
Abstract
Interstrand crosslinks (ICLs) are a highly toxic form of DNA damage. ICLs can interfere with vital biological processes requiring separation of the two DNA strands, such as replication and transcription. If ICLs are left unrepaired, it can lead to mutations, chromosome breakage and mitotic catastrophe. The Fanconi anemia (FA) pathway can repair this type of DNA lesion, ensuring genomic stability. In this review, we will provide an overview of the cellular response to ICLs. First, we will discuss the origin of ICLs, comparing various endogenous and exogenous sources. Second, we will describe FA proteins as well as FA-related proteins involved in ICL repair, and the post-translational modifications that regulate these proteins. Finally, we will review the process of how ICLs are repaired by both replication-dependent and replication-independent mechanisms.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Our genome is constantly exposed to damage caused by both endogenous and exogenous sources. ICLs (interstrand crosslinks) are one of the most cytotoxic lesions because the two Watson and Crick strands of DNA are covalently bound together, causing an obstacle to replication and transcription. ICLs left unrepaired can lead to mutations, chromosome breakage, chromosome missegregation and mitotic catastrophe. To protect the genome from this type of lesion, the cells count on a highly complex repair pathway to detect the lesion, activate the cell cycle checkpoint and repair the ICLs. ICLs can be generated by naturally occurring compounds, such as psoralen and mitomycin C, as well as by chemically synthesized crosslinking agents, such as cisplatin. ICL-forming drugs are widely used as chemotherapeutic drugs against cancer. Reactive aldehydes have been shown to be one of the endogenous sources causing crosslinks. The chemical structure of the resulting ICL depends on the crosslinking agent implicated, and these different structures will lead to different cellular responses. The response to ICLs triggers a complex DDR (DNA damage response) including the Fanconi anemia (FA) pathway as well as the ATR (ATM and Rad3-related)/Chk1 pathway. Therefore, along the FA pathway, several signal transduction events take place mediated by multiple modifications including phosphorylation, ubiquitination and SUMOylation (small ubiquitin-like modifier) events. In order to fully repair the ICL, the FA pathway coordinates different processes including translesion synthesis (TLS), homologous recombination (HR) and nucleotide excision repair (NER). Therefore, there is extensive crosstalk between different DNA repair pathways during ICL repair.
Origin of ICLs
ICLs are formed when the two strands of DNA are covalently bound together through a linker molecule commonly known as a crosslinking agent. The first crosslinking agents to be identified were the nitrogen mustards developed during the warfare of the early twentieth century. A better application for these compounds was found as chemotherapeutic agents, though their mechanism of action was still unknown. Later other compounds such as mitomycin C and cisplatin joined them as chemotherapeutic agents and they were all found to be crosslinking agents [1]. They can react with DNA and give rise to different kinds of products including DNA monoadducts, intrastrand crosslinks and ICLs with variable efficiencies. They also differ in their base specificity for ICL formation and the degree of distortion in the DNA double helix they generate. Some of these compounds, such as psoralens, which are produced by certain plants, may play an important role as environmental sources of ICLs. However, the search for endogenous sources of ICLs has rendered very interesting results in the past few years, suggesting that reactive aldehydes are one of the endogenous crosslinking agents [2]. Some candidates include products derived from lipid peroxidation such as malondialdehyde and crotonaldehyde, but also nitric oxide has been proposed [3].
Nitrogen mustards
The most simple nitrogen mustard and the first to be used as a chemotherapeutic agent is mechlorethamine (bis(2-chloroethyl)methylamine) (Fig. 1a). Nitrogen mustards are bifunctional alkylating agents, thus their chloroethyl moieties can bind two bases on opposite strands of DNA. They bind guanine N7 forming a monoadduct leading then to binding a second guanine on the opposite strand in the sequence GpNpC (Fig. 1a). Other nitrogen mustards, such as melphalan and chlorambucil, substitute the methyl group with aromatic groups and they can also bind adenine N3 [4]. The generated ICLs, which only account to up to 5 % of the products, cause a distortion of the double helix with an unwinding of 2°–6° and a bend of around 10° per ICL because of the shortening needed to accommodate the N7–N7 bond [5, 6]. Recently, nitrogen mustards have been engineered into activatable prodrugs. These new types of aromatic nitrogen mustards generate ICLs only in the presence of H2O2 providing a promising tool for the treatment of tumours in a highly oxidative environment [7].

Schematic representation of the chemical structure of the main crosslinking agents and the ICLs they form. a Mechlorethamine (nitrogen mustard), b mitomycin C, c cisplatin, d psoralen, e BCNU (nitrosourea), f diepoxybutane, g aldehydes [acetaldehyde, acrolein and crotonaldehyde (R = CH3)] and h nitric oxide. Crosslinking agents are shown in red
Mitomycin C
Mitomycin C (MMC) is a natural compound produced by Streptomyces caespitosus. It is unable to bind DNA directly but needs to be metabolically reduced beforehand. This need for a reduction step for its activation makes MMC especially fit as a chemotherapeutic agent since the tumour micro-environment is generally hypoxic [8]. After reduction it specifically reacts with the N2 in guanine in the sequence CpG and its complementary strand to form an ICL (Fig. 1b) [8]. However, it can also form monoadducts through the N7 of guanine [9]. ICLs constitute around 15 % of the products (the rest being 50 % monoadducts and 35 % intrastrand crosslinks) [10]. MMC binds to the N2 of guanines through the minor groove and it causes only a minor distortion of the double helix [11].
Platinum compounds
Cisplatin [cis-diamminedichloroplatinum(II)] was first described as a compound inhibiting bacteria growth in 1965 [12]. It reacts with purine residues to form intrastrand and interstrand crosslinks. The intrastrand crosslinks are formed at sequences GpG and ApG with a preference for the former (65 and 25 % intrastrand crosslinks of total adducts formed, respectively). ICLs are formed with a lower frequency of around 5–8 %. ICLs are formed specifically at GpC sites and bind N7 of guanine (Fig. 1c) [13]. Both crystallographic and NMR structural models have shown cisplatin ICLs to provoke a large distortion in the DNA double helix. These ICLs induce the extrusion of two cytosines in the GC/CG sequence while the platinum locates itself in the minor groove. The double helix suffers an unwinding of 110° and a bent towards the minor groove of 47° (Fig. 2b) [13, 14]. Other platinum compounds that have also been studied and used as chemotherapeutic agents include carboplatin and transplatin. Carboplatin differs from cisplatin in the chloride groups, which are substituted with cyclobutyldicarboxylate. Carboplatin has less reactivity than cisplatin, but it behaves similarly regarding generation of ICLs and their frequency [15]. Transplatin, however, has been shown to induce only minor distortion of the double helix when forming ICLs [16]. Moreover, by substituting the ammine groups in transplatin with the planar bases quinoline or thiazole the frequency of ICLs among the platinated products greatly increased to around 30 % [17].

Structures of various ICLs. a B-DNA and the ICLs formed by b cisplatin, c psoralen, d BCNU and e acetaldehyde and crotonaldehyde viewed from the major groove (left) or the minor groove (right). The crosslinked bases and the crosslinking agents are shown in red. Structures taken from PDB, accession numbers: B-DNA (1-BNA) [140], cisplatin (1A2E) [13], psoralen (204D) [21], BCNU (2MH6) [23], acetaldehyde (2HMD) [29]
Psoralens
Psoralens belong to a family of molecules called furocoumarins. These compounds are produced by at least eight families of plants including Apiaceae and Fabaceae [18]. The planar and hydrophobic nature of these molecules allows them to easily penetrate the cell and intercalate the DNA bases. However, they are unable to form ICLs until irradiated with UVA (ultraviolet light), which induces covalent bonds to thymines on the sequence TpA on opposite strands (Fig. 1d). Psoralens are very effective inducers of ICLs with around 40 % of adducts generated being ICLs. However, a derivative of psoralen, trimethylpsoralen (TMP) can form up to 90 % ICLs [19]. This is due to the incapacity to form intrastrand crosslinks since psoralen must first intercalate the bases on opposing strands of DNA before being photo-activated [20]. The ICLs generated do not bend the DNA double helix and only provoke a minor unwinding of around 25° (Fig. 2c) [21].
Nitrosoureas
Nitrosoureas such as BCNU (1,3-bis(2-chloroethyl)-1-nitrosourea) are able to react with guanine and cytosine after metabolic activation. Their preferred site for binding is the N7 of guanine, although the O6 of guanine can also be attacked. Through this interaction BCNU can form intrastrand crosslinks between adjacent guanines. ICLs are formed in vitro when BCNU attacks O6 of guanine and N3 of cytosine on opposing strands though these adducts are minor products [22]. Other ICLs formed includes the binding of N1 of guanine with N3 of cytosine, which has been observed in vivo in the treatment of brain cancers (Fig. 1e). The structure of this crosslink has been studied through NMR and was found to be well accommodated in the double helix with very minor alterations (Fig. 2d) [23].
Diepoxybutane
1,2,3,4-Diepoxybutane (DEB) is a product of the biotransformation of 1,3-butadiene, a contaminating gas produced in the plastic and rubber industry. DEB is a bifunctional alkylating agent and reacts with DNA to produce monoadducts, ICLs, single-strand breaks and also DNA–protein crosslinks. However, in vivo ICLs are the main product responsible for its cytotoxicity [24]. DEB preferentially reacts with N7 and N1 in guanine, although ICLs are formed through N7–N7 at the sequence GpCpC similarly to the nitrogen mustard mechlorethamine (Fig. 1f) [25]. The bridge formed only contains four carbon atoms, which has been hypothesized to produce a major distortion on DNA. Through gel retardation experiments, it has been shown to lead to a bending of around 34° towards the major groove [26].
