
Abstract
Radiotherapy is often used to treat various types of cancers, but radioresistance greatly limits the clinical efficiency. Recent studies have shown that radiotherapy can lead to ferroptotic cancer cell deaths. Ferroptosis is a new type of programmed cell death caused by excessive lipid peroxidation. The induction of ferroptosis provides a potential therapeutic strategy for radioresistance. As the most common post-transcriptional modification of mRNA, m6A methylation is widely involved in the regulation of various physiopathological processes by regulating RNA function. Dynamic m6A modification controlled by m6A regulatory factors also affects the susceptibility of cells to ferroptosis, thereby determining the radiosensitivity of tumor cells to radiotherapy. In this review, we summarize the mechanism and significance of radiotherapy induced ferroptosis, analyze the regulatory characteristics of m6A modification on ferroptosis, and discuss the possibility of radiosensitization by enhancing m6A-mediated ferroptosis. Clarifying the regulation of m6A modification on ferroptosis and its significance in the response of tumor cells to radiotherapy will help us identify novel targets to improve the efficacy of radiotherapy and reduce or overcome radioresistance.

Similar content being viewed by others


Facts
-
Radiotherapy can induce ferroptosis, a new type of programmed cell death.
-
Inducting ferroptosis provides a potential therapeutic strategy for radioresistance.
-
m6A modification is involved in the regulation of ferroptosis in cancers.
-
Enhancing m6A-mediated ferroptosis is a promising strategy for radiosensitization.
Open questions
-
How does radiotherapy induce ferroptosis?
-
How does m6A modification regulate ferroptosis?
-
Which ferroptosis effector molecules can be regulated via m6A-modification to enhance ferroptosis and radiosensitivity?
-
How to achieve radiosensitization through the regulation of m6A modification?
Introduction
Radiotherapy is a common treatment for many kinds of cancers. However, radioresistance is a major issue, which greatly limits the clinical efficiency and prognosis of cancer patients. Overcoming radioresistance is a major challenge in cancer treatment. Therefore, it is urgent to uncover the potential mechanism leading to radioresistance and find possible solutions. Recently, several studies have shown that radiotherapy can induce ferroptosis in various types of tumors [1,2,3,4]. Ferroptosis is a type of regulated cell death triggered by unrestricted lipid peroxidation [5]. It has been found to play an important role in radiation sensitization [6,7,8]. Induction of ferroptosis may provide a potential therapeutic strategy for clinical radioresistance.
N6-methyladenine (m6A) modification is the most prevalent epitranscriptome modification in mammalian mRNA [9, 10]. It is widely involved in the regulation of various physiological and pathological processes by regulating RNA stability, mRNA splicing, microRNA processing and mRNA translation [11,12,13,14]. There is increasing evidence that m6A modification and m6A regulatory factors regulate the susceptibility of cells to ferroptosis, thereby affecting the radiosensitivity of tumor cells [6, 15,16,17,18]. Therefore, understanding the regulation of m6A modification on ferroptosis and its significance in the response of tumor cells to radiotherapy will help to find novel targets to improve the efficacy of radiotherapy and alleviate or overcome radioresistance. In this review, we will summarize the mechanism and significance of radiotherapy-induced ferroptosis, as well as the regulation of m6A modification on it, and discuss the radiosensitization via enhancing m6A-mediated ferroptosis.
The mechanism and significance of radiotherapy-induced ferroptosis
Ferroptosis and its regulation
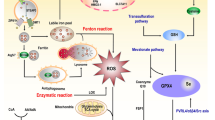
Ferroptosis is a new type of iron-dependent programmed cell death triggered by lipid peroxidation, which is involved in a variety of physiopathological processes [5]. Iron is an essential trace mineral element in almost all organisms, but it can promote lipid peroxidation by catalyzing the production of reactive oxygen species (ROS) through the Fenton reaction [19, 20]. Iron can also indirectly promote ferroptosis by acting as a cofactor in the enzymes that promote lipid oxidation [5, 21, 22]. Lipid biosynthesis and metabolism are closely related to ferroptosis. The peroxidation of lipids, specifically polyunsaturated fatty acids (PUFAs), is the key driver of ferroptosis [23, 24]. Ferroptosis is induced when the peroxidation of phospholipid-PUFAs exceeds the scavenging capacity of the cell antioxidant system. The synthesis of phospholipids containing PUFAs provides substrates for peroxidation [25], which requires acyl-CoA synthetase long-chain family member 4 (ACSL4) to esterify CoA onto long PUFAs [26].
Ferroptosis is regulated by multiple factors, and solute carrier family 7 member 11 (SLC7A11) and glutathione peroxidase 4 (GPX4) are the main regulators (Fig. 1). GPX4 can use reduced glutathione (GSH) as a coenzyme to eliminate lipid peroxides, thereby inhibiting ferroptosis [27,28,29]. GSH depletion promotes the ferroptosis of cancer cells by reducing GPX4 activity [28, 30]. The biosynthesis of GSH requires the rate-limiting precursor cysteine (the reduced form of cystine) as the substrate. The import of extracellular cystine is regulated by cystine/glutamate antiporter (system XC−) [31], which is composed of a heavy chain (SLC3A2) and a light chain (SLC7A11) [32]. SLC7A11 is a key transporter of cysteine and introduces extracellular cystine into the cells. Cysteine is subsequently used to synthesize GSH [33].

