

Abstract
Near-infrared luminophores have many advantages in advanced applications, especially for structures without π-conjugation aromatic rings. However, the fabrication of red clusteroluminogens from nonconjugated polymers is still a big challenge, let alone the near-infrared clusteroluminogens. Here, we develop nonconjugated luminophores with full-spectrum from blue to near-infrared light (470 ~ 780 nm), based on color phenomenon of nonconjugated polyesters synthesized from the amine-initiated copolymerization of epoxides and cyclic anhydrides. We reveal that amines act as initiators attached to polymer chain ends. The formation of various amine-ester complexes in polyesters induces red to near-infrared light, conceptually, amine-ester complexed clusteroluminescence via intra/inter-chain charge transfer. Significantly, emission colors can be easily tuned by the contents and types of amines, microstructures of polyesters, and their concentration. This work provides a low-cost, scalable platform and strategy for the production of high-efficiency, multicolor luminescent materials.
Similar content being viewed by others

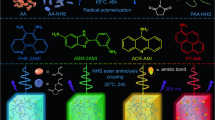
Introduction
Light, as a magnificent and fantastic magician, lights up the colorful world, especially the emerging luminescent materials that play an essential role in the remarkable advancement of modern civilization. Classic artificial luminophores are organic compounds with π-conjugated structures, widely used in interdisciplinary fields such as illumination1,2,3,4, bioimaging5,6,7,8, and sensor9,10,11 owing to high efficiency and multicolor luminescence. Over the past decades, through-bond conjugation12,13,14 has been the prerequisite to guide the design of luminophores by increasing π-conjugated structures. However, conventional π-conjugated molecules suffer from quenching effects at aggregate states, poor processability, complicated synthesis, and environmental hazards12,15,16,17, which limits practical applications.
In recent years, many nonconjugated polymers, including natural and synthetic heteroatom-rich (e.g., O, N, and S) polymers18,19,20,21,22 are observed to show abnormal luminescence, namely clusteroluminescence (CL)23,24,25 manipulated by through-space interactions (TSIs)26,27. These nonconjugated heteroatom-rich polymers have the advantages of easy processing, biocompatibility, etc., and can be mass-produced from renewable, low-cost biomass resources. Nevertheless, lacking universal CL theory to guide the structural design of luminophores, which makes photophysical performances difficult to regulate only with short-wavelength emission (400–500 nm, few at 600 nm) and low quantum yields (QYs)19,21,28,29,30,31,32. Our previous works have disclosed that QYs and wavelengths of ester clusters in polyesters could be regulated by hierarchical structures, which achieved white-to-orange lights33,34. Hence, it is highly desired but a big challenge to develop high-efficiency, red, and even near-infrared (NIR) nonconjugated polymer materials.
Accidentally, our lab observed that nonconjugated polyesters from the copolymerization of epoxides and cyclic anhydrides catalyzed by organic amine-alkyl boron Lewis pairs35 tend to be abnormal dark color and emit red lights under UV light. The intrinsic mechanism of such amazing stable colors and red luminescence was unclear. Herein, we revealed that amines acted as initiators attached to polymer chain ends and formed stable amine-ester complexes, which induce red-to-NIR CL at 600–780 nm through intra-chain/inter-chain charge transfer. Significantly, we proposed an engineering strategy of amine-ester complexes by the initiation of organic amines to transform large-scale, low-cost polyesters into blue-to-NIR luminophores. Meanwhile, CL colors can be easily tuned from blue to NIR by concentration, types of amines, and chain structures of polyesters.
Results
Synthesis and characterization of polyesters
The polyesters P1 and P2 were obtained by the copolymerization of epoxides and cyclic anhydrides, which were catalyzed by heterogeneous inorganic zinc-cobalt (III) double metal cyanide complex (Zn–CoIII DMCC)36 as the control group with no interference of Zn-OH on luminescence33,34 (Supplementary Figs. 1-2). Meanwhile, the polymers of P1-aTEA/DBU/mTBD/TBD and P2-aTEA/DBU/mTBD/TBD catalyzed by organic Lewis pairs35 of triethylborane (TEB) with different amine, triethylamine (TEA), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (mTBD) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), were obtained by using different molar ratios (a%) of amines from 0.5% to 5.0% (Supplementary Figs. 3–6). Then, the chemical structures of all the polyesters were well confirmed, showing that the amines attack the propylene oxide first and attach to polymer chain ends (Supplementary Figs. 7–67), which discloses the initiation mechanisms of amines and paves the way for studying the origin of dark color in amine-initiated polyesters.
