

Abstract
Earth’s habitability is closely tied to its late-stage accretion, during which impactors delivered the majority of life-essential volatiles. However, the nature of these final building blocks remains poorly constrained. Nickel (Ni) can be a useful tracer in characterizing this accretion as most Ni in the bulk silicate Earth (BSE) comes from the late-stage impactors. Here, we apply Ni stable isotope analysis to a large number of meteorites and terrestrial rocks, and find that the BSE has a lighter Ni isotopic composition compared to chondrites. Using first-principles calculations based on density functional theory, we show that core-mantle differentiation cannot produce the observed light Ni isotopic composition of the BSE. Rather, the sub-chondritic Ni isotopic signature was established during Earth’s late-stage accretion, probably through the Moon-forming giant impact. We propose that a highly reduced sulfide-rich, Mercury-like body, whose mantle is characterized by light Ni isotopic composition, collided with and merged into the proto-Earth during the Moon-forming giant impact, producing the sub-chondritic Ni isotopic signature of the BSE, while delivering sulfur and probably other volatiles to the Earth.
Similar content being viewed by others


Introduction
The Earth experienced a protracted accretion history over several tens up to 100 million years, which proceeded by the collision of numerous planetesimals and planetary embryos1,2. A fundamental assumption was that the Earth’s building blocks as a whole were compositionally similar to undifferentiated chondritic meteorites. Researchers have looked among different classes of chondrites for the closest representative of the accreting materials that formed Earth3,4,5,6. However, emerging evidence points to a mismatch in many crucial elemental and isotopic ratios between chondritic meteorites and the accessible Earth, arguing for the possible accretion of additional materials that are chemically and isotopically different from extant meteorite collections7,8,9,10,11. Constraining the nature of these building blocks of Earth is important, because they not only provide fundamental information on terrestrial planet formation, but also help understand how the Earth evolved into its current habitable status.
The late accretion stages, including the Moon-forming giant impact and the late veneer event, likely account for only <10% of Earth’s total mass12, but they represent a critical step for Earth to build its life-essential volatile budgets13,14,15,16,17,18,19,20. Dynamical models of Earth’s growth suggest that the late accretion stages were highly heterogeneous, consisting of a mixture of materials from two genetically distinct reservoirs in the Solar nebula20,21. One end-member may originate from the inner Solar system and contain a reduced, non-carbonaceous component that is probably ‘missing’ in known meteorites11,20,21,22,23,24. The other may be oxidized, carbonaceous chondrite-like material from the outer Solar system17,19,20,21,25. When these materials were added to Earth is still debated19,20,21,22,23,24,25,26,27. The carbonaceous chondrite-like materials are commonly thought to be the source of major volatiles in Earth28,29. Recent high pressure–temperature experiments on metal alloy-silicate partitioning of volatiles (e.g., carbon, sulfur, and nitrogen), however, suggest that Earth’s volatile abundance patterns could have been largely established by impact of a sulfur-rich, differentiated planetary body with minimal contributions from carbonaceous chondrite-like materials15,16. Due to the lack of proper meteorite proxies, the nature of late-stage impactors remains poorly known.
Nickel isotopic compositions of meteorites and terrestrial rocks may hold important clues. Nickel in the bulk silicate Earth (BSE) was mostly derived from late-stage impactors, as that from earlier stages was largely segregated into the core due to its moderately siderophile nature23. Models predict that ~95% of Ni in the BSE was derived from the last ~35% of mass that accreted to Earth23,30. Nickel is non-volatile and partitions compatibly into the mantle dominant phase – olivine – following accretion, so that the BSE can potentially capture the Ni isotopic signature of late-stage accreting materials.
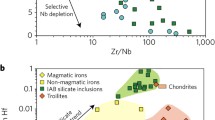
Nickel isotopic variations in meteorites have been well documented as shown in Fig. 1. Mass-independent nucleosynthetic Ni isotope anomalies arise from the heterogeneous distribution of presolar matters in the Solar protoplanetary disk, and thus trace the provenance of Earth’s building blocks. The nucleosynthetic anomalies are present in carbonaceous and ordinary chondrites, with enstatite chondrites largely within error of the BSE23,31,32,33,34,35, supporting the general idea that the late-stage accreting materials mainly originated from an enstatite-like source region in the inner Solar system23. Iron meteorites display similar anomalies, together with different groups of chondrites, forming a dichotomy between carbonaceous and non-carbonaceous meteorites as found in many other isotope systems (e.g., Mo, Cr, Ru, Ti)36. Mass-dependent isotopic variations stem from physico-chemical processes in the Solar nebula and on the planetary parent bodies. Nickel isotopic compositions of enstatite, ordinary, and most carbonaceous chondrites exhibit a common value, expressed as δ60/58Ni (the 60Ni/58Ni ratio in parts per thousand, relative to the SRM986 standard; δ60/58Ni = (60/58Nisample / 60/58NiSRM986 − 1) × 1000), with an average of +0.23 ± 0.11‰ (2 SD, n = 34)32,37,38,39,40. The small isotopic variation observed in the carbonaceous chondrites most likely reflects the heterogeneous distribution of an isotopically light sulfide component41,42, supported by the roughly negative correlation between δ60/58Ni and sulfur content (Fig. 1). It is not surprising that iron meteorites have δ60/58Ni values within the ‘chondritic’ range (Fig. 1), because they represent fragments of the disrupted cores of planetary bodies, and dominate the Ni budget.