Excessive production of lipid peroxides can lead to ferroptosis. GPX4 helps to eliminate lipid peroxides and exerts anti-ferroptotic effects, which requires GSH to provide reducing power. SLC7A11 imports cystine into the cell, providing the substrate for the synthesis of GSH. Ionizing radiation induces the expression of ACSL4, which provides the substrate PUFA-PLs for lipid peroxidation. Ionizing radiation-mediated ROS consumes cellular GSH and promotes lipid oxidation.
The expression and activity of SLC7A11 can be regulated at multiple levels. SLC7A11 is a target of p53-mediated transcriptional repression [34]. Interferon-γ from CD8+ T cells can impair tumor cystine uptake by downregulating SLC3A2 and SLC7A11 [35]. Nuclear factor erythroid factor 2-related factor 2 (Nrf2) and kelch-like ECH-associated protein 1 (KEAP1) signalling can regulate the system XC− and reduce ferroptosis [36]. Nrf2 is a transcription factor that promotes the transcription of SLC7A11 under oxidative stress by binding to the antioxidant response elements in its promotor region [37]. The transcription of SCL7A11 can also be regulated by the tumor suppressor BRCA1-associated protein 1 (BAP1) that encodes a nuclear deubiquitinase to reduce histone 2 A ubiquitination (H2Aub) on chromatin [38]. BAP1-mediated deubiquitination dissociates H2Aub from the SLC7A11 promoter and inhibits the expression of SLC7A11, which subsequently suppresses cystine uptake and induces ferroptosis. In addition to transcriptional regulation, the level of SLC7A11 is also regulated by posttranslational modification and stability. For example, noncanonical deubiquitinase OTU domain-containing ubiquitin aldehyde-binding protein 1 (OTUB1) interacts with SLC7A11 to prevent its degradation [39] and the adhesion molecule CD44 variant (CD44v) acts as a binding partner for stabilizing SLC7A11 [40].
Inhibition of GPX4 and SLC7A11 by corresponding inhibitors can trigger ferroptosis, so they are ferroptosis inducers (FINs). Ferroptosis suppressor protein 1 (FSP1), also known as AIFM2, is a glutathione-independent ferroptosis suppressor, which acts as a coenzyme Q10 oxidoreductase and restores the antioxidant capacity of cells [41]. Many cancer cells are sensitive to ferroptosis [27, 42, 43]. If oxidative damage caused by radiotherapy can to a certain extent lead to ferroptosis, ferroptosis inducers may be used as a radiosensitizer. Recently, ferroptosis has been recognized as an important mechanism of tumor suppression and radioresistance mediated by radiotherapy [44].
Contribution of ferroptosis to radiotherapy
It is well known that radiotherapy can induce DNA double-strand breaks and subsequent unregulated cell death [2, 45]. Further studies found that the response of tumor to radiotherapy involves many other forms of regulated cell death, including apoptosis, necroptosis, and autophagy [46,47,48,49]. In addition to DNA damage, radiotherapy also generates ROS, which can induce oxidative damage of cell components including the lipid membrane [50, 51]. Actually, two decades ago, it was found that ionizing radiation can induce lipid peroxidation [52]. It has also been confirmed that ROS induced by radiotherapy can lead to peroxidation of PUFAs [2, 20, 53]. Excessive production of lipid peroxidation can lead to ferroptosis [5]. In recent years, the contribution of ferroptosis to radiotherapy efficacy or radiosensitization has attracted great attention [54,55,56]. Several studies have confirmed that ferroptosis is an important factor in the radiotherapy-induced cell death response, and ferroptosis inactivation can promote radioresistance [1,2,3,4]. Oxidative stress and ferroptosis caused by ionizing radiation are one of the most important biological effects that destroy tumors [4].
It has been proved that radiotherapy can induce ferroptosis in many cancer models, including non-small cell lung cancer (NSCLC) [1], ovarian cancer [2], fibrosarcoma, adenocarcinoma and glioma [3]. At first, it was found that ionizing radiation could induce ferroptosis in tumor cells of xenograft mice. Ferroptosis agonists enhance the efficacy of radiotherapy, while ferroptosis antagonists have the opposite effect [2]. Further research confirmed that ferroptosis is partly responsible for radiation-induced cancer cell death. Significant genetic and biochemical characteristics of ferroptosis are observed in cancer cells treated with radiation, and ferroptosis inhibitors suppress radiation-induced cell death. Radiation-induced lipid peroxidation can trigger ferroptosis in several cancer types and act in synergy with ferroptosis inducers such as system XC– inhibitor erastin and GPX4 inhibitor RAS-selective lethal 3 (RSL3) [3]. Importantly, this effect is attributed to increased cytoplasmic lipid peroxidation, rather than the enhancement of DNA damage or caspase activation. The application of ferroptosis inducers enhances the antitumor efficacy of radiation in a murine xenograft model and in human patient-derived models [3]. The study also confirmed that radiotherapy can induce ferroptosis in cancer patients, and the increase of ferroptosis in cancer patients is related to better response to radiotherapy and longer survival periods [1]. In addition, the measurements of ferroptosis characteristic indicators, such as the expression of SLC7A11 and GPX4, as well as the intracellular lipid peroxidation and Fe2+ concentration, indicate that high level of ferroptosis increases the radiosensitivity of hepatocellular carcinoma [57].