Endogenous crosslinking agents
Endogenous ICLs are especially difficult to study. Most evidence of endogenous crosslinking agents comes from in vitro studies or assessment of their mutagenicity [3]. Reactive aldehyde has been thought to be the major endogenous crosslinking agent. One such aldehyde is acetaldehyde. It was shown that acetaldehyde poses similar cellular toxicity to FA-deficient cells compared to other crosslinking agents suggesting that the FA pathway is required for the repair of acetaldehyde-derived damage [2]. Acetaldehyde can be derived from the metabolism of ethanol. It is able to react with guanine on DNA and, after a reduction step, form N2-ethyl-2′-deoxyguanosine. Although this compound is the major adduct formed by acetaldehyde, it cannot form ICLs. Two acetaldehyde molecules can also react with guanine to form 1-N2-propano-2′-deoxyguanosine, though the presence of basic molecules such as histones is needed. This compound is also generated by crotonaldehyde and it can exist in a cyclic or open chain configuration. In the open chain form the free aldehyde group can induce ICLs and DNA–protein crosslinks [27]. These ICLs are generally found in a CpG sequence but DNA–protein crosslinks constitute the main type of modification generated by acetaldehyde (Fig. 1g) [28]. These ICLs induced at CpG sequences by either crotonaldehyde or acetaldehyde are located in the minor groove and do not disturb the Watson–Crick pairing of the bases implicated (Fig. 2e) [29].
Another endogenous source of ICL formation is lipid peroxidation from oxidative stress, a process which is promoted by a fat-rich diet in mice and potentially in humans [30]. Lipid peroxidation leads to the production of malondialdehyde (MDA). MDA can react with guanine, adenine and cytosine though the main ICL produced is between the guanines in the sequence CpG [31]. Other products of lipid peroxidation include unsaturated aldehydes such as acrolein and crotonaldehyde. These can also come from exogenous sources such as cigarette smoke and automobile exhaust [32]. These aldehydes can react with nitrogen bases either through the carbonyl group or the double bond. The conjugate addition is followed by cyclization onto the base to generate a monoadduct. The ICL formed is present in CpG sequences as well and does not disturb the structure of the double helix [32, 33].
Nitric oxide (NO) has also been shown to induce ICLs. It generates ICLs between two guanine residues in the CpG sequence, which are bound by a common N2 amine group (Fig. 1h). This reaction might be favoured by the presence of methylated cytosines [34].
The Fanconi anemia pathway
Our knowledge of an ICL repair pathway originates from studies of an autosomal recessive disease called Fanconi anemia (FA). FA is a rare genetic disorder with an incidence of 1/200,000–1/400,000 in the general population [35]. FA is characterized by developmental abnormalities and early bone marrow failure, which leads to aplastic anaemia. FA patients are susceptible to various types of cancer, most often acute myelogenous leukaemia (AML). The mechanism behind bone marrow failure in FA is thought to be related to an excessive inflammatory response and apoptosis mediated by tumour necrosis factor α (TNFα), IFNγ and reactive oxygen species (ROS) [36]. To date, 19 FA genes have been identified. Mutation in these genes accounts for 95 % of the FA patients. Patients are sensitive to ICL-forming agents, such as mitomycin C, due to the cellular failure to repair ICLs. There is growing evidence that the symptoms observed in FA patients are also related to this defect in DNA repair. For instance, the bone marrow failure characteristic of FA patients could be originated from a defect in ICL repair in hematopoietic stem cells exposed to endogenous crosslinking agents such as formaldehyde [37]. This defective hematopoiesis leads to cell death, injury and generates an inflammatory response as previously observed, which further enhances bone marrow failure through apoptosis, production of ROS and inhibition of stem cell function [37, 38].
These 19 genes encode proteins, which together with non-FA proteins as well as proteins from other DNA repair pathways, including homologous recombination (HR), nucleotide excision repair (NER) and translesion synthesis (TLS), coordinate the detection and repair of ICLs as well as activation of the cell cycle checkpoint (Table 1) [35, 39]. The 19 FA proteins can be divided into three groups according to their functions in the pathway: the FA core complex, the FANCD2/FANCI complex and the effector proteins (Table 1).
First, eight FANC proteins (FANCA, FANCB, FANCC, FANCE, FANCF, FANCG, FANCL and FANCM) and three associated proteins (FAAP20, FAAP24 and FAAP100) form the FA core complex. Among other proteins that bind to some components of the core complex as part of the core complex or forming independent complexes, we find BLM (Bloom syndrome helicase), Topo IIIα (topoisomerase IIIα), RPA (replication protein A) and MHF1/2 (histone fold heterodimer) [40–42]. BLM, Topo IIIα and RPA interact with FANCA, FANCC, FANCE, FANCF and FANCG in a complex named BRAFT. This complex potentially plays a role in the FA pathway since BLM deficiency leads to sensitivity to MMC [41, 42]. On the other hand, MHF1/2 bind to FANCM and are recruited to replication forks stalled by ICLs. MHF1/2 are also needed for resistance to ICLs and promote FANCD2 monoubiquitination [40]. However, there is some debate regarding whether FANCM can be considered an FA protein. The controversy arises from the fact that the first FA patient identified with biallelic mutations in FANCM also had alterations on FANCA [43] and also the observation that individuals with homozygous loss of function of FANCM did not display FA symptoms [44]. Despite these observations, FANCM is usually included as an FA protein and a component of the core complex. The FA core complex together with FANCT (UBE2T) [45–47], a ubiquitin-conjugating enzyme, is responsible for monoubiquitination of the FANCD2/FANCI complex. FANCL, an E3 ligase, is the catalytic enzyme carrying out the ubiquitination. While mutations in some of the FA core complex members, such as FANCA, FANCC and FANCG, account for 85 % of the FA patients worldwide, the exact function of these members remains elusive. FANCL and UBE2T are sufficient to monoubiquitinate FANCD2/FANCI complex in vitro [48–50] and, for instance in silkworm; there is an active FA pathway in the absence of the FA core complex [51]. Loss of different FA core complex members causes variable degrees of sensitivity towards crosslinking agents. Some of the FA core complex members are predicted to be entirely helical and have no known conventional domains [52], which makes it difficult to speculate on their molecular functions. Recently, it was shown that a minimal subcomplex containing FANCB, FANCL and FAAP100 is required for robust FANCD2 monoubiquitination in DT40 cells and in vitro [53]. The rest of the FA core complex can be divided into two subcomplexes, FANCA-FANCG-FAAP20 and FANCC-FANCE-FANCF. Their presence facilitates the activity and the recruitment of the whole FA core complex onto DNA [54]. Nonetheless, this function would be redundant with that of the translocase FANCM [55]. The integrity of the FA core complex is also modulated by post-translational modifications, e.g. phosphorylation by ATR/Chk1 and ubiquitination.
Second, the FANCD2/FANCI complex resides at the heart of the FA pathway. It is monoubiquitinated by the FA core complex and is recruited to the ICLs. This is a critical step for the ICL repair. If there is no monoubiquitination, there will be no subsequent repair of the ICL. The function of monoubiquitinated FANCD2/FANCI complex is not fully understood. It is thought to orchestrate the recruitment of the downstream effector proteins to the ICL. In addition to monoubiquitination, the FANCD2/FANCI complex is also regulated by other post-translational modifications that we will discuss extensively in a later section.
Third, FANCD1 (BRCA2), FANCJ (BRIP1), FANCN (PALB2), FANCO (RAD51C), FANCP (SLX4), FANCQ (XPF), FANCR (RAD51) and FANCS (BRCA1) are the effector proteins that contribute to the ICL repair at later stages (Table 1). BRCA1, BRCA2, BRIP1, PALB2, RAD51 and RAD51C have been known for their roles in homologous recombination, which plays an important part in the FA pathway. Mutations in either BRCA1 or BRCA2 lead to higher risk of breast and ovarian cancer [56]. Recently, mutations in other FA genes such as BRIP1, PALB2 and RAD51C have been associated with an intermediate risk of breast cancer [57, 58]. FANCJ was shown to interact with BLM, promoting its stability. This interaction is probably distinct from the BRAFT complex and plays a potential role in the response to replication stress [59, 60]. SLX4 is a nuclease scaffold protein interacting with several nucleases including XPF/ERCC1, MUS81/EME1 and SLX1. However, XPF is thought to be especially important in ICL unhooking.
In addition to the 19 FA proteins and 3 FA associated proteins, there are other non-FA proteins that have been shown to participate in the ICL repair. For example, UHRF1 has been proposed to recognize ICLs in vivo and in vitro [61, 62]. FAN1 (Fanconi-associated nuclease 1) has been shown to be one of the nucleases important for the ICL repair [63–67]. SNM1A is another nuclease that has been demonstrated to participate in the ICL repair [68, 69].
Although the focus of this review is the response to ICLs, alternative roles for the FA proteins are emerging in recent years. There is growing evidence for the role of FA proteins in replication fork protection and recovery after stalling, whether caused by ICLs or other genomic stresses. Monoubiquitinated FANCD2 has been shown to recruit the nuclease FAN1, as well as other FA mediators such as BLM, FANCJ and BRCA2, independently of the core complex, to promote fork recovery and genomic stability [70–72]. Another source of genomic instability are the ultra-fine DNA bridges or UFBs that interlink chromosomes during mitosis. These UFBs are thought to arise from common fragile loci that associate with FANCD2 and FANCI even through mitosis when BLM is also found at the UFBs. These proteins are thought to contribute to the resolution of the UFBs ensuring a correct chromosomal segregation, but the exact mechanism remains unclear [73, 74]. FA proteins have also been associated with the processing of transcription associated DNA: RNA hybrids, also known as R-loops, and the stabilization of replication forks stalled by these structures [75]. Mainly, FANCM was found to resolve R-loops through its translocase activity and, surprisingly, aldehydes were observed to induce R-loops, adding another by-product of their activity to the different adducts already discussed [75].
Post-translational modifications of the FA proteins
Repair of an ICL is a highly complex process involving the FA pathway as well as other repair pathways. Post-translational modifications (PTMs) play an essential role in the regulation of this process. Depending on the type of modification, PTMs can cause protein conformation or surface charge changes or could establish new protein–protein interactions that trigger signal transduction or degradation. There are many PTM events in the FA pathway identified over the past decade (Table 2). One of the keystones is the monoubiquitination of FANCD2 that is required for its localization at the ICLs. However, this modification is preceded by several phosphorylation events on different proteins and mainly mediated through ATR/ATM (ataxia telangiectasia mutated) kinases and downstream target kinases Chk1 and Chk2. Additionally, recently light has been shed on the role of SUMOylation in the FA pathway [76, 77] as well as on the termination events that shut off the pathway in a timely manner once the repair has been completed (Fig. 3). It should be noted that many of the PTMs identified so far for the effector proteins implicated in HR have been linked to DSB (double strand break) repair and not to ICLs. However, these mechanisms could play similar roles in response to crosslinking agents as well.