The ε62Ni (ε62Ni = (62/58Nisample/ 62/58NiSRM986 − 1) × 106 after internal normalization to 61Ni/58Ni), are from literature31,32,33,35,36, and the δ60Ni of chondrites and iron meteorites are from this study and literature37,38,39,40,41,70. The carbonaceous chondrites (CC), enstatite chondrites (EC), and ordinary chondrites (OC) have average δ60Ni values of 0.23 ± 0.14‰ (2 SD), 0.22 ± 0.02‰ (2 SD) and 0.24 ± 0.02‰ (2 SD), respectively. A roughly negative correlation between δ60Ni and sulfur abundance is observed in carbonaceous chondrites (upper panel), which may be caused by the presence of various abundances of sulfides with δ60Ni values as low as −1‰41,42. The gray areas represent the dichotomy between carbonaceous and non-carbonaceous meteorites36. The sulfur abundances are from ref. 71. Meteorite data from this study and literature are presented in Supplementary Table 2. Error bars represent 2 SD.
The Ni isotopic composition of present BSE is poorly constrained. An earlier report of a few ultramafic rocks yielded δ60/58Ni values indistinguishable from the chondritic average, and they concluded that the BSE has a chondritic Ni isotopic composition39. This conclusion is questioned in a recent study combining new and reported peridotite samples40, which suggested that the BSE has a δ60/58Ni ~0.1‰ lower than the chondrite average, a difference that was attributed to Earth’s core formation40,43. Central to this debate is the limited Ni isotope data for terrestrial silicate rocks and scant information on Ni isotope fractionation during igneous and core–mantle differentiations.
Here, we show that the BSE has a sub-chondritic Ni isotopic composition by applying Ni isotope analysis to meteorites and terrestrial rocks. Our first-principles calculations further suggest that the light Ni isotopic signature of the BSE is not a result of core–mantle differentiation. Rather, the signature was established during Earth’s late-stage accretion, via impact and accretion of a highly reduced, Mercury-like impactor that likely originated from the innermost Solar system.
Results and discussion
Non-chondritic Ni isotopic composition of the bulk silicate Earth
Our new high-precision, inter-laboratory analyses on 60 terrestrial silicate rocks demonstrate that the present BSE is unambiguously sub-chondritic. Fertile peridotites, whose major element compositions are closest to the Primitive Mantle (e.g., Mg# = 89.6 ± 1.0; Al2O3 = 3.52 ± 0.60 wt.%)44, have δ60/58Ni values clustering tightly around +0.10 ± 0.07‰ (2 SD; n = 13, Fig. 2 and Supplementary Fig. 2). Peridotites overprinted by mantle metasomatism have Ni isotopic compositions shifted towards either heavier or lighter, but only to a limited degree (Supplementary Note 1). The melting products of mantle have similar or lighter Ni isotopic compositions compared to peridotites (Fig. 2). Komatiites formed by high-degree mantle melting (>45%) record an isotopic signature similar to the fertile peridotites (+0.13 ± 0.09‰, 2 SD, n = 15; Fig. 2). Oceanic basalts (OIBs and MORBs), which are produced by relatively low-degree melting (<25%), have Ni isotopic compositions slightly lighter than peridotites (Fig. 2; Student’s t -test; p < 0.001), with an average δ60/58Ni value of 0.03 ± 0.16‰ (2 SD, n = 15). Eclogites, formed from metamorphism of basalts, display a similar average δ60/58Ni value of 0.02 ± 0.06‰ (2 SD, n = 7; Fig. 2). Whether the difference between oceanic basalts and peridotites implies possible Ni isotope fractionation during partial melting or results from the limited dataset of oceanic basalts deserves further investigations. Nevertheless, the present BSE, as best represented by fertile peridotites reported in this study and literature40, has δ60/58Ni of +0.11 ± 0.06‰, lower than the chondritic average, +0.23 ± 0.11‰ (Student’s t -test, p « 0.001; Supplementary Note 1).

The bands represent the average value for each sample category with 2 SD. The δ60/58Ni of five chondrite samples fall within the chondritic average defined by all published data (0.23 ± 0.11‰). The fertile and non-metasomatized peridotites with chemical compositions most close to the BSE44 have homogeneous Ni isotopic compositions, while those experienced secondary modification have relatively large variations. Therefore, only fertile, non-metasomatized peridotites are used for the average calculation (0.10 ± 0.07‰). Data for chondrites are presented in Supplementary Table 2; Data for peridotites, komatiites, oceanic basalts, and eclogites are presented in Supplementary Tables 4–6. Error bars represent 2 SD.
First-principles calculations on Ni isotope fractionation during core formation
The sub-chondritic Ni isotopic composition of the BSE could have resulted from two possible processes: isotope fractionation associated with Earth’s differentiation, or the accretion of non-chondritic materials.
The former hypothesis is examined using first-principles calculations on Ni isotope fractionation factors (103lnα of 60Ni/58Ni) among Earth’s major Ni-bearing phases: olivine, wadsleyite, ringwoodite, bridgmanite, and Fe–Ni alloy. Limited differences in 103lnα are found between olivine and wadsleyite/ringwoodite in the mantle transition zone and bridgmanite in the lower mantle (e.g., 103lnα < 0.05‰ at 1500 K and <0.03‰ at 2000 K; Fig. 3a), which excludes the possibility of a hidden reservoir enriched in heavy Ni isotopes in the mantle. This lends credence to the use of accessible mantle and mantle-derived samples as representative of the present BSE Ni isotopic signature. Nickel isotope fractionation between Fe–Ni alloys and silicates (e.g., bridgmanite) under core-formation conditions is also negligible (P = 25–130 GPa; Fig. 3b and Supplementary Note 2). Notably, incorporation of sulfur into the Fe–Ni alloy slightly reduces the force constant of Ni, leading to the enrichment of light Ni isotopes in Fe–Ni alloys relative to the silicates (Fig. 3b).