Photophysical properties
The photophysical properties of P1 with only ester groups were systematically characterized by UV–Vis and photoluminescence (PL) spectra. Dominant absorption bands (250–300 nm) and maximum PL peaks (at ~465 nm) indicated typical (n,π*) transition of isolated ester groups33,34,37, which is clearly enhanced by the increased concentration (c) from 10−5 M to 10−1 M (Supplementary Figs. 68–70). The above results suggest that P1 only enhances the intrinsic (n,π*) transition of esters groups with weak TSIs. Then, the color origin in P1-aTEA was explored. Firstly, two control experiments of P1-2.0TEA catalyzed by TEA and TEB-TEA, respectively, showed nearly the same PL spectra (Supplementary Figs. 71–73), which completely excludes the influence of TEB on CL properties, indicating the dominant role of TEA in coloration. As shown in Supplementary Figs. 74–81, in DCM solution, with a% of TEA increasing from 0.5% to 5.0% or c increasing from 10−5 to 10−1 M, the absorption band of P1-aTEA at 300–600 nm appeared and was enhanced, suggesting that complexation between amines and esters induced the new strong TSIs.
To further disclose the origin of new strong TSIs, the PL spectra of P1-aTEA were further investigated under different a% of TEA. In Fig. 1A, besides the short-wavelength blue emission corresponding to the (n,π*) transition of ester groups similar to P1, P1-5.0TEA exhibited an additional long-wavelength PL peak at ~600 nm. It is noteworthy that the ratio of the long-wavelength CL (600 nm) intensity versus the short one (474 nm) increased with the increasing a% of TEA (Fig. 1B, C), suggesting enhancing TSIs between amines and esters. Hence, the fluorescence photographs for P1-aTEA under 365 nm UV-light exhibited significant color change from blue to white, orange, and red when the a% increased from 0.0 to 5.0% (Fig. 1D). Among all the polymers, P1-5.0TEA shows the highest solid-state QY of 15.8% (Supplementary Table 1). Meanwhile, pronounced excitation-dependent emission (EDE) effect from the strong peak at 470 nm to long-wavelength CL at ~600 in P1-0.5TEA indicates relatively weak TSIs, but as a% further increased to 5.0%, the EDE effect was gradually weakened and long-wavelength CL at ~600 nm became dominant (Supplementary Figs. 82–96), which also suggest that increasing the molar ratio of TEA can gain strong TSIs. PL decay curves show that the lifetime of the short-wavelength and long-wavelength emissions are both in the range of 1–10 ns with a slight difference, indicating the nature of fluorescence (Supplementary Figs. 97–110). Further, concentration-dependent PL spectra of P1-aTEA in DCM showed similar CL intensity enhancement on the formation of complexes with c increasing from 10−5 M to 10−1 M (Supplementary Figs. 111–117). These results suggest that the complexes-induced TSIs were formed once the TEA was utilized in the polymerization, and the incremental molar ratio of TEA or solution concentration only increases the number of amine-esters complexes without changing the chemical structures of complexes.

A Normalized photoluminescence (PL) spectra of P1 and P2-5.0TEA in solid at λex = 360 nm. B PL spectra of P1-aTEA in solid at λex = 360 nm. C Plots of intensities ratios versus contents of TEA for P1-aTEA in solid. The intensity ratio is the emissions of 600 nm/474 nm. λex = 360 nm. D Photographs and chemical structures of P1-aTEA in solid films taken under 365 nm UV light.