Diagram of the main posttranslational modification events involved in the activation of response to ICLs. Once the ICL is detected it triggers the ATR/Chk1 pathway leading to the phosphorylation of several components of the FA core complex. ATR and potentially other kinases phosphorylate FANCI and FANCD2 (1) priming the complex for its monoubiquitination. These phosphorylation events lead then to the monoubiquitination of the FANCD2/FANCI complex by FANCL/UBE2T (2), which promotes its recruitment onto chromatin and the action of the effector proteins. On the other hand, the dosage of the FANCD2/FANCI complex on chromatin can be regulated through SUMOylation-dependent polyubiquitination mediated by PIAS1/4, UBC9 and RNF4 (3, 4). Finally, these events can be reversed by the action of a hypothetical deubiquitinase (5), SENP6 (6), the deubiquitinating enzyme USP1/UAF1 complex (7) as well as putative phosphatases (8) still unidentified
Phosphorylation
The FA pathway relies on several phosphorylation events of different proteins in the core complex and the FANCD2/FANCI heterodimer leading to the monoubiquitination of FANCD2 (Fig. 3). Two kinases are at the centre of the DNA damage response: ATM and ATR that phosphorylate several proteins involved in DNA repair, including other kinases such as Chk1 and Chk2. Although ATM phosphorylates FANCD2 on its S222 in vitro and in human cells, this event is not necessary for its monoubiquitination or ICL repair. Rather, the phosphorylation event regulates the S-phase checkpoint, which inhibits DNA replication after IR treatment (Table 2) [78]. However, ATR was found to be required for FANCD2 monoubiquitination upon MMC or IR treatment. The absence of ATR also abrogated FANCD2 foci formation and led to chromosomal abnormalities in cells derived from a patient with Seckel syndrome [79]. Two sites were identified on FANCD2, T691 and S717, which can be phosphorylated by ATR and ATM both in vitro and in human cells, but could not fully account for the phenotype observed in the absence of ATR. These two sites were not essential for FANCD2 monoubiquitination, though if mutated led to an increase in sensitivity to MMC pointing to a role in ICL repair and also affected the intra-S-phase checkpoint (Table 2) [80]. Other components of the ATR pathway can promote the efficient monoubiquitination of FANCD2 and ICL resistance, mainly RAD9 and RAD17 [81]. It was also found that Chk1 and its partner CLASPIN are necessary for efficient FANCD2 monoubiquitination in response to DNA damage in human cells, but whether this happens downstream or independently of the ATR/RAD17/RAD9 pathway remains unknown [81]. The identification of a new phosphorylation site on FANCD2, S331, shed more light on this problem. The lack of phosphorylation on this site also led to sensitivity to MMC and disrupted the interaction with BRCA2 (FANCD1) in human cells. This site is phosphorylated by Chk1 both in vitro and in vivo, which could be a potential explanation for the role of the ATR pathway through Chk1 in promoting FANCD2 monoubiquitination and ICL repair (Table 2) [82].
Phosphorylation of FANCI was also found to be important for FANCD2 monoubiquitination and foci formation. Several conserved sites (at least six) on FANCI with the motif Ser/Thr-Gln predicted to be phosphorylated by ATM or ATR were responsible for the observations in chicken DT40 cells. This phosphorylation event could be a switch for the monoubiquitination and recruitment of FANCD2 onto DNA. However, the monoubiquitination of FANCI was dispensable for monoubiquitination and recruitment of FANCD2 to ICLs [83]. Further evidence for the role of ATR in the response to ICLs came from experiments with chicken DT40 cells lacking expression of ATRIP, an ATR interacting partner needed for its activation. In this case, there was also a reduction in FANCD2 and FANCI monoubiquitination as well as in FANCI phosphorylation after MMC treatment. FANCI was phosphorylated by ATR in vitro and the reaction was enhanced by the presence of FANCD2 and the core complex [84].
Several components of the core complex also undergo phosphorylation although with different effects on the FA pathway. FANCG, for example, is phosphorylated during mitosis in human cells and this was related to the dissociation of the core complex from chromatin once the repair is completed [85]. Later, two residues were identified as responsible for this event, S383 and S387. S387 was phosphorylated (in vitro and in human cells) by Cdc2, which associates with the core complex in mitosis (Table 2) [86].
A more critical role in the FA pathway and ICL repair was found for the phosphorylation of FANCA on S1449. Phosphorylation on this site occurred specifically after DNA damage and not during unperturbed S-phase, unlike FANCD2 monoubiquitination and FANCG phosphorylation, which happen also during S-phase. Lack of phosphorylation on this site reduced FANCD2 monoubiquitination and led to partial sensitivity to MMC. ATR phosphorylated S1449 in vitro and was necessary for the phosphorylation in human cells (Table 2) [87].
Another component of the core complex, FANCE, is also phosphorylated after DNA damage. Chk1 phosphorylates FANCE on T346 and S374 both in vitro and in human cell lines (Table 2) [88]. However, this event is independent of FANCD2 phosphorylation and foci formation. The fact that FANCE phosphorylation was necessary to fully complement FANCE-deficient cells suggests it is still an important event in the FA pathway and ICL repair. FANCE phosphorylation promotes its degradation, thus it was proposed to play a role in the termination of the pathway to complete the repair [88].
Recruitment of the core complex to DNA is usually attributed to FANCM and its phosphorylation could play a critical role in the process. Studies in cell-free Xenopus egg extracts showed that FANCM is hyperphosphorylated during S-phase as well as after DNA damage and this enhances its chromatin localization. ATR and ATM regulate this process. Surprisingly, these events were shown to be favoured by the presence of FANCD2, pointing to a positive feedback-loop taking place (Table 2) [89, 90]. Similar results were obtained in human cell lines where S1045 on FANCM was found to be a target for ATR and necessary for its localization on ICLs as well as the activation of the G2-M checkpoint [90]. There is evidence for a role of ATR physically mediating the recruitment of FANCM to the damaged DNA during replication in human cells. ATR/ATRIP is recruited to stalled replication forks through interaction with RPA, which then interacts with HCLK2. HCLK2 can then recruit the heterodimer formed by FANCM and FAAP24, providing a potential mechanism for the recruitment of the core complex in an ATR-checkpoint signalling dependent manner [91]. On the other hand, when the cell enters M-phase FANCM is hyperphosphorylated and degraded, thus promoting the dissociation of the core complex from chromatin. This phosphorylation-dependent degradation was mediated by β-TRCP and Plk1 (Polo-like kinase 1) in human cells [92].
Among the effector proteins, phosphorylation of those implicated in HR is well established in response to IR and therefore, in DSB repair. However, given that these modifications may also play a role in ICL repair we will discuss some of them (Table 2). Phosphorylation of BRCA1 by ATM was one of the first modifications found that played a role in DSB repair [93]. Later, BRCA1, BRCA2 and PALB2 together with other proteins already discussed (FANCD2, FANCI) were identified as substrates for ATR and ATM in response to DSBs as part of a complex protein network [94]. BRCA1 is a key regulator of HR and several kinases have been linked to its function and regulation. Cdk1 and Plk1 phosphorylate BRCA1, probably in a sequential way, promoting BRCA1 foci formation following DSB in human cells [95, 96]. On the other hand, Chk2 phosphorylation of BRCA1 leads to its degradation and dissociation form DSB in human cells. This allows the nuclease MRE11 to be recruited so that end resection and HR can proceed during S/G2 phase [97, 98]. The BRCT domain of BRCA1 has been characterized as a phosphoprotein-binding domain and one of its binding partners is FANCJ. Phosphorylation of FANCJ on S990 during S/G2 phase was shown to be essential for the interaction with BRCA1 in human cells, controlling the cell cycle as part of the DNA damage checkpoint [99].
Ubiquitination
A central step in the FA pathway is the monoubiquitination of FANCD2 on Lys561 (human) that ensures its recruitment to damaged DNA as well as its interaction with other effector proteins such as BRCA1 (FANCS) (Table 2) [100]. The E3 ubiquitin ligase catalysing this step was found to be FANCL, a member of the core complex [101]. FANCL works together with the E2 conjugating enzyme UBE2T (FANCT) to monoubiquitinate FANCD2 (Fig. 3). UBE2T can also monoubiquitinate itself on K91 decreasing its own activity as a potential regulatory mechanism [102]. FANCL contains three domains: an N-terminal E2-like fold (ELF) domain, a central double RWD domain and a C-terminal RING domain. The RING domain binds to UBE2T while the RWD domain binds to FANCD2 [103]. The ELF domain interacts with ubiquitin and is important for the monoubiquitination of FANCD2 upon DNA damage in chicken DT40 cells (though not in vitro) [104]. The binding partner of FANCD2, FANCI, is also monoubiquitinated in vivo, and is required for the monoubiquitination of FANCD2 and restricts it to K561 [48, 105]. Furthermore, the presence of DNA greatly enhances FANCD2 monoubiquitination but only in the presence of FANCI in vitro, suggesting that the monoubiquitination in vivo occurs on the DNA and when FANCD2 and FANCI are in complex [49]. Finally, FANCD2 is deubiquitinated by the USP1/UAF1 deubiquitinating enzyme complex (Fig. 3) [106, 107].
Ubiquitination also plays a critical role in an alternative model for the recruitment of the core complex. In this study in human cells, RNF8 together with UBC13 promotes K63 polyubiquitination of histone H2A in response to DNA damage and this polyubiquitin is recognized by FAAP20 bringing the core complex onto damaged DNA [108]. In fact, RNF8 and FAAP20 were needed for efficient FANCD2 monoubiquitination after MMC treatment and, thus, for efficient ICL repair [108].