The mechanism of ionizing radiation-induced lipid peroxidation and ferroptosis is not completely clear. First, excessive ROS produced by radiotherapy can promote lipid peroxidation (Fig. 1). Next, ionizing radiation induces ferroptosis partly through upregulating ACSL4 [1]. ACSL4 is a lipid metabolism enzyme required for the synthesis of phospholipids containing PUFAs. ACSL4 deficiency significantly eliminates the radiation-induced ferroptosis and leads to radioresistance. Finally, ionizing radiation also depletes intracellular GSH, which impairs the anti-ferroptosis effect mediated by GPX4 and further promotes ferroptosis [3, 55]. In previous studies, the effect of ionizing radiation on the expression of SLC7A11 was not entirely consistent. As an adaptive response, ionizing radiation induces the expression of ferroptosis inhibitors SLC7A11 and GPX4. Inactivation of SLC7A11 or GPX4 with ferroptosis inducers (FINs) can make radioresistant cancer cells and xenograft tumors sensitive to ionizing radiation, indicating the potential significance of radiotherapy combined with FINs in cancer treatment [1]. However, some studies have reported the inhibitory effect of ionizing radiation on the expression of SLC7A11 [2]. DNA damage-induced ataxia telangiectasia mutated (ATM) kinase activated by radiotherapy transcriptionally inhibits the expression of SLC7A11 to promote tumor ferroptosis. In addition, immune cells, especially CD8+ T cells, may participate in the induction of ferroptosis during radiotherapy. Immunotherapy can make tumors sensitive to radiotherapy by promoting ferroptosis of tumor cells. IFNγ produced by immunotherapy-activated CD8+ T cells can promote tumor ferroptosis and induce radiosensitization. Radiotherapy-activated ATM and IFNγ-induced STAT1 signalling jointly repress SLC7A11 to reduce cystine uptake and enhance tumor lipid peroxidation and ferroptosis [2]. This explains why radiotherapy and immunotherapy can synergistically induce tumor ferroptosis. In addition, other mechanisms mediated by DNA damage, such as mitochondrial DNA stress-activated autophagy, may contribute to ferroptosis in radiotherapy [58].
The regulation of m6A modification on ferroptosis of cancers
m6A modification and its regulators
N6-methyladenine (m6A) is the most common post-transcriptional modification of eukaryotic RNAs [9, 10]. Modification is a dynamic reversible process coordinated by m6A methyltransferases and demethylases (Fig. 2A). m6A methyltransferases, also known as m6A writers, acts in the form of complex and transfer methyl onto the nitrogen atom on amino group at the 6th position of adenine. Known m6A writers include methyltransferase-like (METTL) -3/14/16, Wilms tumor 1-associated protein (WTAP), RNA binding motif protein 15 (RBM15), RBM15B, vir-like m6A methyltransferase associated (VIRMA), and zinc finger CCCH-type containing 13 (ZC3H13). m6A demethylases, such as fat mass and obesity-associated protein (FTO), AlkB homolog 5 (ALKBH5) and ALKBH3, act as m6A erasers to remove m6A from modified RNAs. The m6A writers and erasers cooperate to maintain the dynamic balance of m6A modification. The m6A modification of RNAs can be recognized by the m6A reader proteins to exert different biological functions. Many proteins function as m6A readers, including YT521-B homology (YTH) domain-containing proteins (YTHDC) 1/2, YTH domain-containing families (YTHDF) 1/2/3, insulin-like growth factor 2 mRNA binding proteins (IGF2BP) 1/2/3, heterogeneous nuclear ribonucleoprotein (HNRNP) A2B1 and C, and eukaryotic initiation factor 3 (eIF3).

A The m6A writers and erasers cooperate to maintain a dynamic balance of m6A modification, while the m6A readers recognize m6A modification on RNAs and mediate downstream biological functions. B Several major regulatory factors of ferroptosis regulate ferroptotic cancer cell death through m6A modification.
RNA m6A modification is involved in the regulation of RNA splicing, translation, stability, translocation, and advanced structure, which has broad biological significance [11, 12]. m6A modification and its regulatory genes affect many aspects of tumors [11, 12, 59]. For example, m6A writer METTL3-mediated signalling can promote the development and progression of tumors [60,61,62,63,64,65], and resistance [18] of tumors to chemotherapy and radiotherapy. The m6A demethylation of different signal molecules mediated by demethylase FTO promotes the occurrence, growth, metastasis and progression of tumors [66,67,68,69,70,71,72]. The biological significance of m6A modification is very complicated, largely depending on the targets of the modification [73, 74].
m6A modification is involved in the regulation of ferroptosis
Many studies have linked m6A modification with programmed cell death including ferroptosis [75,76,77,78]. The system XC− plays an important role in the control of ferroptosis. The regulation of system XC− and its components through m6A modification will greatly affect cell’s ferroptosis activity. SLC3A2 and SLC7A11 are two subunits of system XC−, and are the targets of the m6A reader YTHDC2 to execute ferroptosis in lung adenocarcinoma cells [79, 80]. YTHDC2 can disrupt the stability of Homeo box A13 (HOXA13) mRNA in m6A-dependent manner. The latter can regulate the transcription of SLC3A2 subunit of system XC− (Fig. 2B). Therefore, YTHDC2 inhibits SLC3A2 and induces ferroptosis by inhibiting HOXA13 in an m6A-indirect manner [80].