According to our previous reports33, P1 shows a typically loose helix conformation so that the involved TSIs are very weak and result in blue emission from isolated ester groups. However, the dense folding structure of P2 can induce strong TSIs among ester groups and produce orange CL with a maximum emission wavelength (λem) of 570 nm. Inspired by this, we can make stronger complexes by introducing TEA into P2 to generate redshifted CL. The obtained P2-aTEA showed a stronger absorption band at 400–600 in UV–Vis spectra relative to P1-aTEA under equal a% (Supplementary Figs. 118–124); meanwhile, the absorption band had a dramatic concentration-enhanced effect with the concentration increasing from 10−5 M to 10−1 M, which suggests that denser folding structures did favor to the formation of a strong complex. In Fig. 2A, B, Supplementary Figs. 125–138 and Supplementary Table 2, under 365 nm excitation, the PL spectra of P2-aTEA in both solid and concentrated DCM solution (c = 10−1 M) suggest that the short-wavelength emission (470–490 nm) keeps almost no change, but the long-wavelength CL continuously shifts from 600 nm to 660 nm when a% of TEA increased from 0.5% to 5.0%. Moreover, high a% in P2-aTEA induced another new redder CL at 740–750 nm in the NIR region. The plots of PL intensity versus a% further exhibited that blue PL dropped rapidly and NIR CL was intensified steadily with increasing of a% (Fig. 2C). PL decay curves also showed that the lifetimes of all these emissions were from fluorescence (Supplementary Figs. 139–152). In Fig. 2D, the phenomena of excitation-independent emission on red and NIR CL of P2-5.0TEA in concentrated DCM solution (c = 10−1 M) demonstrated the stabilized clusters of amine-ester complexes.

A, B Normalized PL spectra of P2-aTEA in solid and DCM solution, respectively, at λex = 360 nm. The concentration of P2-aTEA in DCM is 10−1 M. C Plots of relative PL intensities of blue PL/NIR CL versus contents of TEA for P2-aTEA in both solid and DCM of 10−1 M at λex = 360 nm. D PL spectra of P2-5.0TEA in DCM solution of 10−1 M. E Concentration-dependent PL spectra of P2-5.0TEA in DCM at λex = 360 nm. Concentration is from 10−5 to 10−1 M. F Plots of relative PL intensities of blue PL, red CL, and NIR CL versus contents of TEA for P2-5.0TEA in DCM of 10−1 M at λex = 360 nm, respectively. Concentration is from 10−5 to 10−1 M. G Fluorescence photographs and chemical structures of P2-aTEA in solid powders taken under 365 nm UV light.
To further investigate the formation of NIR-CL clusters, concentration-dependent PL spectra of P2-aTEA (a% is from 0.5% to 5.0%) in DCM solution with c from 10−5 M to 10−1 M were measured, which shows similar properties (Supplementary Figs. 153–159). Hence, taking P2-5.0TEA with the strongest NIR CL as an example, it exhibited full-spectra emission from blue to NIR only by changing the concentration (Fig. 2E). The plots of relative PL intensity versus c for each emission (blue PL at 470 nm, red CL at 600–660 nm and NIR CL at 740 nm) indicate that there are four stages for the variation of relative PL intensities, which shows three turning points c1 (10−4 M), c2 (10−3 M), and c3 (10−2 M) corresponding to the critical cluster concentration (CCC) of red CL, the concentration at which more esters participate in the complexation and the CCC of NIR CL, respectively. (i) In the first stage below c1, blue PL from isolated esters was dominant with no amine-ester complexes, suggesting a weak TSI in such a dilute solution. (ii) In the range of c1 to c2, the complexes began to form, and the red CL was induced and enhanced with further increasing of c. (iii) In the third stage, once c reached the c2, more isolated esters began to disappear and involve in complexation, causing the decreased relative intensity of the blue PL, then more and more esters involved in complexation with the increased c. (iv) When the c reached c3, saturated clusters of red CL formed, and the increasing c leads to long-range complexes owing to more isolated esters participating in complexation. Then, the long-range complexes started to induce and enhance NIR CL, further weakening the blue PL. The above results not only visualized the clusterization process of NIR CL but also disclosed the existence of long-range complexes between TEA and multiple esters. Finally, clusteroluminogens (CLgens) with white, orange, and red emission colors were obtained (Fig. 2G). The NIR CL in amine-initiated polyesters suggests the efficient role of complexation in generating strong TSI.
