
Abstract
TFE3 and TFEB, as the master regulators of lysosome biogenesis and autophagy, are well characterized to enhance the synaptic protein α-synuclein degradation in protecting against Parkinson’s disease (PD) and their levels are significantly decreased in the brain of PD patients. However, how TFE3 and TFEB are regulated during PD pathogenesis remains largely vague. Herein, we identified that programmed cell death 4 (PDCD4) promoted pathologic α-synuclein accumulation to facilitate PD development via suppressing both TFE3 and TFEB translation. Conversely, PDCD4 deficiency significantly augmented global and nuclear TFE3 and TFEB distributions to alleviate neurodegeneration in a mouse model of PD with overexpressing α-synuclein in the striatum. Mechanistically, like TFEB as we reported before, PDCD4 also suppressed TFE3 translation, rather than influencing its transcription and protein stability, to restrain its nuclear translocation and lysosomal functions, eventually leading to α-synuclein aggregation. We proved that the two MA3 domains of PDCD4 mediated the translational suppression of TFE3 through binding to its 5’-UTR of mRNA in an eIF-4A dependent manner. Based on this, we developed a blood-brain barrier penetrating RVG polypeptide modified small RNA drug against pdcd4 to efficiently prevent α-synuclein neurodegeneration in improving PD symptoms by intraperitoneal injections. Together, we suggest PDCD4 as a PD-risk protein to facilitate α-synuclein neurodegeneration via suppressing TFE3 and TFEB translation and further provide a potential small RNA drug against pdcd4 to treat PD by intraperitoneal injections.
Similar content being viewed by others


Introduction
Transcription factor E3 (TFE3) and transcription factor EB (TFEB) are two major members of the microphthalmia-transcription factor E (MiT/TFE) subfamily and well characterized as master regulators of lysosome biogenesis and autophagy1,2,3,4. TFEB is the first member to be identified as a master trans-activator of lysosomal biogenesis and thus its nuclei-cytoplasmic shuttling, which is largely dependent on its multiple serine phosphorylation modulations by mTORC1, PKCβ, ERK2 and GSK3 kinases, has been extensively evidenced to critically controls its activity5,6,7. More recently, TFE3 has been proved to also bind Coordinated Lysosomal Expression and Regulation (CLEAR) elements, which are highly conserved among the lysosomal biogenesis and autophagy genes, and induce lysosomal biogenesis and autophagy upon activation3. Similar to TFEB, the phosphorylation status of TFE3 is critical for its cytosol and nucleus localization. mTORC1 is the main kinase to phosphorylate TFE3 at Ser321 and thus promotes the interaction of TFE3 with 14-3-3 proteins and their cytosol retention3. Under metabolic stress conditions, such as nutrient deprivation and ER stress, TFE3 and TFEB translocate from the cytoplasm into the nucleus to trigger lysosomal biogenesis and autophagy through mTORC1-dependent and -independent manner8,9. Besides, PPP3/calcineurin can also dephosphorylate TFE3 and TFEB to facilitate their nuclear translocation8,10. Although TFE3 is subject to nuclei-cytoplasmic shuttling and behaves similarly to TFEB3,7,11,12, TFE3 displays more vulnerability than TFEB to form gene fusions with PRCC, ASPSCR1, SFPQ, NONO and CLTS in a subset of renal cell carcinomas (RCC), suggesting that TFE3 and TFEB are distinctly regulated4. Moreover, TFE3 targets include not only autophagic and lysosomal genes, but also ATF4, which is an essential master regulator of the integrated stress response and involved in cellular response to stress, signaling and apoptosis8. Therefore, TFE3 may be more closely linked than TFEB in integrating cooperation between different cellular stress pathways8. However, other regulators of TFE3 independent of phosphorylation modulations remains largely elusive.
Parkinson’s disease (PD) is the second-most-common progressive neurodegenerative disorder with a selective loss of dopaminergic (DA) neurons in the substantia nigra (SN) and the development of neuronal Lewy bodies composed of abnormal α-synuclein aggregates. The proper function of autophagic and lysosomal pathway is required for the removal of excess α-synuclein to alleviate PD symptoms13,14. In PD mouse model and patients, TFEB activity and nuclear levels are responsively reduced, leading to lysosomal depletion and defective autophagy15,16. Importantly, decreased nuclear TFE3 is recently found in the DA neurons of postmortem human PD brains and TFE3 knockdown induces autophagy dysfunction and neurodegeneration of DA neurons in mice, thereby promoting PD development17. Therefore, TFE3 or TFEB upregulation or activation through pharmacological inhibition of mTORC1 can trigger clearance of excess α-synuclein and halt the progression of neurotoxicity17,18,19,20,21,22. However, up to date, the key genes that cause TFE3 or TFEB reduction in PD are still unclear.
Programmed cell death 4 (PDCD4) plays fundamental roles in tumor growth and metastasis, obesity and atherosclerosis23,24,25,26,27,28. It can be ubiquitously expressed in different species including human, mice, rat and chicken and the deduced amino acid sequences are highly conserved among these species29,30,31. However, multiple environmental, nutritional, inflammation-related factors and stresses can affect its levels, supporting that it is a sensor of external and intracellular changes of nutrients, stresses and stimulators32,33,34,35,36,37,38. PDCD4 contains two MA-3 domains (164-275aa, 327-440aa), two phosphorylation sites, two clusters of positively charged residues and two nuclear export signals (NES). Among of them, MA-3 domains of PDCD4 are homologous to the MA-3 domain of eIF4G and compete with it for eIF4A binding39,40. Distinct from eIF4G, PDCD4 binding with eIF4A can inhibit eIF4A’s helicase activity, which is necessary for unwinding structured mRNA and permitting translation initiation complex recruitment to move along the mRNA and locate the translation initiation codon during translation initiation41. Therefore, PDCD4-mediated eIF4A’s helicase activity suppression leads to the halted translation of mRNAs with structured 5’UTR more efficiently than those without42. Apart from the eIF4A-dependent mechanism, PDCD4 can directly bind to mRNA to suppress its translation, such as antiapoptotic proteins Bcl-XL and X chromosome-linked inhibitor of apoptosis (XIAP)43.
In the central nervous system, we reveal that PDCD4 is highly expressed in multiple cell types, including neuron and microglia. Importantly, PDCD4 can suppress brain-derived neurotrophic factor (BDNF) translation in an eIF4A dependent manner to promote major depression disorder pathogenesis37,44. Recently, we also discover that TFEB translation, rather than phosphorylation or protein stability, is significantly suppressed by PDCD4 in an eIF4A dependent manner38. However, PDCD4 role and function in PD pathogenesis is unknown.
Here, we discovered that PDCD4 may be a PD-risk protein and responsively increased to promote aberrant α-synuclein neurodegeneration in mouse model of PD with AAV9-based overexpression of pathogenic human α-synuclein (SYN-PD). Conversely, PDCD4 deficiency significantly enhanced not only TFEB, but also TFE3 activity to trigger lysosomal degradation of α-synuclein and thus improved PD-related motor disfunctions. The protein stability of TFE3 was not influenced by PDCD4. Instead, the two MA-3 domains of PDCD4 were identified to target two types of 5’UTR of TFE3 mRNA and thus suppress their translation in an eIF4A dependent manner. To support PDCD4 as a novel therapeutic target, we constructed a small interfering RNA drug against PDCD4 with RVG polypeptides to cross BBB, and performed intraperitoneal injections to efficiently prevent α-synuclein neurodegeneration in PD mice. Together, we suggest PDCD4 as a novel PD-risk gene to suppress TFE3 and TFEB translation in preventing lysosomal degradation of pathological α-synuclein and further provide a potential small RNA drug against PDCD4 to treat PD by intraperitoneal injections.
Results
PDCD4 is responsively upregulated by different PD-inducing stressors and causes cellular α-synuclein accumulation
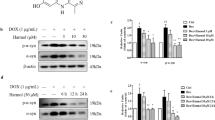
We previously reported that PDCD4 can inhibit global TFEB level to reduce lysosomal biogenesis38. Thus, we hypothesized that PDCD4 might be involved in lysosomal dysfunction-related PD pathogenesis and performed the experiments to observe PDCD4 changes in response to α-synuclein fibrils (PFFs) induced stress. Interestingly, the pathologic α-synuclein treatments with PFFs significantly induced PDCD4 accumulations along with the increased levels of the phosphorylated α-synuclein at serine 129 (p-α-SYN) in both SH-SY5Y and BV2 cells (Fig. 1a, b). Further, in adeno-associated virus (AAV9)-derived α-synuclein-overexpression (AAV9-α-syn) mice, endogenous PDCD4 levels were all remarkably higher in the corpus striatum along with the accumulations of p-α-SYN and total α-synuclein compared with wild type mice (Fig. 1c). These results indicate that PDCD4 is responsively upregulated in vitro and in vivo.