Ubiquitination has also been linked to the regulation of effector proteins implicated in HR. PALB2 and BRCA1 interaction is required for HR during G1 phase. However, PALB2 ubiquitination on its BRCA1 binding motif abrogates this interaction in 293T cells after IR (Table 2) [109].
Recently, the polyubiquitination of FANCG via K63 linkage has been found to mediate its interaction with BRCA1 and play an important role in HR in ICL repair in human cells [110]. Three potential target lysines for this process were identified: K182, K258 and K347 (Table 2) [110].
SUMOylation
Alongside the ubiquitination and phosphorylation events in the FA pathway, several SUMOylation events have started to be discovered in recent years. SUMOylation, therefore, provides a further step of regulation of the already complex cellular response to ICLs. As discussed before, FANCD2 and FANCI form a heterodimer that is both phosphorylated and monoubiquitinated in order to appear on ICLs. A subpopulation of FANCD2 and FANCI is also SUMOylated in response to DNA damage. This SUMOylation is performed by PIAS1/4 and UBC9 on the chromatin-bound complex in human cell lines while it can be reversed by SENP6 (Table 2) [76]. SUMOylated FANCD2/FANCI can then bind RNF4 which polyubiquitinates the complex. This polyubiquitinated complex then interacts with DVC1-p97 promoting its dissociation from chromatin (Fig. 3). Therefore, this mechanism could control the dosage of FANCD2/FANCI on the chromatin avoiding further recruitment of nucleases to DNA and allowing for a dynamic regulation of the pathway [76].
The study of a patient-derived FANCA mutation identified in the clinic has led to the discovery of a regulatory mechanism through its SUMOylation-dependent polyubiquitination and degradation. This mutant form, FANCA-I939S, failed to interact with FAAP20 and this led to its SUMOylation by UBC9, increased polyubiquitination by RNF4 and degradation via the proteasome (Table 2) [77].
SUMOylation of BRCA1 by SUMO2/3 in response to DSB has also been described both in vitro and in vivo in mammalian cell lines [111, 112]. SUMOylation of BRCA1 at its RING and BRCT domains (K32 and K1690, respectively) promotes the binding to other proteins through SIMs (SUMO-interacting motifs), which in this case would enhance accumulation on DNA and its ubiquitin ligase activity [111, 112].
These examples could illustrate a more general mechanism for SUMO signalling as already shown for DNA double-strand break repair [113]. In this case, SUMO modifications target several proteins in a group, such as the core complex or the FANCD2/FANCI complex, thereby promoting interactions through the SUMO-SIMs of the components synergistically. At a later stage, this leads to their polyubiquitination and degradation to ensure the termination of the repair and the progression of the cell cycle.
Recognition of the ICL
When ICLs occur in the cells, the UHRF1 protein is recruited to sites of damage within seconds [61, 62] (Fig. 4, step 1). UHRF1 recognizes ICLs through its SET and RING finger associated (SRA) domain, which was previously known for its role in recognizing hemi-methylated DNA and subsequent recruitment of DNMT1 to maintain the methylation signature in mammalian cells [114–117]. The affinities of UHRF1 to hemi-methylated DNA and to ICLs are similar, suggesting that UHRF1 could interact with hemi-methylated DNA and ICLs through related mechanisms. The recruitment of UHRF1 precedes and is required for proper recruitment of FANCD2 to ICLs [61]. There is about 10 min time lag between UHRF1 and FANCD2 recruitment to ICLs, which leaves room for speculation that recruitment of other proteins or PTM events might take place within this time frame. The exact mechanism of how UHRF1 facilitates FANCD2 recruitment or subsequent repair is still unclear, but might entail a direct protein–protein interaction. It has also been suggested that UHRF1 plays a role as a nuclease scaffold [62]. It is possible that the rapid recruitment of UHRF1 to the ICLs primes the lesion for FA mediated accurate repair later. Due to the diversity in structures of different ICLs (Figs. 1, 2), it is possible that other ICL sensor proteins exist in addition to UHRF1.

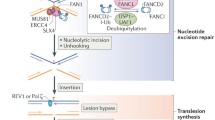
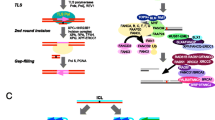
Schematic of ICL repair. 1 UHRF1 is recruited to ICLs through its SRA domain shortly after ICLs are formed in the cell. 2 Single replication fork arrives at the ICL. 3 FANCM/MHF complex mediates the traverse of the replication machinery through the ICL, which allows the replication fork to proceed, and leaves the ICL for later repair. 4, 5 Alternatively, BRCA1 (FANCS) facilitates the unloading of the CMG helicase complex when the second replication fork arrives at the ICL. 6 The replicative polymerase proceeds to the −1 position of the ICL, which leaves an X-shaped structure similar to the traverse mechanism. 7 ATR phosphorylates FANCD2/FANCI complex at multiple sites and FA core complex monoubiquitinates FANCD2/FANCI complex at K561 and K523, respectively. 8 FANCD2/FANCI is recruited to the ICL at the replication fork. 9, 10 Ubiquitinated FANCD2/FANCI complex recruits SLX4/XPF to ICL to unhook the ICL. 11 CtIP and the MRN complex resect the double-strand break ends generated by the incision in the previous step, and BRCA2 facilitates Rad51 filament formation on the ssDNA generated by the resection. 12 Polζ polymerizes new strand of DNA through the unhooked ICL. 13 Rad51 facilitates the strand invasion, which allows extension of the other strand. 14 SLX4 and nucleases resolve the double Holliday junction. 15 NER repair proteins remove the damaged nucleotide
Canonical FA pathway coordinated ICL repair
It is generally believed that the FA pathway is active solely in the S-phase and that the majority of ICLs are repaired in a replication-dependent manner [118]. The repair of ICLs requires coordination of several different DNA damage repair pathways including NER, HR and TLS. It is thought that the FANCD2/FANCI complex and the FA pathway is at the centre orchestrating the order of various events to resolve ICLs. Notably, it has been suggested that the FA pathway antagonizes the NHEJ pathway [119–121], which further emphasizes the importance of the FANCD2/FANCI complex.
There are several models of the replication-coupled ICL repair (Fig. 4). One possibility is that the fork undergoes FANCM/MHF complex-mediated traverse of the ICL, which is independent of the rest of the FA core complex and FANCD2/FANCI complex [19] (Fig. 4, step 3). The ICL traverse mechanism leaves behind the ICL, which has an X-shaped structure similar to that of a stalled replication fork. Due to the structure similarity, it has been implied that it is subsequently repaired by the canonical ICL repair mechanism.
Alternatively, when the replication fork collides with an ICL, the CMG helicase first undergoes BRCA1 (FANCS)-mediated unloading to allow the replicative polymerase ε to approach the ICL [122] (Fig. 4, step 5). The FANCD2/FANCI complex is then recruited to the arrested fork (Fig. 4, step 8), and monoubiquitinated by the FA core complex [123] (Fig. 4, step 9). Whether the FANCD2/FANCI complex is monoubiquitinated prior to recruitment, or on chromatin, remains elusive (Fig. 4, step 7). The K561R mutation of FANCD2 abrogates monoubiquitination and chromatin recruitment of FANCD2/FANCI complex and sensitizes cells to crosslinking agents, [78, 124]. However, it is also possible that the monoubiquitination of FANCD2/FANCI complex is critical for its retention at the ICL rather than the actual recruitment to the ICL. Recent studies show that DNA stimulates the ubiquitination of FANCD2/FANCI in vitro [49, 53, 125], which implies that the interaction between the FANCD2/FANCI complex and DNA may contribute to the regulation of its monoubiquitination in vivo. The monoubiquitinated FANCD2/FANCI complex recruits the XPF/ERCC1/SLX4 nuclease complex (Fig. 4, step 10) and the nucleases carry out incisions at the ICL to unhook the crosslinked bases [126–128]. The TLS polymerase Rev1–polζ complex, which can accommodate bulky DNA substrates, is subsequently recruited to the lesion to carry out insertion through the unhooked ICL base pair (Fig. 4, step 12) [129]. Interestingly, the FA core complex, but not the FANCD2/FANCI complex, regulates recruitment of the Rev1–polζ complex [129]. The double-strand break generated by the incision is resected by CtIP, which is recruited through monoubiquitinated FANCD2 [130, 131], potentially together with the MRN (MRE11-RAD50-NBS1) complex and the nucleases EXO1 and DNA2. MRE11 has been shown to interact with FANCJ, which potentially regulates its nuclease activity ensuring a correct end resection [132]. The DSB is, then, repaired by RAD51-mediated homologous recombination (Fig. 4, step 13) [133]. To complete the repair of the ICL, the unhooked lesion needs to be removed. It is generally thought that the NER pathway is involved in the process (Fig. 4, step 15) [134, 135], though the precise molecular mechanism remains elusive.
Alternative ICL repair pathway in G1
In addition to replication-coupled ICL repair, there is increasing evidence for the existence of a replication-independent ICL repair mechanism. Although it has been understudied, replication-independent ICL repair may play a significant role in maintaining genome integrity in non-cycling cells, e.g. neurons.
It has been shown that ICLs can be removed via the NER pathway independently of replication [136–139]. NER proteins XPA, XPB, XPC and XPF are recruited to psoralen/UVA induced ICLs and monoadducts within minutes in the G1-phase of the cell cycle and this ICL repair process is dependent on Polζ and XPC [137, 138]. It has also been shown that transcription coupled-NER (TC-NER) is crucial for cisplatin-induced ICL repair [136, 139].
The detailed molecular mechanism of the replication-independent ICL repair is still unclear. It is likely that in the absence of arrested replication forks, the activation of ICL repair relies on the proteins recognizing the distorted DNA helix and/or the collision of the RNA transcription machinery with an ICL. There are some contradictions in the literature whether XPC is involved in the replication-independent repair. The ICL-forming agents used in the studies mentioned above differ from study to study, but are still generally considered to pose similar replicative stress. As discussed previously, these crosslinking agents create structurally distinct ICLs, which could give rise to the discrepancies observed.