Amine-ester complexes
Extensive characterization and theoretical calculations were carried out to disclose the complexation and luminescent mechanism of amine-ester complexes. Firstly, the PL spectra of P1-2.0amine initiated by four different organic amines (TEA, DBU, mTBD, and TBD) were measured, and all exhibited the same CL peaks (Fig. 3A and Supplementary Fig. 160), and so were P2-2.0amine (Supplementary Figs. 161–162), indicating that nitrogen on amines plays an important role in red-to-NIR CL. Meanwhile, P1-2.0DBU had the strongest red CL, and P2-2.0DBU also had the strongest red CL and NIR CL. Interestingly, when the molar ratio a% of DBU is increased to 5.0%, P1-5.0DBU also displayed obvious NIR CL at 740 nm, which is absent in P1-5.0TEA (Fig. 3B), suggesting that DBU can form strong amine-ester complexes for NIR CL. Then, time-dependent NMR spectra of various mixtures of DBU and ester-containing structures were measured to reveal the complexation process and mechanism, where ester-containing structures include polymers (P1 and P2) and the corresponding small molecules (dimethyl succinate (DS), dimethyl citraconate (DC), succinic anhydride (SA) and citraconic anhydride (CA)). In Fig. 3C, D, time-dependent 1H- and 13C-NMR spectra of P1@DBU (@ represents the mixing process) with 5:1 molar ratio of structural units of P1 to DBU show that the major changes of atomic chemical shift (ΔδH for hydrogen atoms, ΔδC for carbon atoms) are 2–5 which are adjacent to two different nitrogen atoms on DBU. To confirm which nitrogen atom on DBU has dominant electronic interaction with esters, the plots of the absolute value of chemical shift changes (ΔδH, ΔδC) versus atom positions on DBU for P1@DBU were investigated (Fig. 3E). The maximum ΔδH and ΔδC were located at positions 4 or 5, while positions 2 and 3 had only negligible change. Moreover, NMR spectra of P2@DBU, DS@DBU, DC@DBU, CA@DBU, and SA@DBU all displayed the same trend (Fig. 3F and Supplementary Figs. 163-176), indicating that the nitrogen atom closed to positions 4 and 5 on DBU forms complexes with ester units. Especially, NMR spectra of SA@DBU with various molar ratios of SA to DBU from 0:5 to 5:0 disclosed the molar complexation ratios of DBU to SA is 2:1 (Supplementary Figs. 177–197). When TEA was mixed with ester-containing structures (Supplementary Figs. 198–205), smaller ΔδH and ΔδC of methylene and methyl on TEA relative to DBU were also observed, which also confirmed the structures of amine-ester complexes.

A Normalized PL spectra of P1-2.0TEA, P1-2.0DBU, P1-2.0mTBD, and P1-2.0TBD in solid at λex = 360 nm. Inset: photographs are taken under 365 nm UV light of P1-2.0TEA, P1-2.0DBU, P1-2.0mTBD, and P1-2.0TBD from left to right. B PL spectra of P1-5.0DBU in the solid state were recorded at different excitation wavelengths. C Time-dependent 1H-NMR and D 13C-NMR spectra of P1@DBU were obtained from mixing of P1 and DBU. The molar ratio of structural units of P1 to DBU is 5:1. @ represents the symbol of the mixing process. The h and d represent hours and days, respectively. E, F Plots of the absolute value of chemical shift changes (ΔδH for hydrogen atoms, ΔδC for carbon atoms) versus atom positions on DBU for P1@DBU and DS@DBU, respectively. DS is the model compound of P1. The molar ratio of DS to DBU is 5:1. G Frontier molecular orbitals and H Hole (red color) and electron (blue color) of optimized excited-state geometries of P1@DBU and P1-DBU calculated by TD-DFT method at B3LYP-D3/6–31 G(d,p) level, Gaussian 09 program. HOMO: the highest occupied molecular orbital, LUMO: the lowest unoccupied molecular orbital.
We next disclosed the electronic nature of amine-ester complexes in polyesters via theoretical calculation. Based on a single polymer chain with three repeating units, theoretical calculations were carried out to optimize the excited-state geometries of P1@DBU, P2@DBU, P1-DBU, and P2-DBU respectively (Fig. 3G and Supplementary Fig. 206). For P1@DBU, the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were located on nitrogen atoms of DBU and esters of polyesters, respectively. For DBU-initiated P1-DBU, the HOMO and LUMO were located on ester moieties and the quaternary nitrogen atoms of terminal DBU, respectively. These results suggested that all the electron transitions occur between DBU and ester moieties. Meanwhile, the hole (red color) and electron (blue color) of optimized excited-state geometries of P1@DBU, P2@DBU, P1-DBU, and P2-DBU were further simulated (Fig. 3H and Supplementary Fig. 207). The low-lying excited state with the intra/interchain charge transfer or through-space charge transfer (TSCT) effect38,39 was mapped for both P1@DBU and P1-DBU, further confirming the chemical and electronic structures of amine-ester complexes. Moreover, the physical mixing or chemical bonding methods demonstrated the existence of both interchain and intrachain amine-ester complexes and TSIs.