The protein level of PDCD4, p-α-SYN and α-SYN were detected by western blot in SH-SY5Y (a) or BV2 cells (b) with or without PFFs stimulation. The quantities analyses were then performed in three independent experiments. c The changes of PDCD4, p-α-SYN and α-SYN in the corpus striatum of wild type or AAV9-derived α-syn overexpression-induced PD (AAV9-α-syn) mice (n = 3). The protein levels of PDCD4 and α- SYN were detected by western blot in SH-SY5Y (d) or BV2 cells (e) in the presence or absence of small RNA interference against PDCD4 with PFFs stimulation, the protein levels of p-α-SYN were detected in the presence or absence of small RNA interference against PDCD4 with and without PFFs stimulation. Three independent experiments were calculated. f The changes of PDCD4, p-α-SYN and α- SYN in primary dopamine neurons of wild type or PDCD4-knockout mice (n = 3) with or without PFFs stimulation. g The expression levels of cellular α- SYN were examined by western blot with increasing exogenous PDCD4 in SH-SY5Y cells. Three independent experiments were calculated. (Values are shown as mean ± S.D., unpaired two-tailed t test and one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Furthermore, we found that the downregulations of PDCD4 by small interfering RNA treatments significantly reduced cellular p-α-SYN and α-synuclein levels in PFFs-treated SH-SY5Y and BV2 cells (Fig. 1d, e). In PDCD4 knockout primary dopamine neurons, they also have decreased levels of cellular p-α-SYN and α-synuclein compared with wild type cells with or without PFFs incubation (Fig. 1f). On the contrary, PDCD4 overexpression could significantly induce α-synuclein accumulations in a dose-dependently manner (Fig. 1g). Altogether, these data suggest that PDCD4 is responsively increased in PD mice and its level positively correlated with aberrant α-synuclein accumulation, suggesting that PDCD4 may be a risk protein in inducing PD pathogenesis via promoting aberrant α-synuclein aggregations.
PDCD4 deficiency significantly improves motor dysfunction and pathological change of PD-mice, and its re-expression in the striatum abrogates these effects
As we described above, PD-inducing stressors can stimulate PDCD4 expression and the upregulated PDCD4 induce cellular α-synuclein aggregations, suggesting that PDCD4 may contribute to PD pathogenesis and development. To confirm the role of PDCD4 in PD, we used PDCD4 knockout mice and re-expressed PDCD4 in the striatum of PDCD4 knockout mice to evaluate the effects of PDCD4 deficiency and its re-express on PD-related motor symptoms and pathogenesis in a AAV9-α-syn-induced PD mouse model. We firstly observed that PDCD4-deficient mice significantly improved PD-related motor dysfunctions, illustrated by pole test, beam-walking and rotarod test (Fig. 2a, b). Consistently, the TH (+) neuronal loss in the bilateral substantia nigra pars compacta (SNc) and aberrant accumulations of p-α-SYN and α-synuclein in the striatum of the PD mice were both significantly blocked in response to PDCD4 deletion (Fig. 2c, d, Supplementary Fig. 1a). To further support PDCD4 contribution to PD pathogenesis, we re-expressed PDCD4 in the striatum of PDCD4 knockout mice by stereotaxic injections of AAV9- PDCD4 expression vectors and found that PDCD4 replenishment obviously abrogated PDCD4 deficiency-induced improvements of behavioral deficits, DA neuron loss in the bilateral SNc and the aggregations of p-α-SYN and α-synuclein in PD mice (Fig. 2e–h, Supplementary Fig. 1b). These data indicate that PDCD4 is a PD-risk molecule and its deficiency facilitates aberrant α-synuclein clearance, thereby preventing PD pathogenesis and development.

a–d The impact of PDCD4 knockout on motor dysfunction and pathological change of PD-mice. The diagram of experiments was shown in a: The elements of the chart were from the BioRender.com with the Confirmation of Publication and Licensing Rights (GA26XUCU08); The adeno-associated viruses without or with α-synuclein (AAV9-α-syn) were injected into the substantia of wild mice or PDCD4 knockout mice for 8 weeks to induce PD and then the PD-related motor impairments were examined by the pole test, beam-walking test and rotarod test (WT + ctrl, n = 7; WT+α-syn, n = 9; KO+α-syn, n = 7) (b); The representative immunohistochemical staining image of TH positive neurons in substantia nigra pars compacta (SNc) were shown and then ratio of TH positive neurons in the whole brain sections was calculated (n = 3 per group, select three brain slices from each mouse for statistical analysis, length of scale bars indicates 1 mm and 200 μm respectively) (c); Protein levels of PDCD4, p-α-SYN and α-SYN were determined by western blot in mouse striatum of each group (d). e–h The impact of PDCD4 re-expression in the striatum of PDCD4 knockout mice on motor dysfunction and pathological change of PD-mice. The diagram of PDCD4 re-expression in the striatum of PDCD4 knockout mice was shown in e: The elements of the chart were from the BioRender.com with the Confirmation of Publication and Licensing Rights (GA26XUCU08); The PD-related motor impairments were examined by the pole test, beam-walking test and rotarod test (WT + ctrl, n = 7; WT + α-syn, n = 7; KO + α-syn, n = 6; KO + α-syn + PDCD4, n = 6) (f); The representative immunohistochemical staining image of TH positive neurons in substantia nigra pars compacta (SNc) were shown and the ratio of TH positive neurons in the whole brain sections was calculated (n = 3, select three brain slices from each mouse for statistical analysis, length of scale bars indicates 1 mm and 200 μm respectively) (g); The protein levels of PDCD4, p-α-SYN and α-SYN in the striatum of each group of mice were detected by western blot with indicated antibodies (h). (Values are shown as mean ± S.D., one-way ANOVA and multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
The role of PDCD4 in promoting PD development depends on its suppression on expressions of both TFE3 and TFEB
Next, we sought to further investigate the mechanism of PDCD4 regulation in α-synuclein aggregations during PD pathogenesis. Abundant evidences have demonstrated that lysosomal function and autophagy are predominantly responsible for α-synuclein degradation. We previously reported that PDCD4 can inhibit global level of lysosomal key transcription factors TFEB to reduce lysosome biogenesis in macrophage or MEF cells27,38,45. Since TFE3, another important member in same subfamily, is also involved in PD, we examined changes of both TFEB and TFE3 in PDCD4 knockout PD mice and found that the reductions of both TFE3 and TFEB levels in the striatum of PD mice were significantly reversed in response to PDCD4 deficiency (Fig. 3a). The decrease of Cathepsin D (CTSD), which is transcribed by TFE3 and TFEB and a critical lysosomal enzyme for α-synuclein degradation, was correspondingly blocked as well by PDCD4 deficiency. Conversely, PDCD4 re-expression in the striatum of PDCD4 knockout PD mice abolished the increases of TFE3, TFEB, and CTSD caused by PDCD4 deficiency, suggesting that PDCD4 negatively controls both TFE3 and TFEB (Fig. 3b). Consistently, both TFE3 and TFEB levels were significantly elevated in PDCD4-deficient primary DA neurons together with reduced α-synuclein accumulation (Fig. 3c). Meanwhile, the expressions of TFE3- and TFEB-governed lysosomal genes, such as CTSD and LAMP in PDCD4-deficient primary DA neurons were increased compared to PDCD4 wild type cells (Fig. 3d). Furthermore, PDCD4 overexpression significantly suppressed the levels of TFE3, TFEB and CTSD with increased the levels of p-α-SYN and α-synuclein, while down-regulations of PDCD4 by small interfering RNA blocked their reductions in SH-SY5Y and BV2 cells with or without PFFs incubation (Fig. 3e–g). More importantly, the treatments of lysosome activity inhibitor chloroquine (CQ) abolished the enhanced clearances of p-α-SYN and α-synuclein by PDCD4 knockdown, suggesting that PDCD4 influence on α-synuclein accumulation via TFEB and TFE3 depends on the lysosomal activity of degrading protein (Fig. 3h). Together, these data demonstrates that PDCD4 negatively controls levels of both TFE3 and TFEB.