SLC7A11, the catalytic subunit of system XC−, is the key regulatory target of m6A modification. YTHDC2 destabilizes SLC7A11 mRNA in an m6A-dependent manner [79]. METTL3 can mediate m6A modification of SLC7A11 mRNA (Fig. 2B), which stabilizes SLC7A11 mRNA and promotes its translation, thus enhancing ferroptosis resistance of lung adenocarcinoma [81] and hepatoblastoma [82]. This process may need YTHDF1 [81] or IGF2BP1 [82] as the m6A readers of SLC7A11. IGF2BP1 can enhance SLC7A11 mRNA stability by inhibiting SLC7A11 mRNA deadenylation in an m6A-dependent manner [82]. METTL14 induces m6A modification of SLC7A11 mRNA at 5’-UTR and subsequent YTHDF2-dependent degradation. Hypoxia blocks ferroptosis of hepatocellular carcinoma by inhibiting METTL14 in a HIF-1α-dependent manner [83]. SLC7A11 has been identified as a potential FTO regulatory gene. FTO regulates ferroptosis of papillary thyroid carcinoma cells by mediating m6A demethylation of SLC7A11, thus preventing the progression of thyroid cancer [84]. NF-κB activating protein (NKAP) is an RNA-binding protein that acts as an inhibitor of ferroptosis. NKAP protects glioblastoma cells from ferroptosis by binding to m6A and promoting SLC7A11 mRNA splicing and maturation [85]. m6A-hypomethylation mediates upregulation of fibroblast growth factor receptor 4 (FGFR4) in anti-HER2 resistant breast cancer. FGFR4 inhibition triggers ferroptosis via the β-catenin/TCF4-SLC7A11/FPN1 axis [86].
FSP1 is another important regulatory target of m6A modification. miR-4443 can regulate m6A modification and expression of FSP1 by targeting METTL3, and subsequent FSP1-mediated ferroptosis [87]. Therefore, exosomal miR-4443 promotes cisplatin resistance in NSCLC. High-density lipoprotein-binding protein (HDLBP) binds to and stabilizes ferroptosis-associated lncRNA (lncFAL), which mediates an FSP1-dependent anti-ferroptosis in hepatocellular carcinoma (HCC) [88]. lncFAL interacts with FSP1, inhibiting Trim69-dependent FSP1 polyubiquitination degradation. YTHDF2 could promote lncFAL expression in an m6A-dependent manner. These results support the great potential of targeting FSP1 as a promising therapeutic approach for cancer patients [88]. Moreover, the upregulation of METTL3 induced by fear stress stabilizes FSP1 mRNA through m6A modification, which leads to glioma progression by inhibition of ferroptosis. The data provide a new understanding of the psychological impact on tumor development [89]. Similarly, GPX4 is also regulated by m6A modification. Neutrophil extracellular traps (NETs) mediate m6A modification and regulate sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. The upregulation of ferroptosis depends on the m6A modification of GPX4 induced by METTL3 [90]. METTL16 enhances m6A modification-mediated GPX4 expression and anti-ferroptosis effect to promote breast cancer progression [91]. IGF2BP3 is highly expressed in lung adenocarcinoma and desensitizes ferroptosis in a manner that depends on its binding capacity to m6A-methylated mRNAs encoding anti-ferroptotic factors, including GPX4, SLC3A2, acyl-CoA synthetase long-chain family member 3 (ACSL3), and ferritin heavy chain 1 (FTH1) [92].
Another target modified by m6A methylation is Nrf2. WTAP can promote m6A modification on 3’-UTR of endogenous antioxidant factor Nrf2 mRNA and its stability by binding with m6A reader YTHDF1 on the m6A site of Nrf2 mRNA [93]. Thus, WTAP accelerates bladder cancer progression by targeting Nrf2 through m6A-dependent ferroptosis regulation. It was reported that RNA demethylase ALKBH5 can affect the progression of various tumors. ALKBH5 promotes ferroptosis in hypopharyngeal squamous cell carcinoma by inhibiting the expression of Nrf2 in an m6A-IGF2BP2-dependent manner. ALKBH5 demethylates the 3’-UTR m6A sites of Nrf2 mRNA. m6A modification and the m6A reader IGF2BP2 are necessary to stabilize Nrf2 mRNA [94]. ALKBH5 also suppresses the progression of thyroid cancer by reducing the m6A level of TIAM1 and inducing ferroptosis through m6A-TIAM1-Nrf2/HO-1 axis [13]. Since IGF2BP3 recognizes m6A modification of Nrf2 mRNA and stabilizes it, IGF2BP3 knockdown significantly promotes ferroptosis of hepatocellular carcinoma cells after administration of sorafenib [95].
In addition to directly mediating m6A modification of anti-ferroptotic factors such as SLC7A11 [81, 82], FSP1 [87] and GPX4, m6A regulatory factors also regulate ferroptosis through other different mechanisms. High glucose and high fat (HGHF)-induced ferroptosis in osteoblasts may be the main cause of osteoporosis in diabetes. Osteoblast ferroptosis is activated through the METTL3/ASK1-p38 signalling pathway to promote HGHF-induced diabetic bone loss [96]. METTL14 promotes doxorubicin (DOX)-induced ferroptosis in cardiomyocytes through regulating the KCNQ1OT1-miR-7-5p-TFRC axis. DOX induces the upregulation of METTL14, which catalyzes the m6A modification of the long non-coding RNA KCNQ1OT1. KCNQ1OT1, as a miR-7-5p sponge, can prevent miR-7-5p-mediated degradation of transferrin receptor (TFRC) and subsequent ferroptosis [97]. The m6A writer WTAP-mediated m6A modification on circCMTM3 inhibits hepatocellular carcinoma ferroptosis by recruiting IGF2BP1 to increase the stability of Parkinson’s protein 7 (PARK7) [98]. PARK7 shows antioxidant activity and anti-ferroptosis effect [99].