Besides, the origins of CL from amine-ester complexes were further investigated based on model small molecules. In Fig. 4A and Supplementary Fig. 208, the PL spectra of pure DC and DS only showed optimal PL peaks at 420 ~ 450 nm corresponding to the intrinsic (n,π*) transition of ester groups. Once TEA and DBU with molar ratios of 1/640 were added into bulk DC or DS, obvious redshifted CL at 500 ~ 605 nm appeared for DS@DBU, DC@TEA, and DC@DBU, indicating the formation of stable complexes. Moreover, except for DS@TEA, time-dependent PL intensities for DS@DBU, DC@TEA, DC@DBU clearly displayed that the long-wavelength CL at 500–605 nm enhanced with increasing treatment time (Fig. 4B and Supplementary Figs. 209–212), and the complexation rate of DC@DBU is the fastest with the reddest CL at 605 nm, which further proves that the red CL in P1-amine and P2-amine originates from amine-ester complexes. Meanwhile, increasing molar ratios of TEA or DBU relative to DC or DC from 1/640 to 1/20 also shifted the CL to the long-wavelength range, indicating the process of dynamic complexation (Supplementary Figs. 213–216).

A Normalized PL spectra of DS, DS@TEA, DS@DBU, DC, DC@TEA, DC@DBU in bulk at optimal λex = 360, 380, 440, 360, 560, and 560 nm, respectively. The molar ratios of these ester-containing structures to amines are 640:1. @ represents the mixing process. B Plots of PL intensities versus treatment time for DS@TEA, DS@DBU, DC@TEA, and DC@DBU, respectively. The molar ratios of these ester-containing structures to amines are 640:1. C Temperature-dependent PL spectra and D plots of relative PL intensities of DC@DBU versus the temperature in bulk. The molar ratio of DC to DBU is 20:1. λex = 600 nm. E Normalized PL spectra of P1, P1@TEA, P1@DBU, P2, P2@TEA, P2@DBU in solid at optimal λex = 400, 380, 400, 480, 480 and 580 nm, respectively. These mixtures were obtained by soaking polyesters in amine. F Temperature-dependent PL spectra and G Plots of PL intensities versus temperature of P2-5.0DBU in dimethyl sulfoxide solution of 10−1 M. H PL spectra of P1-5.0DBU and I PL spectra of P2-5.0DBU in solid precipitated in ethanol. Treatments 0, 1, and 2 represent once-precipitated treatment without hydrochloric acid, once- and twice-precipitated treatment under hydrochloric acid of 37 wt% (5 drops of hydrochloric acid per precipitation).
However, the above results cannot clarify the origin of NIR CL. Therefore, temperature-dependent PL spectra of DC@DBU in bulk were recorded to verify the long-range amine-ester complexes corresponding to the NIR CL. In Fig. 4C, D and Supplementary Figs. 217–218, there were two opposite stages for the variation of CL intensity for red CL (λem = 640 nm) and NIR CL (λem = 720 nm). At the first stage, DC@DBU showed original red CL at 640 nm based on amine-ester complexes under 77 K, and then the red CL was enhanced with the temperature increasing from 77 K to 177 K. Meanwhile, NIR CL at 700–800 nm gradually appeared and dominated leading to broadened spectra, because more molecules started to unfreeze and collide with each other to produce long-range complexes. In the second stage, as the temperature further increased from 177 K to 297 K, molecular collisions intensified, and long-range complexes dissociated, resulting in the dominant red CL of short-range complexes. These results indicate that NIR CL was more sensitive to temperature compared with red CL, which should originate from long-range amine-ester complexes.