a The expressions of TFE3, TFEB, CTSD, PDCD4 and α-SYN in the striatum of wild type or Pdcd4-deficient mice were examined by Western blot. The quantities analyses were performed. (n = 4). Values are shown as mean ± S.D., one-way ANOVA and multiple comparison test. b The expressions of TFE3, TFEB, CTSD, PDCD4 and α-syn in the striatum of wild type or Pdcd4-deficient mice with or without Pdcd4 replenishment were examined by Western blot. The quantities analyses were performed. (n = 5); Values are shown as mean ± S.D., one-way ANOVA and multiple comparison test. c The protein levels of TFE3, TFEB, CTSD, PDCD4 and α-SYN in the primary dopamine neurons from wild type or Pdcd4-deficient mice (n = 4) with or without PFFs incubation. The quantities analyses were performed. The values are shown as mean ± S.D., one-way ANOVA and multiple comparison test, d The mRNA levels of CTSD, and LAMP1, which are transcribed by TFE3 and TFEB, were qualitatively measured in the primary dopamine neurons from wild type or Pdcd4-deficient mice with or without PFFs incubation. The quantities analyses were performed. Values are shown as mean ± S.D., one-way ANOVA and multiple comparison test. e The expression changes of TFE3, TFEB, CTSD, PDCD4, p-α-SYN and α-SYN were examined by western blot in SH-SY5Y cells in response to increasing exogeneous PDCD4. Three independent experiments were calculated. (Means ± S.D.; unpaired two-tailed t test). The protein levels of TFE3, TFEB, CTSD, PDCD4 and α-SYN were detected by western blot in SH-SY5Y (f) or BV2 (g) cells in the presence or absence of small RNA interference against PDCD4 with PFFs stimulation. h The protein levels of PDCD4, p-α-SYN and α-SYN were detected by western blot in SH-SY5Y in the presence or absence of small RNA interference against PDCD4 with PFFs and CQ stimulation. Three independent experiments were calculated and used for quantities analysis. (Values are shown as mean ±S.D., one-way ANOVA and multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
To further investigate the requirements of TFE3 and TFEB in the improvement of PD mice in the absence of PDCD4, we silenced TFE3 and TFEB in PDCD4-deficient PD mice via injecting recombinant AAV9s containing TFE3 and TFEB specific siRNAs to the striatum and found that their downregulations abrogated PDCD4 deficiency-caused the improvements of behavioral deficits in PD mice (Fig. 4a, b). The rescue of TH (+) neuronal loss and enhancement of p-α-SYN and α-synuclein clearance by PDCD4 deficiency were also abolished when the increased levels of TFE3 and TFEB in PDCD4-deficient mice were downregulated (Fig. 4c, d, Supplementary Fig. 2a). These data supported that TFE3 and TFEB are responsible for PDCD4 deficiency-mediated aberrant α-synuclein clearance and PD-related motor symptoms improvements. In line with these findings in PD mice, downregulations of TFE3 and TFEB abolished PDCD4 silence-induced the enhancement of p-α-SYN and α-synuclein clearance as well in both SH-SY5Y and BV2 cells with or without PFFs treatment (Fig. 4e, f). The increased levels in response to PDCD4 downregulation were also abrogated when TFE3 and TFEB were silenced (Fig. 4e, f). To further confirm the role of TFE3/TFEB-CTSD axis in α-synuclein degradation, we overexpressed CTSD and found that it reversed PDCD4 overexpression-caused α-synuclein accumulation, indicating that PDCD4 deficiency-mediated the promotion of α-synuclein degradation is dependent on elevations of TFE3/TFEB-CTSD axis (Fig. 4g). Token together, our data demonstrate that the promoting role of PDCD4 in PD development depends on its suppression to both TFE3 and TFEB expression.

a–d The impact of silencing both TFE3 and TFEB expression in the striatum of PDCD4 knockout mice on motor dysfunction and pathological change of PD-mice. The diagram of down-regulating TFE3 and TFEB in the striatum of PDCD4 knockout mice was shown in a: The elements of the chart were from the BioRender.com with the Confirmation of Publication and Licensing Rights (GA26XUCU08); The PD-related motor impairments were examined by the pole test, beam-walking test and rotarod test (b), (WT + ctrl, n = 5; WT + α-syn, n = 5; KO + α-syn, n = 5; KO + α-syn + sitfe3 + sitfeb, n = 4); The representative immunohistochemical staining image of TH positive neurons in substantia nigra pars compacta (SNc) were shown and then ratio of TH positive neurons in the whole brain sections was calculated (n = 3, select three brain slices from each mouse for statistical analysis, length of scale bars indicates 1 mm and 200 μm respectively) (c); The protein levels of PDCD4, p-α-SYN and α-SYN in the striatum of each group of mice were detected by western blot with indicated antibodies (n = 5 per group) (d). The protein levels of TFE3, TFEB, CTSD, PDCD4 and α-SYN were detected by western blot in SH-SY5Y (e) or BV2(p-α-SYN) (f) cells in the presence or absence of small RNA interference against PDCD4 alone or together with TFE3 and TFEB in response to PFFs stimulation. g The protein levels of CTSD, PDCD4 and α-SYN were detected by western blot in 293T cells in the presence or absence of CTSD and PDCD4 recombinant expressing plasmids. Three independent experiments were calculated. (Values are shown as mean ±S.D., one-way ANOVA and multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
The MA3 domains of PDCD4 mediate the suppression of TFE3 translation through an eIF4A dependent manner
As we described above, PDCD4 targets TFE3 and TFEB to control PD progression. Our previous research has investigated the mechanism that PDCD4 suppresses TFEB expression by using PDCD4-deficient and re-expressed mouse embryonic fibroblasts (MEFs)38. Here, we addressed the detail molecule mechanism that PDCD4 regulates TFE3. We detected firstly whether PDCD4 knockout also impacted TFE3 expression in MEFs. As shown in Supplementary Fig. S1, PDCD4 knockout also significantly upregulated the TFE3 expression (Supplementary Fig. 3a) and its downstream targeting genes (such as CTSD and LAMP1) on protein level (Supplementary Fig. 3a, c) or RNA level (Supplementary Fig. 3b) in MEF and its silence by TFE3 specific small RNA (sitfe3) abolished the upregulation of CTSD and LAMP1, suggesting PDCD4 regulation on TFE3 is widely present in multiple types of cells. The nuclear translocation has been evidenced to tightly control its activity of transcribing targeting genes4,9,16. Here, we found that the nuclear TFE3 levels, similar to those of TFEB, were significantly elevated by PDCD4 deficiency (Fig. 5a). In contrast, the increase of nuclear TFE3 was remarkably abrogated with PDCD4 replenishment in PDCD4 knockout MEF cells (Fig. 5a). Moreover, we observed that there is more nuclear translocation of TFE3 in PDCD4-deficient cells via immune-fluorescence (IF) assay, while PDCD4 replenishment prevented nuclear TFE3 elevation (Fig. 5b). Taken together, these data demonstrate that PDCD4 may suppress TFE3 nuclear translocation. Extensive reports demonstrate that mTOR-mediated TFE3 serine S321 phosphorylation can promote its binding to 14-3-3 protein, thereby resulting in sequestration of TFE3 in the cytosol8,46,47. However, we found that TFE3 nuclear distributions of the PDCD4 null cells were further elevated and still possessed much higher abundance compared with those of wild type cells in response to mTORC1 inhibitor, Torin-1 treatment (Fig. 5c). In line with these findings, Torin-1 was incapable of abolishing nuclear TFE3 distinguishments caused by PDCD4 deficiency in MEF cells, whereas TFE3 nuclear levels were all further increased in the presence or absence of PDCD4 with or without exogeneous PDCD4 replenishment upon Torin-1 stimulation (Fig. 5c). Based on these data, we concluded that PDCD4 suppresses TFE3 nuclear localization independent of mTORC1-mediated phosphorylation.

a The levels of cytoplasmic and nuclear TFE3 were determined by western blot in MEFs of wild type and Pdcd4-deficient mice with or without exogenous PDCD4 expression. b The distributions and levels of cytoplasmic and nuclear TFE3 were observed by immuno-fluorescence assay in MEFs from wild type and Pdcd4-deficient mice with or without exogenous PDCD4 expression. c The levels of cytoplasmic and nuclear TFE3 were determined by western blot in MEFs of wild type and Pdcd4-deficient mice with or without exogenous PDCD4 expression in response to Torin 1 (250 nM, 2 h). d The mRNA levels of TFE3 in MEFs form wild type and Pdcd4-deficient mice with or without exogenous PDCD4 expression were evaluated by QPCR. e The wild type or Pdcd4-deficient MEFs were treated with CHX (100 μg/ml) for indicated times and the levels of TFE3 and TFEB were examined by western blot. f The ubiquitination modulations of TFE3 were examined in wild type or Pdcd4-deficient MEFs. g The changes of TFE3 in wild type or Pdcd4-deficient MEFs with or without MG132 (10 μM, 4 h) treatment were detected by western blot. h The protein levels of TFE3, TFEB, CTSD and α-SYN in SH-SY5Y cells in the absence or presence of small RNAs against PDCD4 or eIF4A alone or together. Three independent experiments were calculated and used for quantities analysis. (Values are shown as mean ±S.D., one-way ANOVA and multiple comparison test. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001). i The RNA binding protein immunoprecipitation (RIP) experiments were performed to examined the binding ability of PDCD4 with TFE3 mRNA. j, k The luciferase activities of different types of TFE3 5’-UTR with or without PDCD4 expression were quantitively analyzed in HEK-293T cells. Name the 5’-UTR of TFE3 isoform a and b as 5’-UTR-1, name the 5’UTR of TFE3 isoform c1, c2, c3, d as 5’-UTR-2, 3, 4, 5. l The TFE3 changes in response to different PDCD4 truncations were examined by western blot in wild type or Pdcd4-deficient MEFs.
Further, we noted that the cytosolic TFE3 was also increased in response to PDCD4 deficiency, suggesting that the global TFE3 levels might be suppressed by PDCD4 (Fig. 5a, b). These findings prompted us to propose a possibility of PDCD4 regulation in TFE3 transcription. However, the results showed that mRNA levels of TFE3 were not influenced by PDCD4 in MEFs (Fig. 5d) or in primary DA neuron (Supplementary Fig. 4a), SH-SY5Y (Supplementary Fig. 4b, c), and BV2 (Supplementary Fig. 4d), indicating that TFE3 protein level rather than transcription is regulated by PDCD4. Besides, cycloheximide (CHX)-treated wild type and PDCD4−/− MEFs showed a similar tendency of TFE3 (Fig. 5e), suggesting PDCD4 does not impact TFE3 stability. In addition, the polyubiquitination modulations of TFE3 were not altered by PDCD4 and MG132 treatments didn’t abrogate global TFE3 increase in PDCD4-deficient MEFs cells (Fig. 5f, g), meaning that PDCD4 also has no impact TFE3 ubiquitination-mediated degradation. From these observations, we propose that PDCD4 suppresses TFE3 global protein levels without influencing its transcription or protein stability.
