180–186 (2010).
Howlett, N. G., Taniguchi, T., Durkin, S. G., D’Andrea, A. D. & Glover, T. W. The Fanconi anemia pathway is required for the DNA replication stress response and for the regulation of common fragile site stability. Hum. Mol. Genet. 14, 693–701 (2005).
Okamoto, Y. et al. Replication stress induces accumulation of FANCD2 at central region of large fragile genes. Nucleic Acids Res. 46, 2932–2944 (2018).
Chan, K. L., Palmai-Pallag, T., Ying, S. & Hickson, I. D. Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat. Cell Biol. 11, 753–760 (2009).
Naim, V. & Rosselli, F. The FANC pathway and BLM collaborate during mitosis to prevent micro-nucleation and chromosome abnormalities. Nat. Cell Biol. 11, 761–768 (2009).
García-Rubio, M. L. et al. The Fanconi anemia pathway protects genome integrity from R-loops. PLoS Genet. 11, e1005674 (2015).
Schwab, R. A. et al. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell 60, 351–361 (2015).
Schlacher, K., Wu, H. & Jasin, M. A distinct replication fork protection pathway connects Fanconi anemia tumor suppressors to RAD51-BRCA1/2. Cancer Cell 22, 106–116 (2012).
Suhasini, A. N. et al. Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J. 30, 692–705 (2011).
Tian, Y. et al. Constitutive role of the Fanconi anemia D2 gene in the replication stress response. J. Biol. Chem. 292, 20184–20195 (2017).
Chen, X., Bosques, L., Sung, P. & Kupfer, G. M. A novel role for non-ubiquitinated FANCD2 in response to hydroxyurea-induced DNA damage. Oncogene 35, 22–34 (2016).
Xu, Y. et al. 53BP1 and BRCA1 control pathway choice for stalled replication restart. Elife 6, https://doi.org/10.7554/eLife.30523 (2017).
Kramara, J., Osia, B. & Malkova, A. Break-induced replication: the where, the why, and the how. Trends Genet. 34, 518–531 (2018).
Hirota, K. et al. SUMO-targeted ubiquitin ligase RNF4 plays a critical role in preventing chromosome loss. Genes Cells 19, 743–754 (2014).
Minocherhomji, S. et al. Replication stress activates DNA repair synthesis in mitosis. Nature 528, 286–290 (2015).
Zhang, J. & Walter, J. C. Mechanism and regulation of incisions during DNA interstrand cross-link repair. DNA Repair 19, 135–142 (2014).
Wang, W. Emergence of a DNA-damage response network consisting of Fanconi anaemia and BRCA proteins. Nat. Rev. Genet. 8, 735–748 (2007).
Costantino, L. et al. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science 343, 88–91 (2014).
Dungrawala, H. et al. The replication checkpoint prevents two types of fork collapse without regulating replisome stability. Mol. Cell 59, 998–1010 (2015).
Alcón, P. et al. FANCD2–FANCI is a clamp stabilized on DNA by monoubiquitination of FANCD2 during DNA repair. Nat. Struct. Mol. Biol. 27, 240–248 (2020).
Wang, R., Wang, S., Dhar, A., Peralta, C. & Pavletich, N. P. DNA clamp function of the monoubiquitinated Fanconi anaemia ID complex. Nature 580, 278–282 (2020).
Tan, W. et al. Monoubiquitination by the human Fanconi anemia core complex clamps FANCI:FANCD2 on DNA in filamentous arrays. Elife https://doi.org/10.7554/eLife.54128 (2020).
Bunting, S. F. et al. BRCA1 functions independently of homologous recombination in DNA interstrand crosslink repair. Mol. Cell 46, 125–135 (2012).
Huang, J. et al. The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol. Cell 52, 434–446 (2013).
Eastman, A. Reevaluation of interaction of cis-dichloro(ethylenediamine)platinum(II) with DNA. Biochemistry 25, 3912–3915 (1986).