Afterward, red-to-NIR CL from amine-ester complexes dependent on chain structures was further investigated. P2@TEA and P2@DBU with folding polymer chains displayed redder CL than these of P1@TEA and P1@DBU with helical structures, both in solid and solution states (Fig. 4E and Supplementary Figs. 219-222). Meanwhile, compared with PL spectra of DS@TEA, DS@DBU, DC@TEA, and DC@DBU in Fig. 4A, the PL spectra of P1@TEA, P1@DBU, P2@TEA and P2@DBU showed redder CL, especially P2@DBU displayed distinct NIR CL band at 680–800 nm, suggesting that polymers can form stable amine-ester complexes relative to small molecules. Moreover, temperature-dependent PL spectra of DBU-initiated P2-5.0DBU with three PL peaks (blue light at 470 nm, red CL at 660 nm, and NIR CL at 780 nm) were measured from 293 K to 363 K (Fig. 4F, G). The intensity of blue PL at 470 nm shows almost no change, while the red CL and NIR CL gradually decrease, suggesting that the red-to-NIR CL of polyesters is based on TSI from amine-ester complexes. It is noteworthy that the intensity of NIR CL keeps decreasing from 293 K to 363 K, but the red CL was stabilized above 320 K, demonstrating the higher sensitivity of NIR CL corresponding to long-range amine-ester complexes. Furthermore, red-to-NIR CL was weakened and blue-shifted after eluting terminal amines of P1-5.0DBU, P2-5.0DBU, P1-5.0TEA, and P2-5.0TEA with hydrochloric acid to break complexes (Fig. 4H, I and Supplementary Figs. 223–226), which also prove the important role of amine-ester complexes in the red-to-NIR CL.
Discussion
We prepared a series of nonconjugated polyesters with tunable, blue-to-NIR emission inspired by the appearing color phenomenon during amine-initiated polymerization. Photophysical characterization and theoretical calculation reveal that red-to-NIR CL (600–780 nm) corresponds to amine-ester complexes. Meanwhile, the underlying emission mechanism of these amine-ester complexes is intra/interchain charge transfer. The emission colors can be manipulated by molar ratios and types of amine catalysts, chain microstructures of polyesters and concentration. More importantly, the formation of amine-ester complexes was disclosed by dynamic NMR and photophysical characterization. Meanwhile, NIR CL originating from long-range amine-ester complexes was further confirmed. In general, amine-initiated polyesters with diverse microstructures make it easier to form various amine-ester complexes to induce red-to-NIR CL, suggesting the unique microstructures of polymer chains relative to small molecules in manipulating the photophysical properties (Fig. 5)40,41.

Amine-initiated polyesters with diverse microstructures make it easier to form various amine-ester complexes to induce red-to-NIR CL. Blue and dark-green balls represent ester units and amines, respectively. Blue, pink and red light around aggregates of balls represent the emission color of isolated ester groups, short-range and long-range complexes, respectively.
In conclusion, we not only realize the red-to-NIR CL from nonconjugated polyesters but also provide a sustainable platform to achieve high-efficiency, controllable blue-to-NIR CLgens with deeper sight into CL.
Methods
Materials
All manipulations involving air- and/or water-sensitive compounds were carried out with the standard Schlenk and vacuum line techniques under an argon atmosphere or in a nitrogen-filled glovebox. Succinic anhydride (SA) and citraconic anhydride (CA) were purchased from Macklin. Propylene epoxide (PO) was purchased from Aldrich. Chromatographic purity solvents, including toluene, dichloromethane, tetrahydrofuran (THF), ethanol, and flaky sodium hydroxide (CaH2), were bought from SINOPHARM. Triethyl borane (TEB) in tetrahydrofuran solution (1.0 mol/L), Dimethyl succinate (DS), and dimethyl citraconate (DC) were purchased from the Tokyo Chemical Industry (TCI). Triethylamine (TEA) was purchased from Aldrich, and 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (mTBD) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) were purchased from TCI. PO, TEA, DBU, DS, DC, and toluene were refluxed over CaH2 for 24 h and vacuum-distilled prior to use. In particular, chloroform-d for dynamic NMR characterization of amine-ester complexes was refluxed over CaH2 for 24 h and distilled prior to use. Both the anhydrides (SA and CA) were purified by vacuum sublimation three times. The purified anhydrides were collected under an inert atmosphere and stored in the glovebox for use. Other organic reagents were used without purification.
Synthesis
P1 and P2 were obtained by the copolymerization of PO with SA and CA, respectively, which was catalyzed by heterogeneous inorganic Zn–CoIII DMCC without any organic amines. P1-aTEA/DBU/mTBD/TBD and P2-aTEA/DBU/mTBD/TBD catalyzed by organic Lewis acid-base pairs of triethylborane (TEB) with a different amine such as triethylamine (TEA), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU), 7-methyl-1,5,7-triazabicyclo[4.4.0]dec-5-ene (mTBD) and 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD), were obtained by using different molar ratios (a%) of amines from 0.5% to 5.0% (seeing the Supporting Information for the detailed synthetic procedures)
Data availability
All the data supporting the findings in this work are available within the manuscript and Supplementary Information file and available from the corresponding authors upon request.