PDCD4 is a common suppressor of protein translation in an eIF4A-dependent manner and we speculated that TFE3 might be a novel target of PDCD4. To confirm this, we used small interfering RNAs against eIF4A to downregulate eIF4A and found that eIF4A downregulations abrogated the elevations of TFE3 and its targeting gene CTSD, similarly to TFEB (Fig. 5h). Importantly, the promotions of α-synuclein clearance caused by PDCD4 knockdown was also abolished in response to eIF4A downregulations, indicating that the suppression of TFE3 translation by PDCD4 is dependent on eIF4A (Fig. 5h). Besides, we found that TFE3 mRNA could been detected in PDCD4-eIF4A complex in RNA immunoprecipitation (RIP) experiments (Fig. 5i) and that PDCD4 selectively inhibited translational activities of TFE3 with 5’UTR-1 and 5UTR-2, but not 5’UTR-3, 5’UTR-4 and 5UTR-5 by the luciferase reporter assay (Fig. 5j, k). Furthermore, to identify the critical subregions of PDCD4 in responsible for TFE3 translation suppression, we constructed various truncated PDCD4 to evaluate their inhibitory effects on TFE3. Among of them, we discovered that the two MA3 domains of PDCD4, including MA3-1 and MA3-2, rather than RNA bind regions (RD), were both required for the inhibition of TFE3 translation (Fig. 5l). From these data, we conclude that the MA3 domains of PDCD4 mediates the suppression of TFE3 translation through an eIF4A dependent manner.
The peripheral administration of PDCD4 specific small RNAs with a blood-brain barrier-penetrating RVG(RVG-sipdcd4) significantly ameliorates Parkinson’s disease symptoms
As we identified a novel role of PDCD4 in promoting PD progression via suppressing TFEB and TFE3 translation, and found that PDCD4 knockout mice displayed the significant improvement of neurodegenerations, suggesting PDCD4 may be a promising target for PD therapy. Therefore, we explored the possibility that PDCD4 specific small RNAs (sipdcd4) silences PDCD4 to treats PD. To make peripheral administration of sipdcd4 cross blood brain barrier (BBB) and enter brain, the sipdcd4 was modified with a BBB-penetrating RVG polypeptide (RVG-sipdcd4) as our previous research47 and examine its therapeutic effects in PD. Firstly, we observed that RVG-sipdcd4 was able to efficiently downregulate PDCD4 expression and thus upregulate TFE3, TFEB and CTSD levels in both SH-SY5Y and BV2 cells (Fig. 6a, b). The p-α-SYN and α-synuclein accumulations were also significantly reduced by RVG-sipdcd4 treatment (Fig. 6a, b). These data demonstrate that RVG-sipdcd4 has a high efficacy in silencing PDCD4 in neuronal and microglia by which upregulate the expression of TFE3, TFEB to trigger α-synuclein degradation. Furthermore, we evaluated its effects in PD mice by intraperitoneal injections (Fig. 6c). As expected, RVG-sipdcd4 treatment remarkably improved the PD-related motor dysfunctions in PD mice (Fig. 6d). Moreover, the reductions of TFE3/TFEB and CTSD in the striatum of PD mice were significantly reversed by PDCD4 down-regulation and the aberrant p-α-SYN and α-synuclein accumulations in the striatum were thus decreased (Fig. 6e). The DA neuron loss in the SNc of the PD mice was greatly rescued by the intraperitoneal injections of RVG-sipdcd4 (Fig. 6f). We noted that peripheral administration of RVG-sipdcd4 had no obvious side effects. As shown as in Supplementary Fig. S3, RVG-sipdcd4 therapy didn’t affect mice body weight, liver weight, kidney weight, and spleen weight (Supplementary Fig. 5a–d), and liver or kidney functions or blood glucose illustrated by activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST), aspartate aminotransferase/alanine aminotransferase (S/L), alkaline phosphatase (ALP), total protein (TP), albumin (ALB), globulin (GloB) and albumin/globulin (A/G), lactate dehydrogenase (LDH), blood glucose (GLU), Urea, Crea (Cr) and uric acid (UA) (Supplementary Fig. 5e–q). Collectively, the peripheral administrations of RVG-sipdcd4 significantly ameliorate Parkinson’s disease symptoms without obvious side effects, suggesting RVG-sipdcd4 will be a promising drug for PD therapy.

The protein levels of TFE3, TFEB, CTSD, p-α-SYN and α-SYN in SH-SY5Y (a) or BV2 (b) cells in the absence or presence of RVG-siPdcd4. Three independent experiments were calculated. c–f Intraperitoneal injection of RVG-siPdcd4 to investigate the effect on motor dysfunction and pathological change of PD-mice. The diagram of intraperitoneal injecting RVG-sinc or RVG-sipdcd4 into wild mice was shown in c: The elements of the chart were from the BioRender.com with the Confirmation of Publication and Licensing Rights (GA26XUCU08); The PD-related motor impairments were examined by the rotarod test (RVG-sinc, n = 10; RVG-sinc + α-syn, n = 12; RVG-sipdcd4 + α-syn, n = 12) (d); The protein levels of PDCD4, p-α-SYN and α-SYN in the striatum of each group of mice were detected by western blot with indicated antibodies (n = 5) (e). The representative immunohistochemical staining image of TH positive neurons in substantia nigra pars compacta (SNc) were shown and then ratio of TH positive neurons in the whole brain sections was calculated (n = 3, select three brain slices from each mouse for statistical analysis, length of scale bars indicates 1 mm and 200 μm respectively) (f). (Values are shown as mean ±S.D., one-way ANOVA and multiple comparison test, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001).
Discussion
Here, we establish that PDCD4 may be a novel PD-risk protein to promote excess α-synuclein accumulation and neurodegeneration in PD via co-suppressing TFE3 and TFEB. Abundant evidence supports that TFE3 and TFEB, as the master transactivates of lysosomal and autophagic genes, are critically reduced and thus widely recognized as promising targets for PD treatment2,3,8,15,16,22,48. However, a long-standing question that how they’re responsively decreased remains unsolved. In our study, we propose that PDCD4 is responsively increased in PD pathogenesis to serve as an inducer to cause TFE3 and TFEB reduction. Interestingly, PDCD4 is induced by aberrant α-synuclein to promote PD progression and the resulting elevation of PDCD4 levels can in turn further accelerate aberrant α-synuclein aggregation. Of note, BDNF has been demonstrated to inhibit α-synuclein expression and thus ameliorate PD-related pathology49. Considering that our finding of PDCD4 suppression in BDNF translation, we can’t exclude the possibility that PDCD4 deficiency-mediated improvement of PD mice might be through upregulation of BDNF as well. PDCD4 as a tumor suppressor, can inhibit cell growth and promote cell apoptosis50,51 and this anti-apoptosis function may contribute to prevent DA neuron apoptosis and reverse DA neurons loss in response to oxidative stresses during PD development. Nonetheless, these observations support that the multi-faceted functions of PDCD4 can induce α-synuclein neurodegeneration on the early stage of PD and accelerate PD progression via a positive feedback loop-mediated increase.
As for PDCD4, its own levels are subject to variations on multiple external or intracellular factors, such as nutrient availability, inflammatory stimuli, toxic agents and stresses23,34,36,37,38,52. Considering it, PDCD4 seems like an ideal detector and effector with extremely high sensitivity to dynamically monitor cellular lysosomal and autophagic activity in response to external and intracellular changes. In consistent with this conception, the lysosomal dysfunction occurs earlier than neuronal Aβ-deposition but can prime Aβ accumulation of senile plaque in AD pathogenesis53. This observation significantly updates our opinion that lysosomal and autophagic dysfunction is the result of aberrant aggregations of neuronal protein, such as α-synuclein and Aβ in neurodegenerative disease pathogenesis, such as PD and AD. Currently, lysosomal and autophagic dysfunction is thus more likely to induce PD or AD initiation. As a detector and effector of lysosomal and autophagic function, PDCD4 can raise the risk of PD to suppressing global TFE3 and TFEB translation. Therefore, PDCD4 activity needs tightly controlled. The nuclei-cytoplasmic shuttling of PDCD4 is reported and its cytoplasmic translocation is triggered by starvation to negatively regulate TFE3/TFEB mediated autophagy, thereby preventing it overactivation54. On the other side, cytosolic PDCD4 can be phosphorylated by mTORC1 downstream P70S6K and AKT and thus prone to ubiquitination and degradation through ubiquitin-proteasome pathway55,56. Therefore, mTORC1 activation-dependent PDCD4 degradation may be physiologically important to allow basic lysosomal function and autophagy flux under the nutrient-rich conditions via relieving the TFE3/TFEB translation suppression in the context of mTORC1-mediated TFE3/TFEB cytosolic retention. Together, PDCD4 is responsive to various external and intracellular stresses and thus can tightly control lysosomes and autophagy homeostasis via TFE3 and TFEB to effect PD onset and progression.