Conclusion
Genetic and biochemical studies carried out by various groups have advanced our understanding of ICL repair for the past decade and beyond. 19 FA genes have been identified and the corresponding proteins cooperate with other proteins in HR, NHEJ, TLS and NER to resolve ICLs. However, the more we have learnt, the more questions have arisen. For instance, UHRF1 has been shown to sense ICLs induced by TMP/UVA in vivo. Besides, the recruitment of UHRF1 to ICLs precedes and is required for FANCD2 recruitment. Nonetheless, it is unclear how UHRF1 contributes to the regulation of FANCD2, whether directly or indirectly. Also, it has been well accepted that the FANCD2/FANCI complex is monoubiquitinated prior to its recruitment to ICLs. However, in the crystal structure of the mouse FANCD2/FANCI complex, the ubiquitination sites are embedded in the heterodimer interface. Recent studies show that DNA stimulates the in vitro ubiquitination of the FANCD2/FANCI complex [49, 125], suggesting that the interaction of the complex with DNA might be required for, and thus precede, the ubiquitination event. Such a mechanism could potentially involve a conformational change of the complex upon interaction with DNA, in turn stimulating the ubiquitination, perhaps by allowing the FA core complex access to the target lysines. One of the common features of the FA proteins is the lack of predictable domains, which otherwise could help towards deciphering their exact functions. More structural studies of proteins involved in ICL repair will be required to achieve a full mechanistic understanding of this complicated process. Finally, different crosslinking agents have been thought to cause similar cellular toxicities and the resulting ICLs would be sensed and resolved through the same pathway. However, the nature of the ICLs induced by different crosslinking agents is different. We may need to re-think how we depict the pathways resolving ICLs, which could be more complicated than we currently imagine. For instance, ICLs induced by MMC and TMP/UVA cause a minor distortion of the DNA double helix and can be recognized by XPC and UHRF1, respectively [61, 137]. On the other hand, ICLs caused by cisplatin cause a major distortion of DNA, which may require other sensor proteins or collision of the replication and transcription machineries with the ICL to activate the signal cascade. Thus, different ICLs may lead to activation of different, though related, repair mechanisms.
Abbreviations
- ATM:
-
Ataxia telangiectasia mutated
- ATR:
-
ATM and Rad3-related
- AML:
-
Acute myelogenous leukaemia
- BCNU:
-
1,3-Bis(2-chloroethyl)-1-nitrosourea
- DDR:
-
DNA damage response
- DEB:
-
1,2,3,4-Diepoxybutane
- DSB:
-
Double-strand break
- FA:
-
Fanconi anemia
- HR:
-
Homologous recombination
- ICL:
-
Interstrand crosslink
- IR:
-
Ionizing radiation
- MDA:
-
Malondialdehyde
- MMC:
-
Mitomycin C
- NER:
-
Nucleotide excision repair
- NHEJ:
-
Non-homologous end joining
- NO:
-
Nitric oxide
- PTM:
-
Post-translational modification
- ROS:
-
Reactive oxygen species
- RPA:
-
Replication protein A
- SUMO:
-
Small ubiquitin-like modifier
- TLS:
-
Translesion synthesis
- TMP:
-
Trimethylpsoralen
- TNF-α:
-
Tumour necrosis factor-α
- UAF1:
-
USP1 associated factor 1
- UHRF1:
-
Ubiquitin-like containing PHD and RING finger domains 1
- USP1:
-
Ubiquitin-specific peptidase 1
References
Deans AJ, West SC (2011) DNA interstrand crosslink repair and cancer. Nat Rev Cancer 11(7):467–480
Langevin F, Crossan GP, Rosado IV, Arends MJ, Patel KJ (2011) Fancd2 counteracts the toxic effects of naturally produced aldehydes in mice. Nature 475(7354):53–58
Pang Q, Andreassen PR (2009) Fanconi anemia proteins and endogenous stresses. Mutat Res 668(1–2):42–53
Povirk LF, Shuker DE (1994) DNA damage and mutagenesis induced by nitrogen mustards. Mutat Res 318(3):205–226
Dronkert ML, Kanaar R (2001) Repair of DNA interstrand cross-links. Mutat Res 486(4):217–247
Rink SM, Hopkins PB (1995) A mechlorethamine-induced DNA interstrand cross-link bends duplex DNA. Biochemistry 34(4):1439–1445
Chen W, Han Y, Peng X (2014) Aromatic nitrogen mustard-based prodrugs: activity, selectivity, and the mechanism of DNA cross-linking. Chemistry 20(24):7410–7418
Tomasz M (1995) Mitomycin C: small, fast and deadly (but very selective). Chem Biol 2(9):575–579
Suresh Kumar G, Lipman R, Cummings J, Tomasz M (1997) Mitomycin C-DNA adducts generated by DT-diaphorase. Revised mechanism of the enzymatic reductive activation of mitomycin C. Biochemistry 36(46):14128–14136
Warren AJ, Maccubbin AE, Hamilton JW (1998) Detection of mitomycin C-DNA adducts in vivo by 32P-postlabeling: time course for formation and removal of adducts and biochemical modulation. Cancer Res 58(3):453–461
Norman D, Live D, Sastry M, Lipman R, Hingerty BE, Tomasz M, Broyde S, Patel DJ (1990) NMR and computational characterization of mitomycin cross-linked to adjacent deoxyguanosines in the minor groove of the d (TACGTA). cntdot. d (TACGTA) duplex. Biochemistry 29(11):2861–2875
Rosenberg B, Vancamp L, Krigas T (1965) Inhibition of cell division in Escherichia coli by electrolysis products from a platinum electrode. Nature 205:698–699
Malinge JM, Giraud-Panis MJ, Leng M (1999) Interstrand cross-links of cisplatin induce striking distortions in DNA. J Inorg Biochem 77(1–2):23–29
Coste F, Malinge JM, Serre L, Shepard W, Roth M, Leng M, Zelwer C (1999) Crystal structure of a double-stranded DNA containing a cisplatin interstrand cross-link at 1.63 A resolution: hydration at the platinated site. Nucleic Acids Res 27(8):1837–1846
Blommaert FA, van Dijk-Knijnenburg HC, Dijt FJ, den Engelse L, Baan RA, Berends F, Fichtinger-Schepman AM (1995) Formation of DNA adducts by the anticancer drug carboplatin: different nucleotide sequence preferences in vitro and in cells. Biochemistry 34(26):8474–8480
Paquet F, Boudvillain M, Lancelot G, Leng M (1999) NMR solution structure of a DNA dodecamer containing a transplatin interstrand GN7-CN3 cross-link. Nucleic Acids Res 27(21):4261–4268
Brabec V, Neplechova K, Kasparkova J, Farrell N (2000) Steric control of DNA interstrand cross-link sites of trans platinum complexes: specificity can be dictated by planar nonleaving groups. J Biol Inorg Chem 5(3):364–368
Scott BR, Pathak MA, Mohn GR (1976) Molecular and genetic basis of furocoumarin reactions. Mutat Res 39(1):29–74
Huang J, Liu S, Bellani MA, Thazhathveetil AK, Ling C, de Winter JP, Wang Y, Wang W, Seidman MM (2013) The DNA translocase FANCM/MHF promotes replication traverse of DNA interstrand crosslinks. Mol Cell 52(3):434–446
Brendel M, Ruhland A (1984) Relationships between functionality and genetic toxicology of selected DNA-damaging agents. Mutat Res 133(1):51–85
Spielmann HP, Dwyer TJ, Hearst JE, Wemmer DE (1995) Solution structures of psoralen monoadducted and cross-linked DNA oligomers by NMR spectroscopy and restrained molecular dynamics. Biochemistry 34(40):12937–12953
Wiencke JK, Wiemels J (1995) Genotoxicity of 1,3-bis(2-chloroethyl)-1-nitrosourea (BCNU). Mutat Res 339(2):91–119
O’Flaherty DK, Denisov AY, Noronha AM, Wilds CJ (2014) NMR structure of an ethylene interstrand cross-linked DNA which mimics the lesion formed by 1,3-bis(2-chloroethyl)-1-nitrosourea. ChemMedChem 9(9):2099–2103
Wen Y, Zhang PP, An J, Yu YX, Wu MH, Sheng GY, Fu JM, Zhang XY (2011) Diepoxybutane induces the formation of DNA-DNA rather than DNA-protein cross-links, and single-strand breaks and alkali-labile sites in human hepatocyte L02 cells. Mutat Res 716(1–2):84–91
Zhang XY, Elfarra AA (2006) Characterization of 1,2,3,4-diepoxybutane-2′-deoxyguanosine cross-linking products formed at physiological and nonphysiological conditions. Chem Res Toxicol 19(4):547–555
Millard JT, McGowan EE, Bradley SQ (2012) Diepoxybutane interstrand cross-links induce DNA bending. Biochimie 94(2):574–577
Brooks PJ, Theruvathu JA (2005) DNA adducts from acetaldehyde: implications for alcohol-related carcinogenesis. Alcohol 35(3):187–193
Lorenti Garcia C, Mechilli M, Proietti De Santis L, Schinoppi A, Kobos K, Palitti F (2009) Relationship between DNA lesions, DNA repair and chromosomal damage induced by acetaldehyde. Mutat Res 662(1–2):3–9