m6A modification may regulate ferroptosis through autophagy signaling pathway [100, 101]. The ferroptosis of hepatic stellate cells (HSCs) induced by m6A modification can be attributed to autophagy activation by stabilizing BECN1 mRNA via m6A reader protein YTHDF1 [100]. BECN1 is a key regulator of autophagy, and promotes ferroptosis through the regulation of system XC− activity in cancer cells [102]. Dihydroartemisinin (DHA) increases the autophagy level of HSCs, thus preventing the activation of HSCs via ferroptosis pathway. The up-regulated m6A modification by reducing FTO is required for DHA to activate autophagy and alleviate liver fibrosis by inducing ferroptosis in HSCs [103].
On the contrary, the induction of ferroptosis will lead to changes in m6A modification level and m6A regulator activity. Our recent research has found that oxidative stress induced by the lack of an important antioxidant gene GPX8 causes reprogramming of the m6A epitranscriptome in oral cancer cells [104]. The m6A level of HSCs treated with ferroptosis inducers is enhanced by up-regulating the methyltransferase METTL4 and down-regulating the demethylase FTO [100]. Erastin can induce HSCs ferroptosis, thereby alleviating liver fibrosis in mice, while HSCs-specific inhibition of m6A modification can weaken erastin-induced HSC ferroptosis in murine liver fibrosis. The ferroptosis inducers may be used to prevent liver fibrosis. These studies link m6A with ferroptosis; therefore, targeting m6A to induce ferroptosis may be a promising strategy for ferroptosis-based therapy.
Improving radiosensitivity via m6A-mediated ferroptosis
Radiosensitization through inducing ferroptosis
The main mechanism leading to radioresistance of tumor cells is hypoxia. Hypoxia can also trigger ferroptosis by inducing ROS production and activate hypoxia-inducible factors [55]. Therefore, FINs-mediated ferroptosis of tumor cells may overcome the radioresistance induced by hypoxia [6]. By inducing ferroptosis, tumor cells can be re-sensitive to radiotherapy [6,7,8]. It was found that a ferroptosis-related gene prognostic index may predict biochemical recurrence and radiation resistance of prostate cancer patients receiving radical radiotherapy [105]. Changing the lipid composition of cell membrane by regulating lipid metabolism will affect the sensitivity of tumor cells to radiotherapy. For example, the lack of ACSL4 reduces the efficacy of radiotherapy by inhibiting the synthesis of PUFAs [1].
SLC7A11 is a main inhibitor of ferroptosis and plays a key regulatory role in radioresistance. As mentioned above, radiotherapy can induce ferroptosis of cancer cells [55]. Radiotherapy can also inhibit ferroptosis to induce radiotherapy resistance by inducing the expression of SLC7A11 and GPX4 as a negative feedback regulatory pathway (Fig. 3). The combination of ferroptosis inducer targeting SLC7A11 and radiotherapy synergistically induces ferroptosis and improves the sensitivity of cancer cells to radiotherapy [1, 3]. Several pharmacologic inhibitors of SLC7A11, such as erastin and sulfasalazine, can increase the sensitivity of cancers to radiotherapy or re-sensitize radioresistant cancer cells [1, 3, 106]. Application of ferroptosis inducers can improve the curative effect of radiotherapy. Many proteins regulate ferroptosis and radioresistance through controlling cellular level of SLC7A11. It was reported that RNA-binding motif, single-stranded interacting protein 1 (RBMS1), an RNA-binding protein, directly binds to the translation initiation factor eIF3d to bridge the 3’- and 5’-UTR of SLC7A11, which in turn promotes the translation of SLC7A11 [107]. RBMS1 ablation promotes ferroptosis through inhibiting SLC7A11 translation and SLC7A11-mediated cystine uptake. RBMS1 depletion or inhibition of RBMS1 expression by nortriptyline hydrochloride sensitizes radioresistant lung cancer cells to radiotherapy through promoting ferroptosis [107]. The suppressor of cytokine signaling 2 (SOCS2) promotes ferroptosis and radiosensitization in cancer by enhancing the ubiquitination of SLC7A11 [57]. The expression of SOCS2 is negatively correlated with radiosensitivity of HCC and positively related to ferroptosis. In terms of mechanism, the SH2-domain of SOCS2 can specifically interact with the N-terminal domain of SLC7A11. SOCS2 acts as a bridge to transfer the attached ubiquitin to SLC7A11, and promotes K48-related polyubiquitination degradation of SLC7A11. Therefore, SOCS2 can enhance the ubiquitination degradation of SLC7A11 and promote ferroptosis, which suggests that targeting SOCS2 may improve the efficiency of radiotherapy [57]. In addition, immunotherapy can enhance the efficacy of radiotherapy, which cooperatively inhibits SLC7A11 to induce ferroptosis of tumor cells [2, 35].

The activity of SLC7A11 can be regulated by many regulatory proteins or small molecules.