Unlike TFEB, TFE3 function in activating lysosomal biogenesis and autophagy has recently been investigated. Although they have multiple parallel characteristics, like phosphorylation-dependent nuclei-cytoplasmic shuttling by mTORC1, other regulations of TFE3 are largely unknown8,16. We here reveal a novel regulation mechanism of TFE3 at the translational level by PDCD4 while TFE3 transcription and protein stability are not influenced. Combined with our finding that PDCD4 suppresses TFEB translation, PDCD4 can target both TFE3 and TFEB to regulate cellular lysosomal activities. Previous report points out that TFE3 and TFEB can cooperate with each other to control the innate immune response in activated macrophages8,46. Therefore, the relationships of PDCD4 regulation towards TFE3 and TFEB needs further clarified to better understand how lysosomes and autophagy homeostasis is fine-tuned by PDCD4. Furthermore, whether the expression profiles of TFEB and TFE3 in various tissues and cell types is deferentially regulated by PDCD4 also requires investigation. In addition to TFEB and TFE3, PDCD4 can also target 5’-UTR structured mRNAs with the stable stem-loop of SIN1 and P53 to inhibit their translations. Because there are very few identified target mRNAs of PDCD4, it is presently unclear whether PDCD4 suppresses the translation of all RNAs containing structured 5’-UTRs. Whether there are mechanisms that permit PDCD4 to target only specific RNAs also needs further investigation.
PDCD4 is defined as a co-suppressor of TFE3 and TFEB and may thus be a promising target to enhance α-synuclein degradation for pharmacological PD treatment. It should be of note that there is a limitation in our study that we only examined the PDCD4 changes in the PD mouse model. Therefore, the PDCD4 changes in PD patient needs further investigation to better understand PDCD4 function in PD development in human. Moreover, peripheral PDCD4 has multiple benefits of inhibiting tumors and inflammation-related diseases. The brain-targeted drug for PDCD4 needs developed and we thus designed a small RNA drug with BBB-penetrating RVG polypeptide against PDCD4 (RVG-sipdcd4) to downregulate PDCD4 in neuron and microglia. We previously proved that RVG-sipdcd4 can cross the BBB with effective bioavailability to remarkably decrease both neuronal and microglial PDCD4 expression44. Here, we further confirm that RVG-sipdcd4 can silence PDCD4 and then upregulate TFE3 and TFEB to promote α-synuclein degradation and thus alleviate PD-related motor symptoms in mice. Moreover, it has no obvious side effects on peripheral organs, such as liver and kidney. Collectively, we suggest RVG-sipdcd4 as a novel central nervous system (CNS) delivery drug to downregulate PDCD4 to treat PD.
We identify PDCD4 as a novel PD-risk protein in regulating α-synuclein neurodegeneration via co-suppressing TFE3 and TFEB (Fig. 7). To support PDCD4 as a therapeutic target, we developed a CNS-delivery small RNA drug, RVG-sipdcd4, and further proved its efficacy in improving α-synuclein neurodegeneration and PD-related motor dysfunction via downregulating brain PDCD4 in a mouse model of Parkinson’s disease. Thus, it may be a promising therapeutic approach for PD patients.

The elements of the chart were from the BioRender.com with the Confirmation of Publication and Licensing Rights (GA26XUCU08).
Methods
Animals
Mice used in this study were 6- to 8-week-old and housed in the Shandong University Medical School Animal Care Facility under pathogen-free conditions, according to institutional guidelines. All animal study protocols were approved by the Animal Care and Utilization Committee of Shandong University. PDCD4 knockout (KO) mice were generated through CRISPR/Cas9 system to delete PDCD4 exon 4 by Beijing Biocytogen Co.,Ltd. (Beijing, China). sgRNAs were constructed into the pCS (Puro) sgRNA/Cas9 expression plasmid (Biocytogen, Beijing, China). Two sgRNAs with the best cutting activity were selected for the upstream and downstream target regions. In vitro, the C57BL/6J single fertilized egg was microinjected with transcript sgRNAs and Cas9 mRNA to produce the F0 mice. F0 mice were identified and crossed with C57BL/6J mice to obtain F1 mice. Wild type (WT) mice were born in the same litter. The genotype of mice was determined by DNA sequencing and PCR. The primers used for genotyping were as follows: Forward: 5’- GAAGGAGGGGAAACTGAAACAGGGAG-3’, Reversed: WT-5’-ATCTGAGCAACAGCCATACCACAGAC-3’ and KO-5’- TGCTTATCACCAATCCATGTGTCTCCT-3’. Mice were housed 4-5 per cage and a circadian cycle of 12 h light and 12 h dark with an adequate food-water supply.
Ethics approval and consent to participate
All mouse work conducted in the frame work of this project was approved by the committee responsible for animal ethics of Shandong University (No. ECSBMSSDU2021-2). Mice used in this study were housed in the Shandong University Medical School Animal Care Facility under pathogen-free conditions, according to institutional guidelines.
Consent for publication
All authors consented to publish the manuscript.
Cells and culture conditions
SH-SY5Y, BV2, and HEK-293T cell lines were purchased from Shanghai Cell Bank of Chinese Academy of Sciences (Shanghai, China) and were tested for mycoplasma contamination. SH-SY5Y was cultured in MEM/F12K medium (Cat#KGM415217-500, Keygentec, China). BV2 and HEK-293T were cultured in DMEM medium (Cat#11965-084, Gibco, USA). Mouse embryonic fibroblasts (MEFs) of C57BL/6J mice were prepared from E13.5 embryos and maintained in DMEM. All media were supplemented with 10% heat-inactivated FBS (Cat#10099-141, Gibco, USA), 100 IU/mL penicillin and 100 μg/mL streptomycin (Cat#03-031-1B, Biological Industries, Israel). Cells were all cultured in a humidified cell incubator with an atmosphere of 5% CO2 at 37 °C.
Primary dopaminergic neurons culture was performed according to a previous study57. Sacrifice an E13.5 pregnant mouse (according to institutional guidelines). Place the embryos in a 100 mm Petri dish containing sterile HBSS and wash them by transfer in 3 successive identical baths using forceps. Carefully remove and discard the fore- and hindbrain regions. To the rostral side, cut close to the thalamic region, to the caudal side cut at the isthmus region, remove the superior colliculus. Collect the dissected segments, without the meninges, in a sterile 15 ml tube filled with HBSS. The brain tissue is washed, digested and centrifuged to collect cell precipitates. Incubate the plate coated with poly-L-lysine. The culture medium of Primary dopaminergic neurons is Neurobasal (Cat#21103049, Gibco, USA) with B27(Cat#17504044, Gibco, USA), 100 IU/mL penicillin and 100 μg/mL streptomycin (Cat#03-031-1B, Biological Industries, Israel). No medium change is necessary. Dopaminergic neurons are mature after around 7 days in vitro.
In addition, to block the protein synthesis pathway, cycloheximide (CHX, 100 μg/ml) (Cat# S7418, Selleck, USA) was used for 3 and 6 h; To block the proteasome degradation pathway, cells were treated with MG132 (10 μM) (Cat# S2619, Selleck, USA) for another 4 hours; In order to inhibit mTOR, cells were washed twice with preheated PBS and changed the growth medium containing Torin-1 (250 nM) (Cat#S2827, Selleck, USA) for 2 hours. Three independent experiments have carried out for biological replicates. Primary dopaminergic neurons and SH-SY5Y were stimulated by PFFs (500 ng/mL, Cat#No.SPR324, StressMarq, Canada) for 24 h and BV2 was stimulated by 500 ng/mL PFFs for 12 h.
Plasmid construction
The expression vector pcDNA3.1-Flag-pdcd4 and pEGFP- PDCD4 encodes full-length mouse PDCD4 (NM_011050.4). The following expression vectors with mouse PDCD4 mutants were used: pcDNA3.1-Flag-pdcd4-ΔRBD-1 and pcDNA3.1-Flag-pdcd4-ΔRBD-2 encode RNA-binding domain deficient PDCD4 proteins lacking nucleotide 151-204 or 288-369, respectively. pcDNA3.1-Flag-pdcd4-ΔMA3-1 and pcDNA3.1-Flag-pdcd4-ΔMA3-2 express MA3 domain deficient PDCD4 proteins, which can’t bind with eIF4A, lacking nucleotide 471-906 or 957-1347, respectively. The vectors of pGL3 (Cat#E1771, Promega, USA) contains five isoforms of mouse tfe3(Gene ID: 209446) 5’UTRs. The 5’UTRs are inserted in front the initiation codon of luciferase.
Transfection of plasmid and siRNA
Transient transfection of plasmid DNA was performed using Lipofectamine 3000 (Cat# L3000075, Invitrogen, USA). Cells were transfected at 70–80% confluence in serum-free Opti-MEM (Cat#11058021, Gibco, USA) according to the manufacturer’s instructions. siRNA transfection was performed using INTERFER in (Cat#409-10, Poly plus, France) according to the manufacturer’s protocol. TFE3, TFEB, PDCD4 and eIF4A specific siRNAs and control nonspecific siRNA were purchased from Gene pharma (Shanghai, China). The siRNA sequences are shown in Supplementary Table 1.