Cho Y-JJ, Kozekov ID, Harris TM, Rizzo CJ, Stone MP (2007) Stereochemistry modulates the stability of reduced interstrand cross-links arising from R- and S-alpha-CH3-gamma-OH-1, N2-propano-2′-deoxyguanosine in the 5′-CpG-3′ DNA sequence. Biochemistry 46(10):2608–2621
Folmer V, Soares JC, Gabriel D, Rocha JB (2003) A high fat diet inhibits delta-aminolevulinate dehydratase and increases lipid peroxidation in mice (Mus musculus). J Nutr 133(7):2165–2170
Niedernhofer LJ, Daniels JS, Rouzer CA, Greene RE, Marnett LJ (2003) Malondialdehyde, a product of lipid peroxidation, is mutagenic in human cells. J Biol Chem 278(33):31426–31433
Stone MP, Cho YJ, Huang H, Kim HY, Kozekov ID, Kozekova A, Wang H, Minko IG, Lloyd RS, Harris TM, Rizzo CJ (2008) Interstrand DNA cross-links induced by alpha, beta-unsaturated aldehydes derived from lipid peroxidation and environmental sources. Acc Chem Res 41(7):793–804
Kozekov ID, Nechev LV, Moseley MS, Harris CM, Rizzo CJ, Stone MP, Harris TM (2003) DNA interchain cross-links formed by acrolein and crotonaldehyde. J Am Chem Soc 125(1):50–61
Caulfield JL, Wishnok JS, Tannenbaum SR (2003) Nitric oxide-induced interstrand cross-links in DNA. Chem Res Toxicol 16(5):571–574
Dong H, Nebert DW, Bruford EA, Thompson DC, Joenje H, Vasiliou V (2015) Update of the human and mouse Fanconi anemia genes. Hum Genomics 9(1):32
Dufour C, Svahn J (2008) Fanconi anaemia: new strategies. Bone Marrow Transplant 41(Suppl 2):S90–S95
Pontel LB, Rosado IV, Burgos-Barragan G, Garaycoechea JI, Yu R, Arends MJ, Chandrasekaran G, Broecker V, Wei W, Liu L, Swenberg JA, Crossan GP, Patel KJ (2015) Endogenous formaldehyde is a hematopoietic stem cell genotoxin and metabolic carcinogen. Mol Cell 60(1):177–188
Garaycoechea JI, Patel KJ (2014) Why does the bone marrow fail in Fanconi anemia? Blood 123(1):26–34
Schneider M, Chandler K, Tischkowitz M, Meyer S (2015) Fanconi anaemia: genetics, molecular biology, and cancer—implications for clinical management in children and adults. Clin Genet 88(1):13–24
Yan Z, Delannoy M, Ling C, Daee D, Osman F, Muniandy PA, Shen X, Oostra AB, Du H, Steltenpool J, Lin T, Schuster B, Decaillet C, Stasiak A, Stasiak AZ, Stone S, Hoatlin ME, Schindler D, Woodcock CL, Joenje H, Sen R, de Winter JP, Li L, Seidman MM, Whitby MC, Myung K, Constantinou A, Wang W (2010) A histone-fold complex and FANCM form a conserved DNA-remodeling complex to maintain genome stability. Mol Cell 37(6):865–878
Hemphill AW, Akkari Y, Newell AH, Schultz RA, Grompe M, North PS, Hickson ID, Jakobs PM, Rennie S, Pauw D, Hejna J, Olson SB, Moses RE (2009) Topo IIIalpha and BLM act within the Fanconi anemia pathway in response to DNA-crosslinking agents. Cytogenet Genome Res 125(3):165–175
Meetei AR, Sechi S, Wallisch M, Yang D, Young MK, Joenje H, Hoatlin ME, Wang W (2003) A multiprotein nuclear complex connects Fanconi anemia and Bloom syndrome. Mol Cell Biol 23(10):3417–3426
Singh TR, Bakker ST, Agarwal S, Jansen M, Grassman E, Godthelp BC, Ali AM, Du CH, Rooimans MA, Fan Q, Wahengbam K, Steltenpool J, Andreassen PR, Williams DA, Joenje H, de Winter JP, Meetei AR (2009) Impaired FANCD2 monoubiquitination and hypersensitivity to camptothecin uniquely characterize Fanconi anemia complementation group M. Blood 114(1):174–180
Lim ET, Wurtz P, Havulinna AS, Palta P, Tukiainen T, Rehnstrom K, Esko T, Magi R, Inouye M, Lappalainen T, Chan Y, Salem RM, Lek M, Flannick J, Sim X, Manning A, Ladenvall C, Bumpstead S, Hamalainen E, Aalto K, Maksimow M, Salmi M, Blankenberg S, Ardissino D, Shah S, Horne B, McPherson R, Hovingh GK, Reilly MP, Watkins H, Goel A, Farrall M, Girelli D, Reiner AP, Stitziel NO, Kathiresan S, Gabriel S, Barrett JC, Lehtimaki T, Laakso M, Groop L, Kaprio J, Perola M, McCarthy MI, Boehnke M, Altshuler DM, Lindgren CM, Hirschhorn JN, Metspalu A, Freimer NB, Zeller T, Jalkanen S, Koskinen S, Raitakari O, Durbin R, MacArthur DG, Salomaa V, Ripatti S, Daly MJ, Palotie A, Sequencing Initiative Suomi P (2014) Distribution and medical impact of loss-of-function variants in the Finnish founder population. PLoS Genet 10(7):e1004494
Rickman KA, Lach FP, Abhyankar A, Donovan FX, Sanborn EM, Kennedy JA, Sougnez C, Gabriel SB, Elemento O, Chandrasekharappa SC, Schindler D, Auerbach AD, Smogorzewska A (2015) Deficiency of UBE2T, the E2 ubiquitin ligase necessary for FANCD2 and FANCI ubiquitination, causes FA-T subtype of Fanconi anemia. Cell Rep 12(1):35–41
Virts EL, Jankowska A, Mackay C, Glaas MF, Wiek C, Kelich SL, Lottmann N, Kennedy FM, Marchal C, Lehnert E, Scharf RE, Dufour C, Lanciotti M, Farruggia P, Santoro A, Savasan S, Scheckenbach K, Schipper J, Wagenmann M, Lewis T, Leffak M, Farlow JL, Foroud TM, Honisch E, Niederacher D, Chakraborty SC, Vance GH, Pruss D, Timms KM, Lanchbury JS, Alpi AF, Hanenberg H (2015) AluY-mediated germline deletion, duplication and somatic stem cell reversion in UBE2T defines a new subtype of Fanconi anemia. Hum Mol Genet 24(18):5093–5108
Hira A, Yoshida K, Sato K, Okuno Y, Shiraishi Y, Chiba K, Tanaka H, Miyano S, Shimamoto A, Tahara H, Ito E, Kojima S, Kurumizaka H, Ogawa S, Takata M, Yabe H, Yabe M (2015) Mutations in the gene encoding the E2 conjugating enzyme UBE2T cause Fanconi anemia. Am J Hum Genet 96(6):1001–1007
Alpi AF, Pace PE, Babu MM, Patel KJ (2008) Mechanistic insight into site-restricted monoubiquitination of FANCD2 by Ube2t, FANCL, and FANCI. Mol Cell 32(6):767–777
Sato K, Toda K, Ishiai M, Takata M, Kurumizaka H (2012) DNA robustly stimulates FANCD2 monoubiquitylation in the complex with FANCI. Nucleic Acids Res 40(10):4553–4561
Longerich S, San Filippo J, Liu D, Sung P (2009) FANCI binds branched DNA and is monoubiquitinated by UBE2T-FANCL. J Biol Chem 284(35):23182–23186
Sugahara R, Mon H, Lee JM, Kusakabe T (2012) Monoubiquitination-dependent chromatin loading of FancD2 in silkworms, a species lacking the FA core complex. Gene 501(2):180–187
Walden H, Deans AJ (2014) The Fanconi anemia DNA repair pathway: structural and functional insights into a complex disorder. Annu Rev Biophys 43:257–278
Rajendra E, Oestergaard VH, Langevin F, Wang M, Dornan GL, Patel KJ, Passmore LA (2014) The genetic and biochemical basis of FANCD2 monoubiquitination. Mol Cell 54(5):858–869
Huang Y, Leung JW, Lowery M, Matsushita N, Wang Y, Shen X, Huong D, Takata M, Chen J, Li L (2014) Modularized functions of the Fanconi anemia core complex. Cell Rep 7(6):1849–1857
Meetei AR, Medhurst AL, Ling C, Xue Y, Singh TR, Bier P, Steltenpool J, Stone S, Dokal I, Mathew CG, Hoatlin M, Joenje H, de Winter JP, Wang W (2005) A human ortholog of archaeal DNA repair protein Hef is defective in Fanconi anemia complementation group M. Nat Genet 37(9):958–963
King MC, Marks JH, Mandell JB, New York Breast Cancer Study G (2003) Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 302(5645):643–646
Meindl A, Hellebrand H, Wiek C, Erven V, Wappenschmidt B, Niederacher D, Freund M, Lichtner P, Hartmann L, Schaal H, Ramser J, Honisch E, Kubisch C, Wichmann HE, Kast K, Deissler H, Engel C, Muller-Myhsok B, Neveling K, Kiechle M, Mathew CG, Schindler D, Schmutzler RK, Hanenberg H (2010) Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 42(5):410–414
Turnbull C, Rahman N (2008) Genetic predisposition to breast cancer: past, present, and future. Annu Rev Genomics Hum Genet 9:321–345
Suhasini AN, Brosh RM Jr (2012) Fanconi anemia and Bloom’s syndrome crosstalk through FANCJ-BLM helicase interaction. Trends Genet 28(1):7–13
Suhasini AN, Rawtani NA, Wu Y, Sommers JA, Sharma S, Mosedale G, North PS, Cantor SB, Hickson ID, Brosh RM Jr (2011) Interaction between the helicases genetically linked to Fanconi anemia group J and Bloom’s syndrome. EMBO J 30(4):692–705
Liang CC, Zhan B, Yoshikawa Y, Haas W, Gygi SP, Cohn MA (2015) UHRF1 is a sensor for DNA interstrand crosslinks and recruits FANCD2 to initiate the Fanconi anemia pathway. Cell Rep 10(12):1947–1956
Tian Y, Paramasivam M, Ghosal G, Chen D, Shen X, Huang Y, Akhter S, Legerski R, Chen J, Seidman MM, Qin J, Li L (2015) UHRF1 contributes to DNA damage repair as a lesion recognition factor and nuclease scaffold. Cell Rep 10(12):1957–1966
Kratz K, Schöpf B, Kaden S, Sendoel A, Eberhard R, Lademann C, Cannavó E, Sartori AA, Hengartner MO, Jiricny J (2010) Deficiency of FANCD2-associated nuclease KIAA1018/FAN1 sensitizes cells to interstrand crosslinking agents. Cell 142(1):77–88