SLC7A11 is also regulated by p53. p53 is the most common mutation gene in human cancers, and is also the main effector to radiotherapy. Studies have found that ferroptosis is related to p53-mediated radiosensitization. Radiotherapy-mediated p53 activation promotes irradiation-induced ferroptosis partly through antagonizing irradiation-induced SLC7A11 expression and inhibiting glutathione synthesis [44]. p53 deficiency promotes radioresistance in tumors partly through SLC7A11-mediated ferroptosis inhibition. Ferroptosis inducers that inhibit SLC7A11 can cause radiosensitization of p53-deficient tumor cells, tumor organoids and tumors. Therefore, Ferroptosis inducers combined with radiotherapy can be used to treat p53-mutant cancers [44]. In addition, PKR-like ER kinase (PERK), a sensor of unfolded protein response, facilitates ferroptosis via regulating p53 expression to down-regulate SLC7A11, contributing to the sensitivity of HCC cells to high linear energy transfer carbon ions radiation [108].
Nrf2 induces transcription of antioxidant genes, which plays an important role in resisting oxidative damage [19]. Many genes involved in cellular iron homeostasis are regulated by Nrf2. Since ferroptosis is triggered by unrestricted lipid peroxidation and iron accumulation, Nrf2 inhibition significantly increases sensitivity to ferroptosis [19]. Many Nrf2 target genes are involved in the regulation of ferroptosis [109]. At first, SLC7A11 is one of the downstream target genes of Nrf2. SLC7A11-mediated ferroptosis inhibition contributes to radioresistance. Nrf2 can directly bind to the SLC7A11 promoter region and induce the expression of SLC7A11, thereby promoting radioresistance by inhibiting ferroptosis [110]. Esophageal squamous cell carcinoma patients with high Nrf2 nuclear expression and SLC7A11 expression have poor prognosis and treatment responses [110]. FSP1 has also been identified as a transcriptional target of Nrf2, and acts as the key effector in Nrf2-mediated ferroptosis resistance and radioresistance in KEAP1 deficient lung cancer cells [109, 111]. KEAP1 is frequently mutated or inactivated in lung cancers, while KEAP1 mutant lung cancers are resistant to most therapies including radiotherapy. It is ubiquinone (CoQ)-FSP1 axis that mediates ferroptosis resistance and radioresistance in KEAP1 deficient lung cancer cells. Ferroptosis induced by pharmacological inhibition of the CoQ-FSP1 axis makes KEAP1 deficient lung cancer cells or patient-derived xenograft tumors sensitive to radiation [109, 111]. Inhibition of the Nrf2-antioxidant response element (ARE) pathway can improve the sensitivity of artesunate and eliminate the head and neck cancer resistance to ferroptosis [112]. AGuIX nanoparticles based on gadolinium have been proven to improve the radiosensitivity of cancers, which may regulate the anti-ferroptosis system by inhibiting the Nrf2-GSH-GPX4 signaling pathway [113].
Regulating GPX4 activity also affects ferroptosis and radiosensitivity of cancers. The inhibition of GPX4-mediated ferroptosis and the reduction of lipid peroxidation have been proven to be related to hypoxia-induced radioresistance. Hypoxic NSCLC cells express higher level of angiopoietin-like 4 (ANGPTL4) compared to normoxic cells. The expression level of ANGPTL4 is positively correlated with the radioresistance of NSCLC cells and xenograft tumors [114]. ANGPTL4 derived from the exosomes of hypoxic cells is ingested by adjacent normoxic cells, leading to the radioresistance of these neighbouring cells in a GPX4-dependent manner [114]. Both intracellular and exosomal ANGPTL4 contribute to hypoxia-induced radioresistance of lung cancer. Erastin decreases the radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis [106]. Hyperbaric oxygen can significantly enhance the ferroptosis of oral squamous cell carcinoma (OSCC) cells induced by X-ray, and re-sensitize radioresistant OSCC cells through GPX4/ferroptosis regulation [115].
In addition, the metabolism or related mechanism of iron, lipid, and amino acids also mediate the radioresistance by regulating ferroptosis. Iron metabolism is associated with ferroptosis and radiotherapy efficacy. Iron-saturated holo-Lactoferrin could increase total iron content, which induces ferroptosis in triple-negative breast cancer cells and sensitizes tumor cells to radiotherapy [116]. Stearoyl-CoA desaturase (SCD1) is an enzyme responsible for the formation of oleic acid and palmitoleic acid. It was reported that targeting SCD1 can enhance radiation-induced ferroptosis and immunogenic cell death, thus improving radiation sensitivity [117]. SCD1 inhibitors can induce ferroptosis by reducing the formation of monounsaturated fatty acids. Inhibition of SCD1 makes tumor cells sensitive to radiotherapy and inhibits the growth of esophageal squamous cell carcinoma in vivo. The metabolomics analysis of irradiation-resistant HepG2 cells shows that the intracellular amino acids, especially N-acetylglutamine, increase significantly during the stress of ferroptosis [4]. N-acetylglutamine is a derivative of glutamine, which plays an important role in maintaining redox homeostasis. Glutamine starvation can significantly promote ferroptosis, and vice versa. Bioinformatics analysis based on TCGA data indicates that the glutamine transporter SLC1A5 is an independent prognostic amino acid-ferroptosis gene. The knockdown of SLC1A5 promotes lipid peroxidation and irradiation-mediated oxidative damage. The results indicate that SLC1A5 may be a potential target for radioresistance as an anti-ferroptosis gene [4]. The stem cell characteristics of tumor cells are also related to the resistance of radiotherapy and ferroptosis. It was found that the spheroids with the stem cell-like traits formed by nasopharyngeal carcinoma (NPC) cells exhibit a certain degree of radioresistance and ferroptosis resistance, while itraconazole partially reverses the radioresistance of NPC spheroids through inducing ferroptosis [118].
miRNAs can also affect radiation resistance by regulating ferroptosis. The expression of miR-7-5p in clinically relevant radioresistant cells is up-regulated, and the radioresistance is lost after miR-7-5p knockdown [119]. Knockdown of miR-7-5p increases ROS production and ferroptosis, characterized by increased intracellular Fe2+ amount, up-regulation of ferroptosis marker gene expression, and excessive production of lipid peroxides [120]. These indicate that miR-7-5p controls radioresistance by producing ROS, which can lead to ferroptosis.