Real-time PCR
Total RNA was extracted by TRIzol reagent (Tiangen, Beijing, China) following the manufacturer’s instructions. After the adjustment of RNA concentration, 1% agarose gel was used to check the RNA quality and amount. The purified total RNA (1 µg) was used for reverse transcription by ReverTra Ace qPCR RT Kit (Cat#FSQ-101, Toyobo, Japan). The cDNAs were amplified by the THUNDERBIRD SYBR qPCR Mix (Cat#QPS-201, Toyobo, Japan) on a Step One Plus Real Time PCR System (Bio-Rad, Hercules, CA, USA). The primer sequences are shown in Supplementary Table 2. Three independent experiments have carried out for biological replicates. The housekeeping gene, 18 s or β-actin, was used as an internal control to normalize the variability in expression levels. Relative quantification of gene expression was analyzed by the comparative Ct method (2 –ΔΔCt). 2 –ΔΔCt = [(Ct gene of interest – Ct internal control) sample A − (Ct gene of interest – Ct internal control) sample WT]. Gene expression level in wild type were normalized to 1.
Western blot analysis
Cultured cells or tissue were collected and lysed in RIPA buffer supplemented with 1% Protease Inhibitor and Phosphatase Inhibitor Cocktail (Cat#200-664-3, Sigma-Aldrich, USA). Nuclear and Cytoplasmic Extraction Reagents (Cat#78833, Thermo Fisher Scientific, USA) was used to extract the nuclear and cytoplasmic proteins according to the user’s manual. 30 μg protein were separated on 10% SDS-polyacrylamide gel electrophoresis and then transferred to a 0.45 μm nitrocellulose membranes (Cat#HAWP01300, Millipore, USA). Pre-stained Protein Ladder (Cat#26619, Thermo Fisher Scientific, USA) was used to indicate protein molecular weight. The following primary antibodies were used: anti-PDCD4 (Cat#9535S, CST, USA), anti-CTSD (Cat#21327-1-AP, Proteintech, China), anti-TFE3 (Cat #14779, CST, USA), anti-TFEB (Cat#13372-1-AP, Proteintech, China), anti-α-synuclein (Cat #4179, CST, USA), anti-phospho-α-synuclein(ser129) (Cat #23706, CST, USA), anti-p-P70S6K (Cat#9204, CST, USA), anti-Ubiquitin (Cat#sc-8017, Santa, USA), anti-eIF4A (Cat#2013, CST, USA), anti-GAPDH (Cat#60004-1-Ig, Proteintech, China), anti-Histone-H3 (Cat#17168-1-AP, Proteintech, China), anti-β-actin (Cat#66009-1-Ig, Proteintech, China), anti-Flag (Cat#66008-3-Ig, Proteintech, China), anti-His (Cat#66005-1-Ig, Proteintech, China). Western blot images were acquired by LAS 4000 (GE Healthcare, China) and quantified using ImageJ. Three independent experiments have carried out for biological replicates. For every independent experiment, all blots or gels derive from the same experiment and were parallelly processed.
The Nuclear/Cytosol Fractionation Kit was used to extract the cytoplasmic and nuclear proteins. Briefly, cells were washed 2 times with pre-cooling PBS and resuspended in cytoplasmic extraction reagents I and incubated on ice for 10 min. Cytoplasmic extraction reagents II were added and incubated on ice for 1 min and centrifuged at 4 °C, 16,000 × g, for 5 min. Supernatant was collected as cytoplasmic fraction. The sediments were washed twice with pre-cooling PBS and suspended in nuclear extraction reagent and incubated on ice for 40 min with vortex for 15 s every 10 min. The supernatant of nuclear fraction was harvested by centrifuge at 4 °C, 16,000 × g, for 10 min. Three independent experiments have carried out for biological replicates.
Immunofluorescence
Cells were cultured on circular glass coverslips in 24-well plates and fixed in 4% formaldehyde in PBS for 10 min at room temperature. Cells were incubated with anti-TFE3 antibody (Cat #14779, CST, USA, 1:500) in PBS containing 0.1% Tween-20 and 2% BSA overnight at 4 °C. The secondary antibodies, Alexa 594-conjugated (Cat#ab150088, Abcam, USA, 1:700), were added for 1 h at room temperature. The coverslips were overlaid on slides dripped with SlowFade Diamond Antifade Mountant with DAPI (Cat#S36964, Thermo Fisher Scientific, USA). Images were captured under oil immersion by a confocal laser scanning microscope (Carl Zeiss, LSM780, Oberkochen, Germany) and analyzed using ZEN lite 2012 software package.
All image statistical analyses were performed using Image Pro-Plus Software and Zeiss Auto-measure software. Cell range and nuclear range were circled and the fluorescence intensity of each channel was calculated by Zeiss Auto-measure software. The number of MEFs were more than 30 and the number of PMs were more than 100. Three independent experiments have carried out for biological replicates.
CO-immunoprecipitation
CO-Immunoprecipitation was carried out by lysing cells in TNE buffer (10 mM Tris, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 10% glycerol and protease inhibitors). The cell lysates were centrifuged at 4 °C, 16,000 × g, for 30 min. Supernatants were collected and added TFE3 antibody (Cat #14779, CST, USA, 1:200) or rabbit IgG for 3 h at 4 °C with gentle shaking. Protein A/G beads (Santa Cruz, Dallas, Texas, USA) was added and gently shaken overnight at 4 °C. The beads were rinsed five times on ice with pre-cooling TNE buffer and boiled in 2X sample buffer (Invitrogen, Carlsbad, CA, USA) for SDS-PAGE. Three independent experiments have carried out for biological replicates.
RIP assay
MEF cells were lysed in RNP-IP buffer (150 mM NaCl, 50 mM Tris-Cl pH7.5, 1% NP-40, 0.5% sodium deoxycholate, 0.05% SDS, 1 mM EDTA (pH8.0) with 1× Protease inhibitor and phosphatase inhibitor cocktail (Cat#200-664-3, Sigma-Aldrich, USA) and 40 U/ml RNase Inhibitor (Cat#EO0381, Thermo Fisher Scientific, USA). The supernatant was used for immunoprecipitation with rabbit anti-PDCD4 antibody or rabbit IgG. Proteinase K was used to digest DNA-protein complex and then RNase-free DNase I was added to digest DNA fragments for 15 min at room temperature. RNA was purified by TRIzol extraction. cDNA was synthesized by THUNDERBIRD SYBR qPCR Mix and real-time PCR were performed as described. The primer sequences of TFE3 (TFE3-RIP) for RIP are shown in Supplementary Table 2. Three independent experiments have carried out for biological replicates.
Dual luciferase assays
Luciferase activity was measured with a dual luciferase assay system (Cat#E2920, Promega, Madison, WI, USA) in HEK-293T cells with or without exogenous PDCD4 overexpression and the readout was determined using a microplate luminometer (Centro LB 960; Berthold, Wildbad, Germany). The vectors of pGL3 containing tfe3 5’UTRs have been described in the previous section of plasmid construction. Data was analyzed by GraphPad Prism 8. Three independent experiments have carried out for biological replicates.
Stereotaxic surgery, microinjection
For the establishment of PD mice model, mice (8 week) were anesthetized by pentobarbital sodium (50 mg/kg, i.p.) and deeply anesthetized mice were stereo-tactically injected unilaterally into the left striatum [Chatterjee, 2018 #69]. The liquid of adeno-associated viruses (containing GFP) without (AAV9-Ctrl) or with α-synuclein (AAV9-α-syn) were injected into the corpus striatum of wild mice (WT+ Ctrl, n = 7; WT+α-syn, n = 10) or PDCD4 knockout mice (KO + α-syn, n = 7). For the re-complement of PDCD4, the liquid of adeno-associated viruses (containing GFP) with PDCD4 (AAV9-PDCD4) were injected into the corpus striatum of Pdcd4 knockout mice (KO + α-syn, n = 6). For the interference of TFE3 and TFEB, the liquid of adeno-associated viruses (containing GFP) with sitfe3 and sitfeb (AAV9-sitfe3, AAV9-sitfeb) were injected into the corpus striatum of PDCD4 knockout mice (KO + α-syn, n = 5). After injection, mice fed for 8 weeks and the PD-related motor impairments were examined by the pole test, beam-walking test and rotarod test. After experiment finished, the region of virus infection was detected by GFP fluorescence under fluorescence microscopy (Olympus, Tokyo, Japan), and the expression of α-synuclein and PDCD4 were evaluated by Western Blot. All the AAV9 viruses were derived from ViGene Bioscience Company (Jinan, China). Mice were stereo-tactically injected with a Hamilton micro-syringe with a microinjection pump (KDS 200, KD Scientific) at a rate of 0.2 μl/min with either 2 μl of AAV9 virus, the concentration of each AAV9 virus were listed in Supplementary Table 3. According to the mouse brain atlas of Paxinos and Franklin (Paxinos and Franklin, The Mouse Brain in Stereotaxic Coordinates, Second Edition, 2001) the coordinates from Bregma: AP 1.0 mm and 0.3 mm; ML 2.0 mm and 2.2 mm; DV 3.1 mm were applied for injection.