a Equilibrium fractionation factors (103lnα) between olivine, wadsleyite, ringwoodite, and bridgmanite. b Equilibrium fractionation factors between Fe–Ni (S) alloy and bridgmanite, and between FeNiS melt (Fe92Ni5S3) and silicate melt (Mg30NiSi32O96). c δ60NiBSE vs. distribution coefficient of Ni between metal and silicate (DNimetal–silicate). If the bulk Earth (BE) has a chondritic Ni isotopic composition, the BSE δ60Ni after core formation can be evaluated using the Rayleigh fractionation equation: δ60NiBSE − δ60NiBE = Δ60Nimetal–silicate × lnfBSE, where fBSE is the fraction of Ni in the BSE. The fBSE can be calculated using the mass balance model: fBSE = Mmantle/(Mmantle + D × Mcore). The masses of the mantle and the core are 0.675 and 0.325, respectively. Given that the DNimetal–silicate is likely <4547, core segregation induced Ni isotope difference between metal and silicate cannot account for the light Ni isotopic composition of the BSE. ol olivine, wads wadsleyite, rw ringwoodite, bdg bridgmanite, FeNi Fe–Ni alloy, FeNiS S bearing Fe–Ni alloy or melt.
To directly assess the equilibrium Ni isotope fractionation between silicate and metallic melts during core–mantle differentiation, we performed first-principles molecular dynamic simulations on melt phases of Fe92Ni5S3 and Mg30NiSi32O96 based on the density functional theory. The 103lnα between Fe92Ni5S3 and Mg30NiSi32O96 melts is −0.011‰ at ~38 GPa and 3500 K (Fig. 3b; Supplementary Note 2), further confirming the conclusion based on crystals that core–mantle differentiation does not significantly fractionate Ni isotopes.
Two experimental studies investigated equilibrium Ni isotope fractionation between metal and silicate mineral or melt at low pressures (≤1.3 GPa) and temperatures (≤1623 K)45,46. Both studies predict limited Ni isotope fractionation under core-formation temperatures (<0.01‰ in terms of δ60/58Ni at T > 3000 K), a result that is consistent with our first-principles calculations at higher pressures, implying negligible pressure effect on silicate–metal Ni isotope fractionation. If the bulk Earth (BE) has a chondritic Ni isotopic composition, mass balance calculations using high-pressure metal–silicate Ni elemental partition coefficients47 and isotope fractionation factors obtained from our first-principles calculation demonstrate that core–mantle differentiation cannot explain the sub-chondritic Ni isotopic composition of the BSE (Fig. 3c).
Other possible events including evaporative loss, collision erosion, and core–mantle chemical diffusion can also be discounted as causes for the sub-chondritic Ni isotopic signature of BSE40. Evaporative loss of Ni is unlikely given the relatively refractory nature of Ni. In addition, kinetic isotope fractionation associated with evaporation would lead to a heavy BSE Ni isotopic composition, opposite to observations (Fig. 2). Collisional erosion during Earth’s formation preferentially removed early formed basaltic crust48. The terrestrial oceanic basalts have an average δ60/58Ni (0.03 ± 0.16‰; 2 SD) slightly lower than the BSE value, and thus collisional erosion cannot explain the sub-chondritic Ni isotopic composition of BSE. The presence of a Ni chemical gradient between Earth’s core and mantle may induce diffusive isotope fractionation, due to the faster diffusivity of light isotopes relative to heavy ones. A one-dimensional diffusion model shows that core–mantle chemical diffusion produces ~0.1‰ variation in the silicate part but is restricted to the lowermost two kilometers of the mantle on a time scale of 10 million years (Supplementary Note 3).
The nature of late-stage accreting materials and its implications
Therefore, the Earth’s mantle must have accreted sub-chondritic materials during its growth. In the early stages, metal and silicate melts equilibrate completely in the magma ocean49, so that the proto-BSE likely has a low Ni concentration and a chondritic Ni isotopic composition. Because of the moderately siderophile nature of Ni23 and a possible disequilibrium scenario for the late-stage accretion49,50, the BSE’s sub-chondritic Ni isotopic signature was likely established in the late stages. The late veneer following the main growth stage added the last <0.5% of mass to Earth and contributed <5% of the Ni budget of the BSE18. Hence, it is unlikely to be the event that produced the sub-chondritic Ni isotopic composition of BSE. To account for the observed Ni isotopic value of the present BSE, the late-veneer material accreted to the BSE would have to have had extremely low δ60/58Ni of around −2.5‰ (Supplementary Fig. 8), a value that has not been found in any natural rocks, and would be inconsistent with the Ni isotopic composition of an average carbonaceous chondrite-like material for the late veneer17,19,24,25. The last significant stage of Earth’s accretion was the Moon-forming giant impact, contributing >20% of Ni budget of the BSE49. The Ni isotopic composition of the BSE could have been strongly influenced by the Moon-forming impactor18,23. Assuming a chondritic Ni isotopic composition for the proto-BSE as discussed above, mass balance calculation suggests that materials accreted to the proto-BSE have δ60/58Ni values as low as −0.35‰ (Supplementary Fig. 8). Accordingly, the Moon-forming impactor is unlikely to have a composition represented by chondrites.