Smogorzewska A, Desetty R, Saito TT, Schlabach M, Lach FP, Sowa ME, Clark AB, Kunkel TA, Harper JW, Colaiácovo MP, Elledge SJ (2010) A genetic screen identifies FAN1, a Fanconi anemia-associated nuclease necessary for DNA interstrand crosslink repair. Mol Cell 39(1):36–47
MacKay C, Déclais A-C, Lundin C, Agostinho A, Deans AJ, MacArtney TJ, Hofmann K, Gartner A, West SC, Helleday T, Lilley DMJ, Rouse J (2010) Identification of KIAA1018/FAN1, a DNA repair nuclease recruited to DNA damage by monoubiquitinated FANCD2. Cell 142(1):65–76
Liu T, Ghosal G, Yuan J, Chen J, Huang J (2010) FAN1 acts with FANCI-FANCD2 to promote DNA interstrand cross-link repair. Science 329(5992):693–696
Yoshikiyo K, Kratz K, Hirota K, Nishihara K, Takata M, Kurumizaka H, Horimoto S, Takeda S, Jiricny J (2010) KIAA1018/FAN1 nuclease protects cells against genomic instability induced by interstrand cross-linking agents. Proc Natl Acad Sci USA 107(50):21553–21557
Ishiai M, Kimura M, Namikoshi K, Yamazoe M, Yamamoto K, Arakawa H, Agematsu K, Matsushita N, Takeda S, Buerstedde JM, Takata M (2004) DNA cross-link repair protein SNM1A interacts with PIAS1 in nuclear focus formation. Mol Cell Biol 24(24):10733–10741
Wang AT, Sengerova B, Cattell E, Inagawa T, Hartley JM, Kiakos K, Burgess-Brown NA, Swift LP, Enzlin JH, Schofield CJ, Gileadi O, Hartley JA, McHugh PJ (2011) Human SNM1A and XPF-ERCC1 collaborate to initiate DNA interstrand cross-link repair. Genes Dev 25(17):1859–1870
Chaudhury I, Stroik DR, Sobeck A (2014) FANCD2-controlled chromatin access of the Fanconi-associated nuclease FAN1 is crucial for the recovery of stalled replication forks. Mol Cell Biol 34(21):3939–3954
Lachaud C, Moreno A, Marchesi F, Toth R, Blow JJ, Rouse J (2016) Ubiquitinated Fancd2 recruits Fan1 to stalled replication forks to prevent genome instability. Science 351(6275):846–849
Raghunandan M, Chaudhury I, Kelich SL, Hanenberg H, Sobeck A (2015) FANCD2, FANCJ and BRCA2 cooperate to promote replication fork recovery independently of the Fanconi anemia core complex. Cell Cycle 14(3):342–353
Chan KL, Palmai-Pallag T, Ying S, Hickson ID (2009) Replication stress induces sister-chromatid bridging at fragile site loci in mitosis. Nat Cell Biol 11(6):753–760
Ying S, Hickson ID (2011) Fanconi anaemia proteins are associated with sister chromatid bridging in mitosis. Int J Hematol 93(4):440–445
Schwab RA, Nieminuszczy J, Shah F, Langton J, Lopez Martinez D, Liang CC, Cohn MA, Gibbons RJ, Deans AJ, Niedzwiedz W (2015) The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol Cell 60(3):351–361
Gibbs-Seymour I, Oka Y, Rajendra E, Weinert BT, Passmore LA, Patel KJ, Olsen JV, Choudhary C, Bekker-Jensen S, Mailand N (2015) Ubiquitin-SUMO circuitry controls activated Fanconi anemia ID complex dosage in response to DNA damage. Mol Cell 57(1):150–164
Xie J, Kim H, Moreau LA, Puhalla S, Garber J, Al Abo M, Takeda S, D’Andrea AD (2015) RNF4-mediated polyubiquitination regulates the Fanconi anemia/BRCA pathway. J Clin Invest 125(4):1523–1532
Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim S-TT, Lane WS, Kastan MB, D’Andrea AD (2002) Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 109(4):459–472
Andreassen PR, D’Andrea AD, Taniguchi T (2004) ATR couples FANCD2 monoubiquitination to the DNA-damage response. Genes Dev 18(16):1958–1963
Ho GP, Margossian S, Taniguchi T, D’Andrea AD (2006) Phosphorylation of FANCD2 on two novel sites is required for mitomycin C resistance. Mol Cell Biol 26(18):7005–7015
Guervilly JH, Mace-Aime G, Rosselli F (2008) Loss of CHK1 function impedes DNA damage-induced FANCD2 monoubiquitination but normalizes the abnormal G2 arrest in Fanconi anemia. Hum Mol Genet 17(5):679–689
Zhi G, Wilson JB, Chen X, Krause DS, Xiao Y, Jones NJ, Kupfer GM (2009) Fanconi anemia complementation group FANCD2 protein serine 331 phosphorylation is important for fanconi anemia pathway function and BRCA2 interaction. Cancer Res 69(22):8775–8783
Ishiai M, Kitao H, Smogorzewska A, Tomida J, Kinomura A, Uchida E, Saberi A, Kinoshita E, Kinoshita-Kikuta E, Koike T, Tashiro S, Elledge SJ, Takata M (2008) FANCI phosphorylation functions as a molecular switch to turn on the Fanconi anemia pathway. Nat Struct Mol Biol 15(11):1138–1146
Shigechi T, Tomida J, Sato K, Kobayashi M, Eykelenboom JK, Pessina F, Zhang Y, Uchida E, Ishiai M, Lowndes NF, Yamamoto K, Kurumizaka H, Maehara Y, Takata M (2012) ATR-ATRIP kinase complex triggers activation of the Fanconi anemia DNA repair pathway. Cancer Res 72(5):1149–1156
Qiao F, Moss A, Kupfer GM (2001) Fanconi anemia proteins localize to chromatin and the nuclear matrix in a DNA damage- and cell cycle-regulated manner. J Biol Chem 276(26):23391–23396
Mi J, Qiao F, Wilson JB, High AA, Schroeder MJ, Stukenberg PT, Moss A, Shabanowitz J, Hunt DF, Jones NJ, Kupfer GM (2004) FANCG is phosphorylated at serines 383 and 387 during mitosis. Mol Cell Biol 24(19):8576–8585
Collins NB, Wilson JB, Bush T, Thomashevski A, Roberts KJ, Jones NJ, Kupfer GM (2009) ATR-dependent phosphorylation of FANCA on serine 1449 after DNA damage is important for FA pathway function. Blood 113(10):2181–2190
Wang X, Kennedy RD, Ray K, Stuckert P, Ellenberger T, D’Andrea AD (2007) Chk1-mediated phosphorylation of FANCE is required for the Fanconi anemia/BRCA pathway. Mol Cell Biol 27(8):3098–3108
Sobeck A, Stone S, Landais I, de Graaf B, Hoatlin ME (2009) The Fanconi anemia protein FANCM is controlled by FANCD2 and the ATR/ATM pathways. J Biol Chem 284(38):25560–25568
Singh TR, Ali AM, Paramasivam M, Pradhan A, Wahengbam K, Seidman MM, Meetei AR (2013) ATR-dependent phosphorylation of FANCM at serine 1045 is essential for FANCM functions. Cancer Res 73(14):4300–4310
Collis SJ, Ciccia A, Deans AJ, Horejsi Z, Martin JS, Maslen SL, Skehel JM, Elledge SJ, West SC, Boulton SJ (2008) FANCM and FAAP24 function in ATR-mediated checkpoint signaling independently of the Fanconi anemia core complex. Mol Cell 32(3):313–324
Kee Y, Kim JM, D’Andrea AD (2009) Regulated degradation of FANCM in the Fanconi anemia pathway during mitosis. Genes Dev 23(5):555–560
Cortez D, Wang Y, Qin J, Elledge SJ (1999) Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 286(5442):1162–1166
Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, Shiloh Y, Gygi SP, Elledge SJ (2007) ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 316(5828):1160–1166
Chabalier-Taste C, Brichese L, Racca C, Canitrot Y, Calsou P, Larminat F (2016) Polo-like kinase 1 mediates BRCA1 phosphorylation and recruitment at DNA double-strand breaks. Oncotarget 7(3):2269–2283
Johnson N, Cai D, Kennedy RD, Pathania S, Arora M, Li YC, D’Andrea AD, Parvin JD, Shapiro GI (2009) Cdk1 participates in BRCA1-dependent S phase checkpoint control in response to DNA damage. Mol Cell 35(3):327–339
Parameswaran B, Chiang HC, Lu Y, Coates J, Deng CX, Baer R, Lin HK, Li R, Paull TT, Hu Y (2015) Damage-induced BRCA1 phosphorylation by Chk2 contributes to the timing of end resection. Cell Cycle 14(3):437–448
Lee JS, Collins KM, Brown AL, Lee CH, Chung JH (2000) hCds1-mediated phosphorylation of BRCA1 regulates the DNA damage response. Nature 404(6774):201–204
Yu X, Chini CC, He M, Mer G, Chen J (2003) The BRCT domain is a phospho-protein binding domain. Science 302(5645):639–642
Garcia-Higuera I, Taniguchi T, Ganesan S, Meyn MS, Timmers C, Hejna J, Grompe M, D’Andrea AD (2001) Interaction of the Fanconi anemia proteins and BRCA1 in a common pathway. Mol Cell 7(2):249–262
Meetei AR, de Winter JP, Medhurst AL, Wallisch M, Waisfisz Q, van de Vrugt HJ, Oostra AB, Yan Z, Ling C, Bishop CE, Hoatlin ME, Joenje H, Wang W (2003) A novel ubiquitin ligase is deficient in Fanconi anemia. Nat Genet 35(2):165–170
Machida YJ, Machida Y, Chen Y, Gurtan AM, Kupfer GM, D’Andrea AD, Dutta A (2006) UBE2T is the E2 in the Fanconi anemia pathway and undergoes negative autoregulation. Mol Cell 23(4):589–596
Hodson C, Cole AR, Lewis LP, Miles JA, Purkiss A, Walden H (2011) Structural analysis of human FANCL, the E3 ligase in the Fanconi anemia pathway. J Biol Chem 286(37):32628–32637