Intraperitoneal injection therapy
The RVG-9dR (with 9 D-arginine at the C-end) used in the experiment was synthesized by Jill Biochemical Co., Ltd (Shanghai, China) using High Performance Liquid Chromatography (HPLC). RVG-9dR can bind to acetylcholine receptors on the surface of nerve cells and specifically enter nerve cells by crossing the blood-brain barrier through endocytosis. The peptide purity was above 95%, and the dry powder tube could be stored in a refrigerator at −80 °C, and dissolved in sterile triple distilled water during use. The specific amino acid sequence is as follows: RVG-9dR: YTIWMPENPRPGTPCDIFTNSRGKRASNGGGGRRRRRRRRR. Mix 50 ug of siPdcd4 in a molar ratio of siRNA: RVG-9dR = 1:10 to 200 μ in a 5% glucose solution (the silencing concentration of siRNA in vivo experiments was 2 mg/kg). Inject RVG-9dR/siPdcd4 into mice via intraperitoneal injection using a 1 ml syringe, and inject it every other day for two months.
Pole test
The pole test was employed to evaluate fine motor coordination, balance, and bradykinesia. The pole test was performed according to a previous study with minor modification58. Briefly, a rough-surfaced plastic rod (8 mm in diameter, 50 cm in height) was used to make the pole. The mouse was placed on the top of the pole facing head up, and the time taken to reach the bottom of the pole was recorded. The mice received three trials at 1-minute intervals, and the average times were analyzed.
Beam-walking
The beam-walking test was performed according to a previous study with minor modifications59. The beam-walking apparatus consisted of a long strip of wood (length of 100 cm, width of 0.6 cm). The beam was suspended at a height of 60 cm, with the other end attached to an enclosed box. The mice were placed at the initial 20 cm of the beam and were trained to cross the beam to the enclosed box within 60 s. Then, the time taken to cross the beams and the number of gliding foots off the beam in each trial were assessed. For each testing day, the times of the three trials were averaged per mouse.
Rotarod test
A Rotarod apparatus (IITC Life Science, CA, USA) was used to assess motor coordination and balance. The mice were placed on the Rotarod and the rotating speed were set up as 4 rpm to 40 rpm in 300 s. The latency time of each mouse to fall off the Rotarod was recorded. The mouse was tested three times with an interval of at least 30 min. For each testing day, the times of the three trials were averaged per mouse.
Tissue processing, immunohistochemistry and immunofluorescence
The anesthetized mice were given cardiac irrigation with heparinized saline (0.15 M, 15 ml) followed by fresh ice-cold 4% paraformaldehyde (in 0.1 M phosphate buffer (PB), pH 7.4, 100 ml). Brains were removed and postfixed in the same solution at 4 °C for 24 h and then transferred to increasing concentrations of sucrose (10, 20, and 30%, in 0.1 M PB, pH 7.4) for another 2 days. The frozen fixed brains were cut using a cryostat (Leica CM-3050-S, chamber temperature −20 °C) into 20 μm serial coronal sections containing the entire substantia nigra.
For immunohistochemistry, the staining was performed by washing three times in PBS (0.1 M, pH 7.4). Non-specific binding was blocked by immersion in 5% BSA in 0.1 M PBS with 0.3% Triton X-100 at room temperature for 1 h. Sections were incubated with rabbit anti-mouse TH antibody (Cat#58844, CST, USA, 1:500) diluted in 5% BSA/PBS for overnight at 4 °C. To visualize the primary antibody, a biotinylated secondary antibody to rabbit IgG was applied for 1 h followed by staining with diaminobenzidine-HCl (DAB).
For immunofluorescence, the slices were incubated with primary antibodies (TH, 1:500, Cat#58844, CST, USA; TFE3, 1:500, Cat #14779, CST, USA) in PBS containing 1% BSA, 0.3% TritonX-100 overnight at 4 °C. After three washes with PBS, slices were incubated with Secondary antibodies (Alexa Fluor 594, Goat anti-rabbit IgG, 1:500, abcam, ab150080; Alexa Fluor 488, Goat anti-rabbit IgG, 1:500, abcam, ab150077) in PBS containing 1% BSA for 2 h at room temperature and dark environment. After staining, slides were visualized and captured using a confocal microscope.
Sonication of the PFF
Before treating cells with PFF, use diagenode’s water wave ultrasound to treat PFF for 30 s, pause for 30 s, and use after 20 cycles of treatment.
Serum analysis
Analysis of liver, kidney function and related serum data were performed by NanJing JianCheng Bioengineering Institute company (Jinan, China).
Statistical analysis
Mean values, standard deviation values, and statistical significance were analyzed by GraphPad Prism 8. Differences between groups were analyzed with Student’s t test (unpaired), one-way or two-way ANOVA followed by Tukey’s test (for multiple comparisons). P-values < 0.05 was considered to be statistically significant; *p < 0.05, **p < 0.01, ***p < 0.001; ns not significant. Results are presented followed by at least three independent experiments of biological replicates.
Data availability
If you need any materials, please contact professor Lining Zhang or Yan Li (E-mail: zhanglining@sdu.edu.cn, liyan2015@sdu.edu.cn).
References
Settembre, C. et al. TFEB links autophagy to lysosomal biogenesis. Science 332, 1429–1433 (2011).
Sardiello, M. et al. A gene network regulating lysosomal biogenesis and function. Science 325, 473–477 (2009).
Martina, J. A. et al. The nutrient-responsive transcription factor TFE3 promotes autophagy, lysosomal biogenesis, and clearance of cellular debris. Sci. Signal 7, ra9 (2014).
Dang, T. T. & Back, S. H. Translation Inhibitors Activate Autophagy Master Regulators TFEB and TFE3. Int. J. Mol. Sci. 22, 12083 (2021).
Martina, J. A., Chen, Y., Gucek, M. & Puertollano, R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy 8, 903–914 (2012).
Settembre, C. et al. A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J. 31, 1095–1108 (2012).
Roczniak-Ferguson, A. et al. The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci. Signal 5, ra42 (2012).
Martina, J. A., Diab, H. I., Brady, O. A. & Puertollano, R. TFEB and TFE3 are novel components of the integrated stress response. EMBO J. 35, 479–495 (2016).
Puertollano, R., Ferguson, S. M., Brugarolas, J. & Ballabio, A. The complex relationship between TFEB transcription factor phosphorylation and subcellular localization. EMBO J. 37, e98804 (2018).
Medina, D. L. et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat. Cell Biol. 17, 288–299 (2015).
Bronisz, A. et al. Microphthalmia-associated transcription factor interactions with 14-3-3 modulate differentiation of committed myeloid precursors. Mol. Biol. Cell 17, 3897–3906 (2006).
Martina, J. A. & Puertollano, R. Rag GTPases mediate amino acid-dependent recruitment of TFEB and MITF to lysosomes. J. Cell Biol. 200, 475–491 (2013).
Dehay, B. et al. Lysosomal impairment in Parkinson’s disease. Mov. Disord. 28, 725–732 (2013).
Ebrahimi-Fakhari, D. et al. Distinct roles in vivo for the ubiquitin-proteasome system and the autophagy-lysosomal pathway in the degradation of α-synuclein. J. Neurosci. 31, 14508–14520 (2011).
Decressac, M. et al. TFEB-mediated autophagy rescues midbrain dopamine neurons from α-synuclein toxicity. Proc. Natl Acad. Sci. USA 110, E1817–E1826 (2013).
Raben, N. & Puertollano, R. TFEB and TFE3: Linking Lysosomes to Cellular Adaptation to Stress. Annu. Rev. Cell Dev. Biol. 32, 255–278 (2016).
He, X. et al. TFE3-Mediated Autophagy is Involved in Dopaminergic Neurodegeneration in Parkinson’s Disease. Front. Cell Dev. Biol. 9, 761773 (2021).
Dehay, B. et al. Pathogenic lysosomal depletion in Parkinson’s disease. J. Neurosci. 30, 12535–12544 (2010).
Rusmini, P. et al. Trehalose induces autophagy via lysosomal-mediated TFEB activation in models of motoneuron degeneration. Autophagy 15, 631–651 (2019).
Martini-Stoica, H., Xu, Y., Ballabio, A. & Zheng, H. The Autophagy-Lysosomal Pathway in Neurodegeneration: A TFEB Perspective. Trends Neurosci. 39, 221–234 (2016).
Yao, R. Q., Ren, C., Xia, Z. F. & Yao, Y. M. Organelle-specific autophagy in inflammatory diseases: a potential therapeutic target underlying the quality control of multiple organelles. Autophagy 17, 385–401 (2021).
Lu, J., Wu, M. & Yue, Z. Autophagy and Parkinson’s Disease. Adv. Exp. Med. Biol. 1207, 21–51 (2020).
Matsuhashi, S., Manirujjaman, M., Hamajima, H. & Ozaki, I. Control Mechanisms of the Tumor Suppressor PDCD4: Expression and Functions. Int J. Mol. Sci. 20, 2304 (2019).