Vandenberg, C. J. et al. BRCA1-independent ubiquitination of FANCD2. Mol. Cell 12, 247–254 (2003).
Garcia-Higuera, I. et al. Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol. Cell 7, 249–262 (2001).
Long, D. T., Joukov, V., Budzowska, M. & Walter, J. C. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol. Cell 56, 174–185 (2014).
Houghtaling, S. et al. Epithelial cancer in Fanconi anemia complementation group D2 (Fancd2) knockout mice. Genes Dev. 17, 2021–2035 (2003).
Dubois, E. L. et al. A Fanci knockout mouse model reveals common and distinct functions for FANCI and FANCD2. Nucleic Acids Res. 47, 7532–7547 (2019).
Parmar, K., D’Andrea, A. & Niedernhofer, L. J. Mouse models of Fanconi anemia. Mutat. Res. 668, 133–140 (2009).
Van den Berg, C. L. et al. Pharmacokinetics of hydroxyurea in nude mice. Anti-Cancer Drugs 5, 573–578 (1994).
Durkin, S. G. & Glover, T. W. Chromosome fragile sites. Annu. Rev. Genet. 41, 169–192 (2007).
Wu, R. A. et al. TRAIP is a master regulator of DNA interstrand crosslink repair. Nature 567, 267–272 (2019).
Sonneville, R. et al. TRAIP drives replisome disassembly and mitotic DNA repair synthesis at sites of incomplete DNA replication. Elife 8, https://doi.org/10.7554/eLife.48686 (2019).
Budzowska, M., Graham, T. G., Sobeck, A., Waga, S. & Walter, J. C. Regulation of the Rev1–pol ζ complex during bypass of a DNA interstrand cross-link. EMBO J. 34, 1971–1985 (2015).
McVey, M., Khodaverdian, V. Y., Meyer, D., Cerqueira, P. G. & Heyer, W. D. Eukaryotic DNA polymerases in homologous recombination. Annu. Rev. Genet. 50, 393–421 (2016).
Yamaguchi-Iwai, Y. et al. Homologous recombination, but not DNA repair, is reduced in vertebrate cells deficient in RAD52. Mol. Cell. Biol. 18, 6430–6435 (1998).
Garribba, L. et al. Folate stress induces SLX1- and RAD51-dependent mitotic DNA synthesis at the fragile X locus in human cells. Proc. Natl Acad. Sci. USA 117, 16527–16536 (2020).
Rickman, K. A. et al. Distinct roles of BRCA2 in replication fork protection in response to hydroxyurea and DNA interstrand cross-links. Genes Dev. 34, 832–846 (2020).
Luebben, S. W., Kawabata, T., Johnson, C. S., O’Sullivan, M. G. & Shima, N. A concomitant loss of dormant origins and FANCC exacerbates genome instability by impairing DNA replication fork progression. Nucleic Acids Res. 42, 5605–5615 (2014).
Flach, J. et al. Replication stress is a potent driver of functional decline in ageing haematopoietic stem cells. Nature 512, 198–202 (2014).
Alvarez, S. et al. Replication stress caused by low MCM expression limits fetal erythropoiesis and hematopoietic stem cell functionality. Nat. Commun. 6, 8548 (2015).
Flach, J. & Milyavsky, M. Replication stress in hematopoietic stem cells in mouse and man. Mutat. Res. 808, 74–82 (2018).
Ahuja, A. K. et al. A short G1 phase imposes constitutive replication stress and fork remodelling in mouse embryonic stem cells. Nat. Commun. 7, 10660 (2016).
Dingler, F. A. et al. Two aldehyde clearance systems are essential to prevent lethal formaldehyde accumulation in mice and humans. Mol. Cell 80, 996–1012.e9 (2020).
Shen, X. et al. A surge of DNA damage links transcriptional reprogramming and hematopoietic deficit in Fanconi anemia. Mol. Cell 80, 1013–1024.e6 (2020).