Miles JA, Frost MG, Carroll E, Rowe ML, Howard MJ, Sidhu A, Chaugule VK, Alpi AF, Walden H (2015) The Fanconi anemia DNA repair pathway is regulated by an interaction between ubiquitin and the E2-like fold domain of FANCL. J Biol Chem 290(34):20995–21006
Smogorzewska A, Matsuoka S, Vinciguerra P, McDonald ER, Hurov KE, Luo J, Ballif BA, Gygi SP, Hofmann K, D’Andrea AD, Elledge SJ (2007) Identification of the FANCI protein, a monoubiquitinated FANCD2 paralog required for DNA repair. Cell 129(2):289–301
Cohn MA, Kowal P, Yang K, Haas W, Huang TT, Gygi SP, D’Andrea AD (2007) A UAF1-containing multisubunit protein complex regulates the Fanconi anemia pathway. Mol Cell 28(5):786–797
Nijman SM, Huang TT, Dirac AM, Brummelkamp TR, Kerkhoven RM, D’Andrea AD, Bernards R (2005) The deubiquitinating enzyme USP1 regulates the Fanconi anemia pathway. Mol Cell 17(3):331–339
Yan Z, Guo R, Paramasivam M, Shen W, Ling C, Fox D 3rd, Wang Y, Oostra AB, Kuehl J, Lee DY, Takata M, Hoatlin ME, Schindler D, Joenje H, de Winter JP, Li L, Seidman MM, Wang W (2012) A ubiquitin-binding protein, FAAP20, links RNF8-mediated ubiquitination to the Fanconi anemia DNA repair network. Mol Cell 47(1):61–75
Orthwein A, Noordermeer SM, Wilson MD, Landry S, Enchev RI, Sherker A, Munro M, Pinder J, Salsman J, Dellaire G, Xia B, Peter M, Durocher D (2015) A mechanism for the suppression of homologous recombination in G1 cells. Nature 528(7582):422–426
Zhu B, Yan K, Li L, Lin M, Zhang S, He Q, Zheng D, Yang H, Shao G (2015) K63-linked ubiquitination of FANCG is required for its association with the Rap80-BRCA1 complex to modulate homologous recombination repair of DNA interstrand crosslinks. Oncogene 34(22):2867–2878
Morris JR, Boutell C, Keppler M, Densham R, Weekes D, Alamshah A, Butler L, Galanty Y, Pangon L, Kiuchi T, Ng T, Solomon E (2009) The SUMO modification pathway is involved in the BRCA1 response to genotoxic stress. Nature 462(7275):886–890
Vialter A, Vincent A, Demidem A, Morvan D, Stepien G, Venezia ND (1810) Rio PG (2011) Cell cycle-dependent conjugation of endogenous BRCA1 protein with SUMO-2/3. Biochim Biophys Acta 4:432–438
Psakhye I, Jentsch S (2012) Protein group modification and synergy in the SUMO pathway as exemplified in DNA repair. Cell 151(4):807–820
Bostick M, Kim JK, Esteve PO, Clark A, Pradhan S, Jacobsen SE (2007) UHRF1 plays a role in maintaining DNA methylation in mammalian cells. Science 317(5845):1760–1764
Avvakumov GV, Walker JR, Xue S, Li Y, Duan S, Bronner C, Arrowsmith CH, Dhe-Paganon S (2008) Structural basis for recognition of hemi-methylated DNA by the SRA domain of human UHRF1. Nature 455(7214):822–825
Hashimoto H, Horton JR, Zhang X, Bostick M, Jacobsen SE, Cheng X (2008) The SRA domain of UHRF1 flips 5-methylcytosine out of the DNA helix. Nature 455(7214):826–829
Qian C, Li S, Jakoncic J, Zeng L, Walsh MJ, Zhou MM (2008) Structure and hemimethylated CpG binding of the SRA domain from human UHRF1. J Biol Chem 283(50):34490–34494
Raschle M, Knipscheer P, Enoiu M, Angelov T, Sun J, Griffith JD, Ellenberger TE, Scharer OD, Walter JC (2008) Mechanism of replication-coupled DNA interstrand crosslink repair. Cell 134(6):969–980
Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A (2010) Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell 39(1):25–35
Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ (2010) Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 329(5988):219–223
Raschle M, Smeenk G, Hansen RK, Temu T, Oka Y, Hein MY, Nagaraj N, Long DT, Walter JC, Hofmann K, Storchova Z, Cox J, Bekker-Jensen S, Mailand N, Mann M (2015) DNA repair. Proteomics reveals dynamic assembly of repair complexes during bypass of DNA cross-links. Science 348(6234):1253671
Long DT, Joukov V, Budzowska M, Walter JC (2014) BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol Cell 56(1):174–185
Knipscheer P, Raschle M, Smogorzewska A, Enoiu M, Ho TV, Scharer OD, Elledge SJ, Walter JC (2009) The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 326(5960):1698–1701
Taniguchi T, Garcia-Higuera I, Andreassen PR, Gregory RC, Grompe M, D’Andrea AD (2002) S-phase-specific interaction of the Fanconi anemia protein, FANCD2, with BRCA1 and RAD51. Blood 100(7):2414–2420
Longerich S, Kwon Y, Tsai MS, Hlaing AS, Kupfer GM, Sung P (2014) Regulation of FANCD2 and FANCI monoubiquitination by their interaction and by DNA. Nucleic Acids Res 42(9):5657–5670
Klein Douwel D, Boonen RA, Long DT, Szypowska AA, Raschle M, Walter JC, Knipscheer P (2014) XPF-ERCC1 acts in Unhooking DNA interstrand crosslinks in cooperation with FANCD2 and FANCP/SLX4. Mol Cell 54(3):460–471
Yamamoto KN, Kobayashi S, Tsuda M, Kurumizaka H, Takata M, Kono K, Jiricny J, Takeda S, Hirota K (2011) Involvement of SLX4 in interstrand cross-link repair is regulated by the Fanconi anemia pathway. Proc Natl Acad Sci USA 108(16):6492–6496
Kuraoka I, Kobertz WR, Ariza RR, Biggerstaff M, Essigmann JM, Wood RD (2000) Repair of an interstrand DNA cross-link initiated by ERCC1-XPF repair/recombination nuclease. J Biol Chem 275(34):26632–26636
Budzowska M, Graham TG, Sobeck A, Waga S, Walter JC (2015) Regulation of the Rev1-pol zeta complex during bypass of a DNA interstrand cross-link. EMBO J 34(14):1971–1985
Murina O, von Aesch C, Karakus U, Ferretti LP, Bolck HA, Hanggi K, Sartori AA (2014) FANCD2 and CtIP cooperate to repair DNA interstrand crosslinks. Cell Rep 7(4):1030–1038
Unno J, Itaya A, Taoka M, Sato K, Tomida J, Sakai W, Sugasawa K, Ishiai M, Ikura T, Isobe T, Kurumizaka H, Takata M (2014) FANCD2 binds CtIP and regulates DNA-end resection during DNA interstrand crosslink repair. Cell Rep 7(4):1039–1047
Suhasini AN, Sommers JA, Muniandy PA, Coulombe Y, Cantor SB, Masson JY, Seidman MM, Brosh RM Jr (2013) Fanconi anemia group J helicase and MRE11 nuclease interact to facilitate the DNA damage response. Mol Cell Biol 33(11):2212–2227
Long DT, Raschle M, Joukov V, Walter JC (2011) Mechanism of RAD51-dependent DNA interstrand cross-link repair. Science 333(6038):84–87
Mouw KW, D’Andrea AD (2014) Crosstalk between the nucleotide excision repair and Fanconi anemia/BRCA pathways. DNA Repair (Amst) 19:130–134
Wood RD (2010) Mammalian nucleotide excision repair proteins and interstrand crosslink repair. Environ Mol Mutagen 51(6):520–526
Enoiu M, Jiricny J, Scharer OD (2012) Repair of cisplatin-induced DNA interstrand crosslinks by a replication-independent pathway involving transcription-coupled repair and translesion synthesis. Nucleic Acids Res 40(18):8953–8964
Muniandy PA, Thapa D, Thazhathveetil AK, Liu ST, Seidman MM (2009) Repair of laser-localized DNA interstrand cross-links in G1 phase mammalian cells. J Biol Chem 284(41):27908–27917
Sarkar S, Davies AA, Ulrich HD, McHugh PJ (2006) DNA interstrand crosslink repair during G1 involves nucleotide excision repair and DNA polymerase zeta. EMBO J 25(6):1285–1294
Wang X, Peterson CA, Zheng H, Nairn RS, Legerski RJ, Li L (2001) Involvement of nucleotide excision repair in a recombination-independent and error-prone pathway of DNA interstrand cross-link repair. Mol Cell Biol 21(3):713–720
Drew HR, Wing RM, Takano T, Broka C, Tanaka S, Itakura K, Dickerson RE (1981) Structure of a B-DNA dodecamer: conformation and dynamics. Proc Natl Acad Sci USA 78(4):2179–2183
Acknowledgments
We apologize to authors whose work was not cited in this publication due to space limitations. We thank reviewers for helpful comments. The Cohn laboratory is supported by Grant UF100717 from the Royal Society (M.A.C.), Grant C5255/A18085 from Cancer Research UK (CR-UK) through the CRUK Oxford Centre (M.A.C.), Grant MR/N021002/1 from the MRC (M.A.C.), an MRC-Clarendon Scholarship (D.L.M.), the Taiwanese Government (C.C.L) and a Goodger Scholarship (C.C.L).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution 4.0 International License (http://creativecommons.org/licenses/by/4.0/), which permits unrestricted use, distribution, and reproduction in any medium, provided you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made.
About this article

Cite this article
Lopez-Martinez, D., Liang, CC. & Cohn, M.A. Cellular response to DNA interstrand crosslinks: the Fanconi anemia pathway. Cell. Mol. Life Sci. 73, 3097–3114 (2016). https://doi.org/10.1007/s00018-016-2218-x
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00018-016-2218-x