Radiosensitization through m6A regulation
Concurrent radiotherapy and chemotherapy is the most common treatment after surgery [121]. Radiotherapy is an effective treatment for many kinds of cancers, and radioresistance is the main reason for local treatment failure. However, the potential mechanism and valuable markers of radioresistance have not been well established [17]. m6A modification plays an important role in gene expression regulation. Although m6A modification is involved in the development of tumor, its role in therapeutic resistance is still unclear. Understanding the effect of m6A modification on radiation response is of great significance for finding new targets and improving tumor treatment. Considering that m6A modification is involved in the regulation of ferroptosis and ferroptosis contributes to radiosensitivity, m6A methylation will regulate the efficacy of radiotherapy. In fact, some studies have shown that m6A RNA modification contributes to the regulation of radiotherapy resistance [6, 15, 16]. The dysregulated expression of many m6A enzymes, including demethylase FTO, methyltransferase METTL3 and WTAP, mediates the development of resistance of cancer cells to chemotherapy and radiotherapy [122, 123].
It was reported that m6A demethylase FTO enhances the radioresistance of NPC via promoting deubiquitylase OTUB1-mediated anti-ferroptosis [17]. The OTUB1 can mediate ferroptosis via the stabilization of SLC7A11 in human cancer [39]. The inhibition of OTUB1 on ferroptosis depends on the interaction between OTUB1 and SLC7A11 [17]. FTO, as an m6A demethylase, erases the m6A modification of the OTUB1 transcript, which up-regulates the expression of OTUB1 and leads to radiotherapy resistance of NPC [17]. The expression of FTO in radioresistant NPC tissues and cells is significantly higher than that of its parental radiosensitive tissues and cells. Accelerating ferroptosis by FTO inhibitor or ferroptosis inducer overcomes the radioresistance of NPC patient-derived xenografts [17]. This is the first report that m6A regulator can promote tumor resistance to radiotherapy by suppressing radiation-induced ferroptosis (Fig. 4), suggesting that m6A regulator may serve as a potential therapeutic target and prognostic biomarker. It was reported that FTO can also enhance the radiotherapy resistance of cervical squamous cell carcinoma through regulating expression of β-catenin by reducing m6A levels in its mRNA transcripts [124].

The demethylation of OTUB1 transcript by FTO promotes the expression of OTUB1, which inhibits ferroptosis through binding to SLC7A11 and stabilizing it.
Methyltransferase METTL3-mediated m6A modification plays a critical role in the development and maintenance of radioresistance. Cell response to ultraviolet-induced DNA damage can induce RNA m6A modification, which is regulated by METTL3 and FTO. METTL3 knockdown impairs the repair of irradiation-induced DNA damage and improves therapeutic sensitivity, suggesting the importance of m6A modification in the irradiation-mediated DNA damage response [125]. The expression of METTL3 is increased in glioma stem-like cells (GSCs), which plays an important role in the maintenance and radioresistance of GSCs by regulating m6A modification of SOX2 mRNA [126]. METTL3-mediated m6A mRNA contributes to the resistance of pancreatic cancer and NSCLC to radiotherapy [18, 127]. After carbon ion radiotherapy, the level of METTL3 and its mediated m6A modification in NSCLC cells is increased. METTL3-mediated mRNA m6A modification inhibits the decay of H2A histone family member X (H2AX) mRNA and enhances its expression, thus facilitating DNA damage repair and cell survival [127].
Several studies have also proved the regulatory effect of m6A readers on radioresistance. For example, YTHDC2 promotes radiotherapy resistance of NPC cells by activating the IGF1R/ATK/S6 signalling axis. YTHDC2 is consistently highly expressed in radioresistant NPC cells, and its expression is associated with the therapeutic effect of radiotherapy. YTHDC2 can bind to insulin-like growth factor 1 receptor (IGF1R) mRNA and promote translation initiation of IGF1R mRNA, which in turn activates the IGF1R-AKT/S6 signalling pathway [128]. YTHDF3 accelerates the translation of the DNA repair protein RAD51 homologue 4 (RAD51D) in an m6A-dependent manner, thereby mediating effect of hepatocyte nuclear factor 1-alpha (HNF1α) on radioresistance of cervical cancer. HNF1α is significantly up-regulated in radioresistant cervical cancer, thus promoting the resistance of cervical cancer cells to radiation. HNF1α enhances the transcription of YTHDF3, and YTHDF3 subsequently promotes m6A modification of RAD51D mRNA [129]. We must note that most of the above studies have shown to a large extent that radiotherapy can be sensitized through m6A regulation, but only a few studies have attributed radiosensitization to ferroptosis mediated by m6A modification [17].