Hodskinson, M. R. et al. Alcohol-derived DNA crosslinks are repaired by two distinct mechanisms. Nature 579, 603–608 (2020).
Escribano-Díaz, C. et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol. Cell 49, 872–883 (2013).
Hirota, K. et al. The POLD3 subunit of DNA polymerase δ can promote translesion synthesis independently of DNA polymerase ζ. Nucleic Acids Res. 43, 1671–1683 (2015).
Seki, S. et al. A requirement of FancL and FancD2 monoubiquitination in DNA repair. Genes Cells 12, 299–310 (2007).
Hoa, N. N. et al. Relative contribution of four nucleases, CtIP, Dna2, Exo1 and Mre11, to the initial step of DNA double-strand break repair by homologous recombination in both the chicken DT40 and human TK6 cell lines. Genes Cells 20, 1059–1076 (2015).
Gao, S. et al. An OB-fold complex controls the repair pathways for DNA double-strand breaks. Nat. Commun. 9, 3925 (2018).
Xue, Y., Li, Y., Guo, R., Ling, C. & Wang, W. FANCM of the Fanconi anemia core complex is required for both monoubiquitination and DNA repair. Hum. Mol. Genet. 17, 1641–1652 (2008).
Feng, S. et al. Ewing tumor-associated antigen 1 interacts with replication protein A to promote restart of stalled replication forks. J. Biol. Chem. 291, 21956–21962 (2016).
Acknowledgements
We thank W. Wang for his advice about the paper and antibodies, J. Huang for BRCA1- and FAN1-expressing plasmids and L. Zuo for the SLX4-expressing plasmid. We thank the Imaging Core at the National Center for Protein Sciences at Peking University. This work was supported in part by the Beijing Outstanding Young Scientist Program (BJJWZYJH01201910001005) to Q.L.; the National Natural Science Foundation of China (81672773 and 31870807) to D.X. and Rong Guo; a China Postdoctoral Science Foundation grant (no. 2018M641078) and the National Natural Science Foundation of China (grant no. 31900928) to Y.X.; JSPS KAKENHI (grant no. JP16H06306 to S.T. and JP19H04267 to H.S.) and the JSPS Core-to-Core Program to S.T.
Author information
Authors and Affiliations
Contributions
X.X. performed the comet assays, a portion of the drug-sensitivity assays and chromosome spread, the immunoprecipitations, FISH and the mouse experiments. Y.X. performed the immunofluorescence experiments, UFBs staining, most of the drug-sensitivity assays and DNA combing assays and a portion of the MiDAS assays, chromosome spread and the fractionation assays. Ruiyuan Guo and R.X. performed the fractionation assays, a portion of the MiDAS experiments and the DNA combing assays. C.F. generated the plasmids for expressing truncated SLX4 and BRCA1. M.X. performed a portion of the drug-sensitivity assays. H.S. and S.T. generated FANCC-knockout TK6 cells and provided some DT40 knockout cells. M.T. generated most FA-deficient DT40 cells. Q.L., Rong Guo and D.X. designed experiments and interpreted the results. X.X. and D.X. wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Structural & Molecular Biology thanks the anonymous reviewers for their contribution to the peer review of this work. Beth Moorefield was the primary editor on this article and managed its editorial process and peer review in collaboration with the rest of the editorial team.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Extended data
Extended Data Fig. 1 FA-deficient cells are hypersensitive to persistent replication stress.
a, Cisplatin sensitivity of the fancC- DT40 cells or chicken FancC (chFancC)-complemented fancC- cells assessed using MTT staining. The mean and s.d. from three independent experiments are shown. b, HU, APH and cisplatin sensitivity of the fancL-/- DT40 cells or human FancL (huFancL)-complemented fancL-/- cells assessed using MTT staining or by colony formation assay. The mean and s.d. from three independent experiments are shown. c, HU sensitivity of the FancL-/- or FancLkf/kf mouse GM by colony formation assay. The mean and s.d. from three independent experiments are shown except the experiment of FancLkf/kf mouse GM in the right panel where n = 2. Statistical source data are provided in Source data.