Instead, we hypothesize that the sub-chondritic Ni isotopic composition of the BSE resulted from the impact and accretion of the sulfide-rich mantle of a highly reduced, differentiated planetary body. It has long been recognized that accretion of planetary embryos that were already differentiated into cores and mantles contributed significantly to the growth of Earth51,52,53. The Moon-forming impactor has been suggested to be a sulfur-rich, differentiated planetary body15,16,27; but uncertainties remain as to whether it is a highly reduced, Mercury-like impactor23,24,27, or a relatively more oxidized body20,21,26. Mercury is the most reduced planet in the inner Solar system and has an abnormally high abundance of sulfides in its mantle54,55,56, whereas oxidized planetary embryos have sulfur segregated into their cores57. This is because sulfur is highly siderophile at high oxygen fugacity (fO2) and partitions into the metallic core, but becomes lithophile and enters into the silicate melt as sulfide species under low fO2 (e.g., five units below the iron-wüstite buffer; IW-5)58,59. Magmatic sulfides are the only major Ni-bearing phases that are isotopically much lighter than silicates (δ60/58Nisulfide down to −1‰)41. Rocks with high sulfide/silicate ratios have light Ni isotopic compositions, which is most evident in magmatic Ni-sulfide deposits where the bulk δ60/58Ni values are negatively correlated with the sulfur content41,42. Therefore, when small planetary embryos (the proto-impactor) were formed in a sulfur-rich early Solar nebula and differentiated into core and mantle under highly reduced environment similar to the Mercury (mean IW-5.4)58, the mantle would be sulfur-rich and have a light Ni isotopic composition (Fig. 4). By contrast, in the large proto-Earth, core–mantle differentiation proceeded under much higher pressure and likely more oxidizing condition (>IW-3; Fig. 4)60, in which sulfur behaves as a siderophile element61,62, leading to a sulfur-poor mantle. In this case, limited silicate–metal Ni isotope fractionation is expected (see discussion above), and thus the proto-Earth mantle likely has a chondritic Ni isotopic composition. During the Moon-forming impact, the impactor’s core merged directly into the proto-Earth’s core due to its limited emulsification, while the remaining parts of the impactor were incorporated into the Earth’s mantle49,50. The Mercury-like impactor’s sulfur-rich mantle would have been completely dissolved in the planet-wide, more oxidizing terrestrial magma ocean63, and produced the sub-chondritic Ni isotopic signature of Earth’s mantle (Fig. 4).

a The small, proto-impactor from the highly reduced innermost region of the Solar system differentiated into a core, a sulfur-rich mantle, and likely a sulfide layer at the mantle–core boundary, because sulfur behaves more lithophile at low fO2 and pressure58,59,61,62. The impactor’s mantle likely had a sub-chondritic Ni isotopic composition due to the enrichment of sulfides. Differentiation on the large, proto-Earth partitioned sulfur dominantly into the core because sulfur is more siderophile at relatively high fO2 and pressure58,59,61,62. Therefore, the mantle of the proto-Earth had a Ni isotopic composition close to the chondritic value. b The Moon-forming giant impact would have completely melted the Earth, forming a planet-wide, more oxidizing magma ocean with high solubility of sulfur60. The sulfides in the impactor’s mantle were dissolved in the terrestrial magma ocean, producing a sub-chondritic Ni isotopic composition for the Earth’s mantle. c The late veneer following the main growth stage added carbonaceous chondrite-like materials to the Earth with limited effects on the Ni isotopic systematics of the BSE as discussed in the text.
Our findings imply that, instead of an outer Solar system origin20,21, the Moon-forming impactor may represent a ‘missing’ Earth’s building component originated from a highly reduced reservoir in the inner Solar system. While the absence of nucleosynthetic Ni isotopic anomalies in enstatite chondrites is consistent with an inner Solar system provenance for the Moon-forming impactor23, the sub-chondritic mass-dependent Ni isotopic composition of the BSE further points towards a sulfide-rich, Mercury-like impactor, likely from closer to the Sun. This is in accordance with variations in nucleosynthetic isotope anomalies of other elements in meteorites, suggesting that the Earth possesses the most s-process enriched materials from the inner Solar system11,22,24,64. A most recent study found s-process enriched ruthenium (Ru) isotopic signatures in Eoarchean rocks, supporting the idea that the pre-late veneer Earth incorporated building materials from the innermost region of the Solar system, most likely through the Moon-forming giant impact24. The impactor might be sulfur-rich, such that the highly siderophile Ru was partially retained in the mantle without being completely extracted to the core. Later addition of a carbonaceous chondrite-like late veneer with s-process Ru deficits from the outer Solar system ultimately built up the modern mantle Ru isotopic composition24. The proposed late-stage accretion of the highly reduced, Mercury-like planetary body may not only explain the broad geochemical similarity between Earth and Moon27, but also account for the volatile abundance patterns in the BSE16. Our study highlights the importance of inner planets, e.g., Mercury and Venus, in searching for the Earth’s ‘missing’ building blocks that are not present in extant meteorite collections. Future studies on achondrites from the inner Solar system and samples from Venus and Mercury as well as experimental work will shed more light on these issues.
Methods
Nickel isotope analyses
We undertook an inter-laboratory comparison of geological reference materials using different analytical protocols in two labs: Indiana University (IU) and China University of Geosciences, Beijing (CUGB). Despite different double spike solutions and different column chemistry methods, the results of standards from two labs agree with each other within analytical uncertainty (Supplementary Table 1). A comparison of our data with published values is shown in Supplementary Fig. 1.