References
Kabe, R., Notsuka, N., Yoshida, K. & Adachi, C. Afterglow organic light-emitting diode. Adv. Mater. 28, 655–660 (2016).
Kabe, R. & Adachi, C. Organic long persistent luminescence. Nature 550, 384–387 (2017).
Abdurahman, A. et al. Understanding the luminescent nature of organic radicals for efficient doublet emitters and pure-red light-emitting diodes. Nat. Mater. 19, 1224–1229 (2020).
Du, S. et al. Hot exciton mechanism and aie effect boost the performance of deep-red emitters in non-doped OLEDs. Adv. Mater. 35, 2303304 (2023).
Cheng, H.-B., Li, Y., Tang, B. Z. & Yoon, J. Assembly strategies of organic-based imaging agents for fluorescence and photoacoustic bioimaging applications. Chem. Soc. Rev. 49, 21–31 (2020).
Li, Y. et al. Novel NIR-II organic fluorophores for bioimaging beyond 1550 nm. Chem. Sci. 11, 2621–2626 (2020).
Wang, Y. et al. High performance of simple organic phosphorescence host–guest materials and their application in time-resolved bioimaging. Adv. Mater. 33, 2007811 (2021).
Xiao, F. et al. Guest-host doped strategy for constructing ultralong-lifetime near-infrared organic phosphorescence materials for bioimaging. Nat. Commun. 13, 186 (2022).
Wu, S., Min, H., Shi, W. & Cheng, P. Multicenter metal–organic framework-based ratiometric fluorescent sensors. Adv. Mater. 32, 1805871 (2020).
Xue, K. et al. A sensitive and reliable organic fluorescent nanothermometer for noninvasive temperature sensing. J. Am. Chem. Soc. 143, 14147–14157 (2021).
Ye, F.-Y., Hu, M. & Zheng, Y.-S. Advances and challenges of metal ions sensors based on AIE effect. Coord. Chem. Rev. 493, 215328 (2023).
Hong, Y., Lam, J. W. Y. & Tang, B. Z. Aggregation-induced emission: phenomenon, mechanism and applications. Chem. Commun. 9, 4332–4353 (2009).
Milián-Medina, B. & Gierschner, J. π-Conjugation. WIREs Comput. Mol. Sci. 2, 513–524 (2012).
Ye, G. et al. A bionic paired hydrogen-bond strategy for extending organic π-conjugation to regulate emission. J. Mater. Chem. C 9, 9142–9146 (2021).
Yuan, W. Z. et al. Changing the behavior of chromophores from aggregation-caused quenching to aggregation-induced emission: development of highly efficient light emitters in the solid state. Adv. Mater. 22, 2159–2163 (2010).
Huang, Y. et al. Reducing aggregation caused quenching effect through co-assembly of PAH chromophores and molecular barriers. Nat. Commun. 10, 169 (2019).
Zhang, X., Liu, H., Zhuang, G., Yang, S. & Du, P. An unexpected dual-emissive luminogen with tunable aggregation-induced emission and enhanced chiroptical property. Nat. Commun. 13, 3543 (2022).
Pastor-Pérez, L., Chen, Y., Shen, Z., Lahoz, A. & Stiriba, S.-E. Unprecedented blue intrinsic photoluminescence from hyperbranched and linear polyethylenimines: polymer architectures and pH-effects. Macromol. Rapid Commun. 28, 1404–1409 (2007).
Zhao, E. et al. Poly[(maleic anhydride)-alt-(vinyl acetate)]: a pure oxygenic nonconjugated macromolecule with strong light emission and solvatochromic effect. Macromolecules 48, 64–71 (2015).
Wang, Q. et al. Reevaluating protein photoluminescence: remarkable visible luminescence upon concentration and insight into the emission mechanism. Angew. Chem. Int. Ed. 58, 12667–12673 (2019).
Zhang, Z. et al. Oxygen and sulfur-based pure n-electron dendrimeric systems: generation-dependent clusteroluminescence towards multicolor cell imaging and molecular ruler. Sci. China Chem. 64, 1990–1998 (2021).
Zhang, Z. et al. Manipulation of clusteroluminescence in carbonyl-based aliphatic polymers. Aggregate 3, e278 (2022).
Zhang, H. et al. Why do simple molecules with “Isolated” phenyl rings emit visible light.?. J. Am. Chem. Soc. 139, 16264–16272 (2017).