Extended Data Fig. 2 Generation of FANCC-/- TK6 cells and FANCA-/- HCT116 cells.
a, Schematics showing FANCC knockout in TK6 cells using CRISPR. b, Western-blotting showing protein level of FANCC in wild-type and FANCC-/- TK6 cells. c, Schematic representation of the generation of FANCA-/- HCT116 cells using CRISPR. Guide sequences are highlighted in blue. PAM sequences are indicated by red lines. The red arrow indicates a putative cleavage site. Red dashes indicate deleted bases. d, Immunoblots showing the expression levels of FANCA in knockout cells. Uncropped images of the are provided in Source data.
Extended Data Fig. 3 Low doses of HU induce chromosome loss over time, but not a significant increase of chromosome aberration in FANCC-deficient cells.
a, b, Chromosome aberrations in wild-type and FANCC-deficient DT40 (a) and TK6 (b) cells over time. Cells were persistently treated with 100 μM (a) or 40 μM (b) HU. The mean and s.d. from three independent experiments are shown. For each sample from each experiment, 100 metaphase cells were scored. c, Replicated experiments of chromosome number analysis in wild-type and FANCC-/- TK6 cells over time. Cells were cultured with medium containing 40 μM HU for 10 days. 100 metaphases were counted for each sample. Statistical source data are provided in Source data.
Extended Data Fig. 4 FANCD2 and FANCL act in the same BIR pathway as BRCA1 for restart of stalled forks.
a, Experimental workflows for MiDAS measurement. b, f, Images (left panel) and quantifications (right panel) showing MiDAS (EdU foci, green) rates in FANCD2- (b) and FANCL- (f) depleted BRCA1-/- cells. Scale bar, 5 μm. The mean and s.d. of three independent experiments are shown. c, g, j, Stalled replication fork restart rates in FANCD2- (c), FANCL- (g) and POLD3- (j) depleted wild-type or BRCA1-/- cells. The mean and s.d. from three independent experiments are shown. d, h, Comet assays measuring DSBs in FANCD2- (d) and FANCL- (h) depleted BRCA1-/- cells. The mean and s.e.m. are shown; the numbers of cells examined are indicated and data are representative of three independent experiments. *** P < 0.001, ** P < 0.01, * P < 0.05, ns P > 0.05, two-tailed Student’s t-test. e, i, k, Immunoblots showing the knockdown efficiency of FANCD2 (e), FANCL (i) and POLD3 (k) in wild-type or BRCA1-/- HCT116 cells. Uncropped images of the immunoblots and statistical source data including the precise P values are provided in Source data.
Extended Data Fig. 5 BRCA1 and SLX4 interact each other through their N-terminal regions.
a, schematic representation of the different BRCA1 deletion mutants (left) and their ability to coimmunoprecipitate with GFP-SLX4 from HEK293 extracts (right). b, c, Immunoprecipitation and Western blotting to assess whether the various deletion mutants of BRCA1 described in (a) coimmunoprecipitated with GFP-SLX4. S protein-FLAG-Streptavidin binding peptide (SFB)-tagged BRCA1 mutants and GFP-SLX4 were co-expressed in HEK293 cells. d-g, Immunoprecipitation and Western blotting to assess whether the various deletion mutants of SLX4 described in Fig. 4b coimmunoprecipitated with BRCA1. SLX4 mutants were fused with a SFB tag and expressed in HEK293. Uncropped images of the immunoblots are provided in Source data.
Extended Data Fig. 6 ICLs, but not mono-adducts, induce accumulation of DSBs during repair.
Cells were exposed to UV (365-nm, 12 W; 10 cm from the cells) for the indicated times after incubation with psoralen (10 μg/ml) or angelicin (10 μg/ml) for 30 min and were harvested for use in comet assays after 8 h. The mean and s.e.m. are shown; the numbers of cells examined are indicated and data are representative of three independent experiments. **** P < 0.0001, two tailed Student’s t-test. Statistical source data including the precise P values are provided in Source data.