Ni isotope analyses at IU:
Sample powders were digested in a mixture of distilled HF + HNO3 + HCl. After complete dissolution, aliquots of sample solutions containing 1.5 μg Ni were spiked with a 61Ni –62Ni double spike to reach an optimal spike–sample ratio of 64:36. The mixtures were refluxed on a hotplate to ensure sample–spike equilibration before column chemistry. Separation of Ni from the matrices was achieved using a three-stage, cation exchange chromatography procedure using Bio-Rad 200–400 mesh AG 50W-X8 resin. Briefly, the first column applies mixture of 20% 10 M HCl and 80% acetone to separate Ni from Fe, Mn, and Cr. The second column uses 15% 10 M HCl and 85% acetic acid to separate Ni from elements such as Mg, Al, Ca, and Ti, and the last column further purifies Ni using 0.9 M HNO3 to remove Na and K. The Ni isotopic ratios were measured using Nu Plasma II MC-ICPMS at Indiana University.
Ni isotope analyses at CUGB:
Sample powders were dissolved and spiked follow the same protocol at IU, but with a different 61Ni–62Ni spike solution to obtain optimal ratio of 62Nispike/58Nisample = 1.3. The spike–sample solution was then passed through four-stage column chemistry. The step 1 column uses AG 50W-X8 and AG 1-X8 resins to separate Ni from Fe and Ca. Step 2 column uses AG 50W-X8 resin to separate Ni from Mg, Ti, and Al in a media of 0.15 M HNO3 and 4 M HF. A third column uses 0.5 M HCl containing 95% acetone to remove Mn; and the last column further separates Ni from the residual matrix using 0.5 M HCl + 95% acetone + 0.1 M DMG. The Ni isotopic ratios were determined on Nu Plasma III MC-ICPMS at the Laboratory of Surficial Environmental Geochemistry, Institute of Earth Sciences, China University of Geosciences (Beijing).
First-principles calculations
Ab initio calculations were performed using the software “Quantum Espresso”65, which is based on the density functional theory (DFT), plane wave, and pseudopotentials. The generalized gradient approximation (GGA) was adopted to describe the exchange-correlation functional. The pseudopotential for magnesium was generated using von Barth and Car’s methods with a cutoff radius 2.5 Bohr. The electron configurations are 3s23p0, 3s13p1, 3s13p0.53d0.5, 3s13p0.5, and 3s13d1 with decreasing weights of 1.5, 0.6, 0.3, 0.3, and 0.2, respectively. The pseudopotentials for nickel, silicon, and oxygen were generated by the method in Troullier and Martins66. The cutoff radius are 1.45 Bohr with the electron configuration of 2s22p4 for oxygen and 1.47 Bohr with the electron configuration of 3s23p43d0 for silicon. The cutoff radius for nickel is 2.1 Bohr with the electron configuration of 4s23d84p0. The pseudopotential for Fe was generated using the Vanderbilt method67 with a valence configuration of 3s23p63d6.54s14p0 and a cutoff radius of 2.0 Bohr for Fe.
We first optimized all crystal structures of Ni-bearing minerals using the variable cell shape molecular dynamics method68 with different k-point grids dependent on the sizes of unit cells (Supplementary Table 7). The energy cutoff for plane wave and charge density are set to 70 Ry and 700 Ry, respectively. The residual forces converge within 10−4 Ry/Bohr. After the relaxed structures were obtained, we then calculated vibrational frequencies using the finite displacement method as implemented in the open-source code PHONOPY69. Consequently, the reduced partition function ratios β of 60Ni/58Ni for all phases can be calculated from the equation: \(\beta _A = \frac{{Q_h}}{{Q_l}} = \mathop {\prod}\nolimits_i^{3N} {\frac{{u_{ih}}}{{u_{il}}}\frac{{e^{ - \frac{1}{2}u_{ih}}}}{{1 - e^{ - u_{ih}}}}\frac{{1 - e^{ - u_{il}}}}{{e^{ - \frac{1}{2}u_{il}}}}}\).
In order to directly estimate the equilibrium Ni isotope fractionation between silicate and metallic melts, we conducted first-principles molecular dynamics (FPMD) simulations on Mg30Ni2Si32O96 and Fe92Ni5S3 melts based on the DFT using the Vienna ab initio simulation package (VASP). The GGA was adopted for the exchange-correlation functional and the projector-augmented-wave (PAW) pseudopotentials were used. The energy cutoff for the plane wave was 600 eV. The Brillouin zone summations over the electronic states were performed at gamma point. The FPMD simulations were performed in the NVT thermodynamic ensemble with a fixed temperature of 3000 K and the Nosé thermostat was used. The time step was set to be 1 fs, and the total running time is up to 60 ps. The initial liquid configurations were prepared by conducting simulations on the structures at 6000 K. The cell parameter of the cubic box is 11.15 Å for Mg30Ni2Si32O96 silicate melt and 10.05 Å for Fe92Ni5S3 melt. The simulated statistical pressures of Mg30Ni2Si32O96 and Fe92Ni5S3 are 37.6 GPa and 38.5 GPa at 3000 K (Supplementary Fig. 5), respectively. After equilibration, we extracted 66 snapshots from the FPMD trajectory every 200 steps and only optimized the atomic positions of Ni with fixed cubic boxes. This strategy makes the Ni atoms in each snapshot are at the local equilibrium positions. Then we estimated the force constant matrix of Ni atoms in all snapshots using the small displacement method based on the harmonic approximation. The force constants <F > of the Ni atom in Mg30Ni2Si32O96 and Fe92Ni5S3 melts are the cumulative averages in the time domain. According to the high-temperature approximation of the Urey equation, it can be written as: \(\beta=1+(\frac{{1}}{{m_l}}-\frac{{1}}{{m_h}})\frac{{\hslash}^2}{{8}{{k_{B}}^{2}}{{T}^2}}<{F}> \)
where ml and mh are the masses of light and heavy isotopes, respectively. The equilibrium Ni isotope fractionation between Mg30Ni2Si32O96 and Fe92Ni5S3 melts can be derived from the <F> difference between these two melts. The resulting <F> of Ni in Mg30Ni2Si32O96 and Fe92Ni5S3 melts at ~38 GPa are 272.1 ± 7.5 and 214.4 ± 2.8 N/m (Supplementary Fig. 6), respectively. Therefore, the 103lnα between Mg30Ni2Si32O96 and Fe92Ni5S3 melts is 0.016 ± 0.003 ‰ at 3000 K and 0.011 ± 0.002 ‰ at 3500 K.