Wang, Q. et al. Tumor suppressor Pdcd4 attenuates Sin1 translation to inhibit invasion in colon carcinoma. Oncogene 36, 6225–6234 (2017).
Santhanam, A. N., Baker, A. R., Hegamyer, G., Kirschmann, D. A. & Colburn, N. H. Pdcd4 repression of lysyl oxidase inhibits hypoxia-induced breast cancer cell invasion. Oncogene 29, 3921–3932 (2010).
Yang, H. S., Knies, J. L., Stark, C. & Colburn, N. H. Pdcd4 suppresses tumor phenotype in JB6 cells by inhibiting AP-1 transactivation. Oncogene 22, 3712–3720 (2003).
Wang, Q. et al. Programmed cell death-4 deficiency prevents diet-induced obesity, adipose tissue inflammation, and insulin resistance. Diabetes 62, 4132–4143 (2013).
Jiang, Y. et al. Deficiency of programmed cell death 4 results in increased IL-10 expression by macrophages and thereby attenuates atherosclerosis in hyperlipidemic mice. Cell Mol. Immunol. 13, 524–534 (2016).
Azzoni, L. et al. Differential transcriptional regulation of CD161 and a novel gene, 197/15a, by IL-2, IL-15, and IL-12 in NK and T cells. J. Immunol. 161, 3493–3500 (1998).
Schlichter, U., Burk, O., Worpenberg, S. & Klempnauer, K. H. The chicken Pdcd4 gene is regulated by v-Myb. Oncogene 20, 231–239 (2001).
Göke, A. et al. DUG is a novel homologue of translation initiation factor 4G that binds eIF4A. Biochem Biophys. Res Commun. 297, 78–82 (2002).
van den Bosch, M. W. M., Palsson-Mcdermott, E., Johnson, D. S. & O’Neill, L. A. J. LPS induces the degradation of programmed cell death protein 4 (PDCD4) to release Twist2, activating c-Maf transcription to promote interleukin-10 production. J. Biol. Chem. 289, 22980–22990 (2014).
Kumar, R., Poria, D. K. & Ray, P. S. RNA-binding proteins La and HuR cooperatively modulate translation repression of PDCD4 mRNA. J. Biol. Chem. 296, 100154 (2021).
Moustafa-Kamal, M. et al. The mTORC1/S6K/PDCD4/eIF4A Axis Determines Outcome of Mitotic Arrest. Cell Rep. 33, 108230 (2020).
Ruan, Q. et al. The microRNA-21-PDCD4 axis prevents type 1 diabetes by blocking pancreatic beta cell death. Proc. Natl Acad. Sci. USA 108, 12030–12035 (2011).
Sheedy, F. J. et al. Negative regulation of TLR4 via targeting of the proinflammatory tumor suppressor PDCD4 by the microRNA miR-21. Nat. Immunol. 11, 141–147 (2010).
Li, Y. et al. Programmed cell death 4 as an endogenous suppressor of BDNF translation is involved in stress-induced depression. Mol. Psychiatry 26, 2316–2333 (2021).
Chen, X. et al. Programmed cell death 4 modulates lysosomal function by inhibiting TFEB translation. Cell Death Differ. 28, 1237–1250 (2021).
Suzuki, C. et al. PDCD4 inhibits translation initiation by binding to eIF4A using both its MA3 domains. Proc. Natl Acad. Sci. USA 105, 3274–3279 (2008).
Chang, J. H. et al. Crystal structure of the eIF4A-PDCD4 complex. Proc. Natl Acad. Sci. USA 106, 3148–3153 (2009).
Yang, H. S. et al. The transformation suppressor Pdcd4 is a novel eukaryotic translation initiation factor 4A binding protein that inhibits translation. Mol. Cell Biol. 23, 26–37 (2003).
Yang, H. S. et al. A novel function of the MA-3 domains in transformation and translation suppressor Pdcd4 is essential for its binding to eukaryotic translation initiation factor 4A. Mol. Cell Biol. 24, 3894–3906 (2004).
Liwak, U. et al. Tumor suppressor PDCD4 represses internal ribosome entry site-mediated translation of antiapoptotic proteins and is regulated by S6 kinase 2. Mol. Cell Biol. 32, 1818–1829 (2012).
Jia, Y. et al. The brain targeted delivery of programmed cell death 4 specific siRNA protects mice from CRS-induced depressive behavior. Cell Death Dis. 12, 1077 (2021).
Song, X. et al. Tumor suppressor gene PDCD4 negatively regulates autophagy by inhibiting the expression of autophagy-related gene ATG5. Autophagy 9, 743–755 (2013).
Pastore, N. et al. TFEB and TFE3 cooperate in the regulation of the innate immune response in activated macrophages. Autophagy 12, 1240–1258 (2016).
Wada, S. et al. The tumor suppressor FLCN mediates an alternate mTOR pathway to regulate browning of adipose tissue. Genes Dev. 30, 2551–2564 (2016).
Napolitano, G. & Ballabio, A. TFEB at a glance. J. Cell Sci. 129, 2475–2481 (2016).
Cao, Q. et al. Suppression of abnormal α-synuclein expression by activation of BDNF transcription ameliorates Parkinson’s disease-like pathology. Mol. Ther. Nucleic Acids 29, 1–15 (2022).
Bitomsky, N., Wethkamp, N., Marikkannu, R. & Klempnauer, K. H. siRNA-mediated knockdown of Pdcd4 expression causes upregulation of p21(Waf1/Cip1) expression. Oncogene 27, 4820–4829 (2008).
Wedeken, L., Singh, P. & Klempnauer, K. H. Tumor suppressor protein Pdcd4 inhibits translation of p53 mRNA. J. Biol. Chem. 286, 42855–42862 (2011).
Wang, Q. & Yang, H. S. The role of Pdcd4 in tumour suppression and protein translation. Biol. Cell 110, 169–177 (2018).
Lee, J. H. et al. Faulty autolysosome acidification in Alzheimer’s disease mouse models induces autophagic build-up of Aβ in neurons, yielding senile plaques. Nat. Neurosci. 25, 688–701 (2022).
Palamarchuk, A. et al. Akt phosphorylates and regulates Pdcd4 tumor suppressor protein. Cancer Res. 65, 11282–11286 (2005).
Dorrello, N. V. et al. S6K1- and betaTRCP-mediated degradation of PDCD4 promotes protein translation and cell growth. Science 314, 467–471 (2006).
Schmid, T. et al. Translation inhibitor Pdcd4 is targeted for degradation during tumor promotion. Cancer Res 68, 1254–1260 (2008).
Gaven, F., Marin, P. & Claeysen, S. Primary culture of mouse dopaminergic neurons. J. Vis. Exp., 91, e51751 (2014).
Luo, D., Zhao, J., Cheng, Y., Lee, S. M. & Rong, J. N-Propargyl Caffeamide (PACA) Ameliorates Dopaminergic Neuronal Loss and Motor Dysfunctions in MPTP Mouse Model of Parkinson’s Disease and in MPP(+)-Induced Neurons via Promoting the Conversion of proNGF to NGF. Mol. Neurobiol. 55, 2258–2267 (2018).
Gomez-Soler, M. et al. Triglyceride Form of Docosahexaenoic Acid Mediates Neuroprotection in Experimental Parkinsonism. Front. Neurosci. 12, 604 (2018).
Acknowledgements
We thank Advanced Medical Research Institute/Translational Medicine Core Facility of Advanced Medical Research Institute of Shandong University for consultation and instrument availability that supported this work.
Funding
This research was supported by the National Natural Science Foundation of China (82101606, 82270605, 82173105, 81771775, 81971471), Youth Fund of the National Natural Science Foundation of China (82201592), Key Research Invention Program of Shandong Province (No. 24 2019JZZY011113), Youth Fund of the Natural Science Foundation of Shandong Province (No. ZR2022QC180) and Shandong Province higher education youth innovation team development plan (2023KJ343).
Author information
Authors and Affiliations
Contributions
Conceptualization and design: Baihui Cao and Xiaotong Chen; Methodology: Baihui Cao, Xiaotong Chen, Yubin Li, Tian Zhou; Investigation: Baihui Cao, Nuo Chen, Yaxin Guo and Ming Zhao; Funding acquisition: Lining Zhang and Yan Li; Project administration: Baihui Cao, Xiaotong Chen and Chun Guo; Supervision: Yongyu Shi, Qun Wang, Xuexiang Du, Lining Zhang and Yan Li; Writing: Baihui Cao, Xiaotong Chen, Lining Zhang and Yan Li; All authors read and approved the final manuscript.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License, which permits any non-commercial use, sharing, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if you modified the licensed material. You do not have permission under this licence to share adapted material derived from this article or parts of it. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc-nd/4.0/.
About this article

Cite this article
Cao, B., Chen, X., Li, Y. et al. PDCD4 triggers α-synuclein accumulation and motor deficits via co-suppressing TFE3 and TFEB translation in a model of Parkinson’s disease. npj Parkinsons Dis. 10, 146 (2024). https://doi.org/10.1038/s41531-024-00760-9
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41531-024-00760-9
- Springer Nature Limited