Extended Data Fig. 7 FANCD2 and POLD3 act in the same pathway to repair ICLs.
a, Cisplatin sensitivity of various DT40 knockout cells. b, Graphic showing the cisplatin and MMC sensitivity of POLD3-depleted PD20 or FANCD2-complemented PD20 cells. The mean and s.d. from three independent experiments are shown. Statistical source data are provided in Source data.
Extended Data Fig. 8 Generation of FancL knockout mice.
a, Schematic representation of the wild-type and targeted genomic DNA in the FancL gene. Arrows indicate locations targeted by primers for genomic PCR. b, c, Genomic PCR analysis to show that exon 6 is replaced by a knockout-first cassette or is undetectable in the FancLkf/kf (b) or FancL-/- (c) mice. d, mRNA level of FancL in FancLkf/kf mice. The mean and s.d. from three independent experiments are shown. e, Observed and expected birth numbers of FancLkf/kf mice. f, Weight of wild-type and FancLkf/kf mice at 3 or 8 weeks of age. The mean and s.d. are shown. * P < 0.05, ns P > 0.05, two tailed Student’s t-test; n = 10 biologically independent mice. Uncropped images of the gels and statistical source data including the precise P values are provided in Source data.
Extended Data Fig. 9 Chromosome number analysis after MMC or APH treatment.
Cells were persistently treated with APH (40 nM for 6 days) or pulsed with MMC (40 ng/mL for 1 day). 100 metaphases for each sample in each independent experiment were counted. Experiments were replicated three times. These experiments were carried out together with that in Fig. 1f and the same control samples were used. Statistical source data are provided in Source data.
Supplementary information
Source data
Source Data Fig. 1
Statistical source data and calculation of statistical values.
Source Data Fig. 2
Statistical source data and calculation of statistical values.
Source Data Fig. 2
Unprocessed western blots and/or gels.
Source Data Fig. 3
Statistical source data and calculation of statistical values.
Source Data Fig. 3
Unprocessed western blots and/or gels.
Source Data Fig. 4
Statistical source data and calculation of statistical values.
Source Data Fig. 4
Unprocessed western blots and/or gels.
Source Data Fig. 5
Statistical source data and calculation of statistical values.
Source Data Fig. 6
Statistical source data and calculation of statistical values.
Source Data Fig. 7
Statistical source data and calculation of statistical values.
Source Data Extended Data Fig. 1
Statistical source data and calculation of statistical values.
Source Data Extended Data Fig. 2
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 3
Statistical source data and calculation of statistical values.
Source Data Extended Data Fig. 4
Statistical source data and calculation of statistical values.
Source Data Extended Data Fig. 4
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 5
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 6
Statistical source data and calculation of statistical values.
Source Data Extended Data Fig. 7
Statistical source data and calculation of statistical values.
Source Data Extended Data Fig. 8
Statistical source data and calculation of statistical values.
Source Data Extended Data Fig. 8
Unprocessed western blots and/or gels.
Source Data Extended Data Fig. 9
Statistical source data and calculation of statistical values.
Rights and permissions
About this article

Cite this article
Xu, X., Xu, Y., Guo, R. et al. Fanconi anemia proteins participate in a break-induced-replication-like pathway to counter replication stress. Nat Struct Mol Biol 28, 487–500 (2021). https://doi.org/10.1038/s41594-021-00602-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41594-021-00602-9
- Springer Nature America, Inc.
This article is cited by
-
Replication stress as a driver of cellular senescence and aging
Communications Biology (2024)
-
FANCD2 promotes mitotic rescue from transcription-mediated replication stress in SETX-deficient cancer cells
Communications Biology (2022)
-
The emergence of a unified mechanism in the Fanconi anemia pathway
Genome Instability & Disease (2021)