Data availability
All data in this study are included in the supplementary information files and are available from the corresponding author.
References
Halliday, A. N. Mixing, volatile loss and compositional change during impact-driven accretion of the Earth. Nature 427, 505–509 (2004).
Nimmo, F. & Kleine, T. Early differentiation and core formation: processes and timescales. in The Early Earth: Accretion and Differentiation (eds Badro, J. & Walter, M.) 83-102 (John Wiley & Sons, 2015).
Fitoussi, C. & Bourdon, B. J. S. Silicon isotope evidence against an enstatite chondrite Earth. Science 335, 1477–1480 (2012).
Clayton, R. N., Mayeda, T. K. & Rubin, A. E. Oxygen isotopic compositions of enstatite chondrites and aubrites. J. Geophys. Res. 89, C245-C249 (1984).
Javoy, M. et al. The chemical composition of the Earth: enstatite chondrite models. Earth Planet. Sci. Lett. 293, 259–268 (2010).
Allègre, C. J., Poirier, J.-P., Humler, E. & Hofmann, A. W. The chemical composition of the Earth. Earth Planet. Sci. Lett. 134, 515–526 (1995).
Burkhardt, C. et al. A nucleosynthetic origin for the Earth’s anomalous 142 Nd composition. Nature 537, 394 (2016).
Campbell, I. H. & O’Neill, H. S. C. J. N. Evidence against a chondritic Earth. Nature 483, 553 (2012).
Drake, M. J. & Righter, K. Determining the composition of the Earth. Nature 416, 39 (2002).
Nielsen, S. G., Prytulak, J., Wood, B. J. & Halliday, A. N. Vanadium isotopic difference between the silicate Earth and meteorites. Earth Planet. Sci. Lett. 389, 167–175 (2014).
Render, J., Fischer-Gödde, M., Burkhardt, C. & Kleine, T. The cosmic molybdenum-neodymium isotope correlation and the building material of the Earth. Geochem. Perspect. Lett. 3, 170–178 (2017).
Canup, R. M. & Asphaug, E. Origin of the Moon in a giant impact near the end of the Earth’s formation. Nature 412, 708–712 (2001).
Albarede, F. Volatile accretion history of the terrestrial planets and dynamic implications. Nature 461, 1227–1233 (2009).
Schönbächler, M., Carlson, R., Horan, M., Mock, T. & Hauri, E. Heterogeneous accretion and the moderately volatile element budget of Earth. Science 328, 884–887 (2010).
Grewal, D. S., Dasgupta, R., Sun, C., Tsuno, K. & Costin, G. Delivery of carbon, nitrogen, and sulfur to the silicate Earth by a giant impact. Sci. Adv. 5, eaau3669 (2019).
Li, Y., Dasgupta, R., Tsuno, K., Monteleone, B. & Shimizu, N. Carbon and sulfur budget of the silicate Earth explained by accretion of differentiated planetary embryos. Nat. Geosci. 9, 781 (2016).
Wang, Z. & Becker, H. Ratios of S, Se and Te in the silicate Earth require a volatile-rich late veneer. Nature 499, 328–331 (2013).
Walker, R. J. et al. In search of late-stage planetary building blocks. Chem. Geol. 411, 125–142 (2015).
Braukmüller, N., Wombacher, F., Funk, C. & Münker, C. Earth’s volatile element depletion pattern inherited from a carbonaceous chondrite-like source. Nat. Geosci. 12, 564–568 (2019).
Hopp, T., Budde, G. & Kleine, T. Heterogeneous accretion of Earth inferred from Mo-Ru isotope systematics. Earth Planet. Sci. Lett. 534, 116065 (2020).
Budde, G., Burkhardt, C. & Kleine, T. Molybdenum isotopic evidence for the late accretion of outer Solar System material to Earth. Nat. Astron. 3, 736–741(2019).
Fischer-Gödde, M. & Kleine, T. Ruthenium isotopic evidence for an inner Solar System origin of the late veneer. Nature 541, 525 (2017).
Dauphas, N. The isotopic nature of the Earth’s accreting material through time. Nature 541, 521 (2017).
Fischer-Gödde, M. et al. Ruthenium isotope vestige of Earth’s pre-late-veneer mantle preserved in Archaean rocks. Nature 579, 240–244 (2020).
Varas-Reus, M. I., König, S., Yierpan, A., Lorand, J.-P. & Schoenberg, R. Selenium isotopes as tracers of a late volatile contribution to Earth from the outer Solar System. Nat. Geosci. 12, 779–782 (2019).
O’Neill, H. S. C. The origin of the Moon and the early history of the Earth—a chemical model. Part 2: The Earth. Geochim. et Cosmochim. Acta 55, 1159–1172 (1991).