Zhang, H. et al. Clusterization-triggered emission: uncommon luminescence from common materials. Mater. Today 32, 275–292 (2020).
Zhao, Z. et al. Revisiting an ancient inorganic aggregation-induced emission system: an enlightenment to clusteroluminescence. Aggregate 2, e36 (2021).
Hoffmann, R. Interaction of orbitals through space and through bonds. Acc. Chem. Res. 4, 1–9 (1971).
Zhang, H. & Tang, B. Z. Through-space interactions in clusteroluminescence. JACS Au 1, 1805–1814 (2021).
Zhou, Q. et al. Clustering-triggered emission of nonconjugated polyacrylonitrile. Small 12, 6586–6592 (2016).
Liu, B. et al. Fluorescent linear CO2-derived poly(hydroxyurethane) for cool white LED. J. Mater. Chem. C 5, 4892–4898 (2017).
Wang, Y. et al. Emission and emissive mechanism of nonaromatic oxygen clusters. Macromol. Rapid Commun. 39, 1800528 (2018).
Huang, Q. et al. Significantly red-shifted emissions of nonconventional AIE polymers containing zwitterionic components. Macromol. Rapid Commun. 42, 2100174 (2021).
Ji, X. et al. Anionic polymerization of nonaromatic maleimide to achieve full-color nonconventional luminescence. Nat. Commun. 13, 3717 (2022).
Chu, B. et al. Aliphatic polyesters with white-light clusteroluminescence. J. Am. Chem. Soc. 144, 15286–15294 (2022).
Chu, B. et al. Altering chain flexibility of aliphatic polyesters for yellow-green clusteroluminescence in 38 % quantum yield. Angew. Chem. Int. Ed. 61, e202114117 (2022).
Hu, L.-F. et al. Highly active organic lewis pairs for the copolymerization of epoxides with cyclic anhydrides: metal-free access to well-defined aliphatic polyesters. Macromolecules 51, 3126–3134 (2018).
Hu, L. et al. Alternating copolymerization of isobutylene oxide and cyclic anhydrides: a new route to semicrystalline polyesters. Macromolecules 54, 6182–6190 (2021).
Zhang, Z. et al. The role of amide (n,π*) transitions in polypeptide clusteroluminescence. Cell Rep. Phys. Sci. 3, 100716 (2022).
Shao, S. et al. Blue thermally activated delayed fluorescence polymers with nonconjugated backbone and through-space charge transfer effect. J. Am. Chem. Soc. 139, 17739–17742 (2017).
Shao, S. & Wang, L. Through-space charge transfer polymers for solution-processed organic light-emitting diodes. Aggregate 1, 45–56 (2020).
Jing, Y.-N., Li, S.-S., Su, M., Bao, H. & Wan, W.-M. Barbier hyperbranching polymerization-induced emission toward facile fabrication of white light-emitting diode and light-harvesting film. J. Am. Chem. Soc. 141, 16839–16848 (2019).
Liu, B. et al. Polymerization-induced emission. Mater. Horiz. 7, 987–998 (2020).
Acknowledgements
We gratefully acknowledge the financial support of the National Science Foundation of China (No. 51973190, received by X.Z.), Zhejiang Provincial Department of Science and Technology (No. 2020R52006, received by X.Z.), National Science Foundation of China (No. 22205197, received by H.Z.) and the support of the Youth Talent Excellence Program of ZJU-Hangzhou Global Scientific and Technological Innovation Center (to H.Z.).
Author information
Authors and Affiliations
Contributions
B.C. conceived the idea. B.C., H.Z., and X.Z. designed the experiments. B.C. and X.L. performed the synthesis. B.C., X.L., and B.L. did the photophysical measurements and analyzed the data. Z.X. conducted theoretical calculations. B.C., Z.Z., C.Z., J.S., Q.Y., H.Z., X.Z., and B.Z.T. took part in the discussion and gave important suggestions. B.C., H.Z., and X.Z. co-wrote the paper. All authors approved the final version of the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewers for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chu, B., Liu, X., Xiong, Z. et al. Enabling nonconjugated polyesters emit full-spectrum fluorescence from blue to near-infrared. Nat Commun 15, 366 (2024). https://doi.org/10.1038/s41467-023-44505-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-023-44505-3
- Springer Nature Limited
This article is cited by
-
Multiple hydrogen-bonding induced nonconventional red fluorescence emission in hydrogels
Nature Communications (2024)