Wade, J. & Wood, B. J. The oxidation state and mass of the Moon-forming impactor. Earth Planet. Sci. Lett. 442, 186–193 (2016).
Alexander, C. O. D. et al. The provenances of asteroids, and their contributions to the volatile inventories of the terrestrial planets. Science 337, 721–723 (2012).
Marty, B. The origins and concentrations of water, carbon, nitrogen and noble gases on Earth. Earth Planet. Sci. Lett. 313, 56–66 (2012).
Brasser, R., Dauphas, N. & Mojzsis, S. Jupiter’s influence on the building blocks of Mars and earth. Geophys. Res. Lett. 45, 5908–5917 (2018).
Regelous, M., Elliott, T. & Coath, C. D. Nickel isotope heterogeneity in the early Solar System. Earth Planet. Sci. Lett. 272, 330–338 (2008).
Steele, R. C., Coath, C. D., Regelous, M., Russell, S. & Elliott, T. Neutron-poor nickel isotope anomalies in meteorites. Astrophys. J. 758, 59 (2012).
Tang, H. & Dauphas, N. 60Fe–60Ni chronology of core formation in Mars. Earth Planet. Sci. Lett. 390, 264–274 (2014).
Burkhardt, C. et al. In search of the Earth‐forming reservoir: mineralogical, chemical, and isotopic characterizations of the ungrouped achondrite NWA 5363/NWA 5400 and selected chondrites. Meteorit. Planet. Sci. 52, 807–826 (2017).
Tang, H. & Dauphas, N. Abundance, distribution, and origin of 60Fe in the solar protoplanetary disk. Earth Planet. Sci. Lett. 359, 248–263 (2012).
Nanne, J. A., Nimmo, F., Cuzzi, J. N. & Kleine, T. Origin of the non-carbonaceous–carbonaceous meteorite dichotomy. Earth Planet. Sci. Lett. 511, 44–54 (2019).
Cameron, V., Vance, D., Archer, C. & House, C. H. A biomarker based on the stable isotopes of nickel. Proc. Natl Acad. Sci. 106, 10944–10948 (2009).
Chernonozhkin, S. M., Goderis, S., Costas-Rodríguez, M., Claeys, P. & Vanhaecke, F. Effect of parent body evolution on equilibrium and kinetic isotope fractionation: a combined Ni and Fe isotope study of iron and stony-iron meteorites. Geochim. et Cosmochim. Acta 186, 168–188 (2016).
Gall, L., Williams, H. M., Halliday, A. N. & Kerr, A. C. Nickel isotopic composition of the mantle. Geochim. et Cosmochim. Acta 199, 196–209 (2017).
Klaver, M., Ionov, D. A., Takazawa, E. & Elliott, T. The non-chondritic Ni isotope composition of Earth’s mantle. Geochim. et Cosmochim. Acta 268, 405–421 (2020).
Gueguen, B., Rouxel, O., Ponzevera, E., Bekker, A. & Fouquet, Y. Nickel isotope variations in terrestrial silicate rocks and geological reference materials measured by MC‐ICP‐MS. Geostand. Geoanalytical Res. 37, 297–317 (2013).
Hofmann, A. et al. Comparing orthomagmatic and hydrothermal mineralization models for komatiite-hosted nickel deposits in Zimbabwe using multiple-sulfur, iron, and nickel isotope data. Miner. Depos. 49, 75–100 (2014).
Elliott, T. & Steele, R. C. The isotope geochemistry of Ni. Rev. Miner. Geochem. 82, 511–542 (2017).
Lyubetskaya, T. & Korenaga, J. Chemical composition of Earth’s primitive mantle and its variance: 1. Method and results. J. Geophys. Res. 112, B3 (2007).
Guignard, J. et al. Nickel isotope fractionation during metal-silicate differentiation of planetesimals: experimental petrology and ab initio calculations. Geochim. et Cosmochim. Acta 269, 238–256 (2020).
Lazar, C., Young, E. D. & Manning, C. E. Experimental determination of equilibrium nickel isotope fractionation between metal and silicate from 500 °C to 950 °C. Geochim. et Cosmochim. Acta 86, 276–295 (2012).
Righter, K. Prediction of metal–silicate partition coefficients for siderophile elements: an update and assessment of PT conditions for metal–silicate equilibrium during accretion of the Earth. Earth Planet. Sci. Lett. 304, 158–167 (2011).
O’Neill, H. S. C. & Palme, H. Collisional erosion and the non-chondritic composition of the terrestrial planets. Philosophical transactions of the royal society A: mathematical. Phys. Eng. Sci. 366, 4205–4238 (2008).
Rubie, D. C. et al. Heterogeneous accretion, composition and core–mantle differentiation of the Earth. Earth Planet. Sci. Lett. 301, 31–42 (2011).
Rudge, J. F., Kleine, T. & Bourdon, B. Broad bounds on Earth’s accretion and core formation constrained by geochemical models. Nat. Geosci. 3, 439 (2010).
Taylor, S. & Norman, M. Accretion of differentiated planetesimals to the Earth. in Origin of the Earth (eds Newsom, H. & Jones, J.) 29–43 (Oxford Univ. Press, New York, 1990).
Morbidelli, A., Lunine, J. I., O’Brien, D. P., Raymond, S. N. & Walsh, K. J. Building terrestrial planets. Annu. Rev. Earth Planet. Sci. 40, 251–275 (2012).
Fitoussi, C., Bourdon, B. & Wang, X. The building blocks of Earth and Mars: a close genetic link. Earth Planet. Sci. Lett. 434, 151–160 (2016).
McCubbin, F. M., Riner, M. A., Vander Kaaden, K. E. & Burkemper, L. K. Is Mercury a volatile‐rich planet? Geophys. Res. Lett. 39, 9 (2012).
Manthilake, G. et al. Thermal conductivity of FeS and its implications for Mercury’s long‐sustaining magnetic field. J. Geophys. Res. 124, 2359–2368 (2019).
Sprague, A. L., Hunten, D. M. & Lodders, K. Sulfur at Mercury, elemental at the poles and sulfides in the regolith. Icarus 118, 211–215 (1995).
Cartier, C. & Wood, B. J. The role of reducing conditions in building Mercury. Elements 15, 39–45 (2019).
Namur, O., Charlier, B., Holtz, F., Cartier, C. & McCammon, C. Sulfur solubility in reduced mafic silicate melts: implications for the speciation and distribution of sulfur on Mercury. Earth Planet. Sci. Lett. 448, 102–114 (2016).
Kilburn, M. & Wood, B. Metal–silicate partitioning and the incompatibility of S and Si during core formation. Earth Planet. Sci. Lett. 152, 139–148 (1997).
Wood, B. J., Walter, M. J. & Wade, J. J. N. Accretion of the Earth and segregation of its core. Nature 441, 825 (2006).
Boujibar, A. et al. Metal–silicate partitioning of sulphur, new experimental and thermodynamic constraints on planetary accretion. Earth Planet. Sci. Lett. 391, 42–54 (2014).
Rose-Weston, L., Brenan, J. M., Fei, Y., Secco, R. A. & Frost, D. J. Effect of pressure, temperature, and oxygen fugacity on the metal-silicate partitioning of Te, Se, and S: implications for earth differentiation. Geochim. et Cosmochim. Acta 73, 4598–4615 (2009).
Rubie, D. C. et al. Highly siderophile elements were stripped from Earth’s mantle by iron sulfide segregation. Science 353, 1141–1144 (2016).
Ek, M., Hunt, A. C., Lugaro, M. & Schönbächler, M. The origin of s-process isotope heterogeneity in the solar protoplanetary disk. Nat. Astron. 4, 273–281 (2020).
Giannozzi, P. et al. QUANTUM ESPRESSO: a modular and open-source software project for quantum simulations of materials. J. Phys. 21, 395502 (2009).
Troullier, N. & Martins, J. L. Efficient pseudopotentials for plane-wave calculations. II. Operators fast iterative diagonalization. Phys. Rev. B 43, 8861 (1991).
Vanderbilt, D. Soft self-consistent pseudopotentials in a generalized eigenvalue formalism. Phys. Rev. B 41, 7892 (1990).
Wentzcovitch, R. M. Invariant molecular-dynamics approach to structural phase transitions. Phys. Rev. B 44, 2358 (1991).
Togo, A. & Tanaka, I. First principles phonon calculations in materials science. Scr. Mater. 108, 1–5 (2015).
Steele, R. C., Elliott, T., Coath, C. D. & Regelous, M. Confirmation of mass-independent Ni isotopic variability in iron meteorites. Geochim. et Cosmochim. Acta 75, 7906–7925 (2011).
Braukmueller, N., Wombacher, F., Hezel, D. C., Escoube, R. & Muenker, C. The chemical composition of carbonaceous chondrites: implications for volatile element depletion, complementarity and alteration. Geochim. et Cosmochim. Acta 239, 17–48 (2018).
Acknowledgements
We deeply thank Laura Wasylenki for accessing the analytical facilities at Indiana University. We thank Edward Ripley for providing the komatiite samples, Jan Render, Patrick Frings, Michael Henehan and Yaoling Niu for constructive discussion. Funding: S.J.W. was funded by the National Key R&D Program of China (2019YFA0708404), the National Nature Science Foundation of China (41973010), the 111 Project of the Ministry of Science and Technology, China (BP0719021), and the Fok Ying-Tong Education Foundation of China. Z.Q.W. and W.Z.W. were funded by National Nature Science Foundation of China (41925017 and 41721002), and the Strategic Priority Research Program (B) of the Chinese Academy of Sciences (grant XDB18000000). S.C.H. was supported by NSF EAR-1942042. The computations were conducted partly in the Supercomputing Center of the University of Science and Technology of China.
Author information
Authors and Affiliations
Contributions
S.J.W. and W.Z.W. contributed equally to this work. S.J.W. conceived and designed the project, interpreted the data, and drafted the manuscript. W.Z.W performed the theoretical simulations. J.M.Z and S.J.W performed the experimental data analyses. Z.Q.W., J.G.L., G.L.H., F.Z.T., S.C.H., H.J.W., Y.J.W., G.L.W., and W.H.L participated in the discussion of the experimental results. All the authors contributed to the overall scientific interpretation and edited the manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Peer review information Nature Communications thanks Mario Fischer-Gödde, Yuan Li and the other, anonymous reviewer for their contribution to the peer review of this work.
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, SJ., Wang, W., Zhu, JM. et al. Nickel isotopic evidence for late-stage accretion of Mercury-like differentiated planetary embryos. Nat Commun 12, 294 (2021). https://doi.org/10.1038/s41467-020-20525-1
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-020-20525-1
- Springer Nature Limited
This article is cited by
-
Early planetesimal differentiation and late accretion shaped Earth’s nitrogen budget
Nature Communications (2024)
-
Sulfur isotopic signature of Earth established by planetesimal volatile evaporation
Nature Geoscience (2021)
-
Active methanogenesis during the melting of Marinoan snowball Earth
Nature Communications (2021)