Since the radiosensitivity of tumor cells can be regulated by m6A modification, targeting m6A regulators has shown great potential in tumor therapy [130, 131]. Many studies have attempted to identify small molecule inhibitors associated with m6A. Some compounds have been reported as potential METTL3 inhibitors [132, 133]. The anti-HIV drug elvitegravir has also been identified as a novel inhibitor of METTL3, which can inhibit the metastasis of esophageal carcinoma [134]. STM2457 is the first METTL3 catalytic inhibitor with high affinity for METTL3 [135]. It can reduce overall m6A levels and mRNA translation efficiency, and inhibit the growth of acute myeloid leukemia (AML). STM2457 exhibits a synergistic effect with PD-1 antibody in colorectal cancer [136]. Some small molecules can inhibit demethylase activity of m6A eraser proteins. Several FTO inhibitors have been identified, such as rhein [137], N-CDPCB [138], CHTB [139], and meclofenamic acid [140] and its derivatives [141, 142]. For example, rhein can disrupt the binding of FTO to m6A RNAs by competitively binding to the FTO catalytic domain [137]. Meclofenamic acid specifically inhibits FTO by competitively binding to FTO sites of m6A-modified oncogenic mRNAs, thus effectively inhibiting cancer cell proliferation [140]. Some small molecule inhibitors of FTO can exert effective anti-tumor effects by making cancer cells sensitive to the cytotoxicity of T cells [143]. A compound with anti-ALKBH5 catalytic activity has been identified to inhibit the proliferation of leukemia cells [144]. BTYNB has been shown to block the interaction between m6A reader IGF2BP1 and its substrate RNAs [145], leading to cancer cell cycle arrest [146]. Cucurbitacin B and 7773 block different RNA binding domains of IGF2BP1 respectively and regulate different downstream targets [147, 148]. CWI1-2 and JX5 have been identified as IGF2BP2 inhibitors, which selectively disrupt the binding of IGF2BP2 to its m6A modified target RNAs and exhibit good anti-leukemia activity [149, 150].
Before these m6A related inhibitors enter clinical application, further comprehensive research is needed, especially to strengthen in vivo research and clinical trials. Currently, only a few studies have conducted similar work. For example, the treatment of mice with R-2-hydroxyglutarate (R-2HG), a FTO inhibitor, can inhibit FTO demethylase activity, thus increasing the overall m6A levels and leading to anti-tumor effects in vivo [151]. METTL3 inhibitor STM2457 impairs engraftment and prolongs survival in AML mouse model [135]. METTL3 inhibitor STC-15 has entered the first stage of clinical trials [131]. In addition, the role or significance of these inhibitors in radiotherapy and radioresistance is not yet well understood. It is still unclear whether the role of these inhibitors is related to the ferroptosis. Due to the context-dependent epigenetic characteristics of cancers, it is also necessary to improve the target selectivity of inhibitors. However, some m6A related inhibitors have been shown to inhibit cancer progression, indicating that m6A could be a target for cancer therapy.
Conclusions
Radiotherapy is widely used to treat cancer, but often leads to resistance. The well-known mechanisms of radiation resistance include activation of DNA repair and inhibition of cell apoptosis. Therefore, it is valuable to make cancer cells sensitive to radiation by alternative cell death pathways [3]. Recently, it has been confirmed that lipid peroxidation can trigger ferroptosis, which is also a major feature of cell death induced by radiotherapy and contributes to radiosensitivity [1,2,3,4]. The combination of ferroptosis inducers and radiotherapy will greatly improve the efficacy of radiotherapy. Importantly, many anti-tumor drugs have been found to cause ROS accumulation, oxidative stress, and subsequent ferroptosis in tumor cells. For example, cisplatin can trigger ferroptosis by directly consuming intracellular GSH and inhibiting GPX4 [152]. Temozolomide induces ferroptosis by promoting the expression of SLC7A11 [153]. Sorafenib can inhibit system XC− or regulate HIF-1α/SLC7A11 pathway to promote ferroptosis [154, 155]. Therefore, they can be used as ferroptosis inducers. This can help us further understand the role of concurrent chemotherapy during radiotherapy and guide us in selecting appropriate chemotherapy drugs. As the most common post-transcriptional modification, m6A methylation regulates ferroptosis through different effectors and affects the sensitivity of tumor cells to radiotherapy. This makes various m6A regulatory molecules a promising therapeutic target in radiotherapy. Radiosensitization through m6A-mediated ferroptosis may be an alternative method for improving the efficacy of radiotherapy in the future.
Data availability
Correspondence and requests for data should be addressed to DY.
References
Lei G, Zhang Y, Koppula P, Liu X, Zhang J, Lin SH, et al. The role of ferroptosis in ionizing radiation-induced cell death and tumor suppression. Cell Res. 2020;30:146–62.
Lang X, Green MD, Wang W, Yu J, Choi JE, Jiang L, et al. Radiotherapy and immunotherapy promote tumoral lipid oxidation and ferroptosis via synergistic repression of SLC7A11. Cancer Discov. 2019;9:1673–85.
Ye LF, Chaudhary KR, Zandkarimi F, Harken AD, Kinslow CJ, Upadhyayula PS, et al. Radiation-induced lipid peroxidation triggers ferroptosis and synergizes with ferroptosis inducers. ACS Chem Biol. 2020;15:469–84.
Yuan Z, Liu T, Huo X, Wang H, Wang J, Xue L. Glutamine transporter SLC1A5 regulates ionizing radiation-derived oxidative damage and ferroptosis. Oxid Med Cell Longev. 2022;2022:3403009.
Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–72.
Shriwas O, Mohapatra P, Mohanty S, Dash R. The impact of m6A RNA modification in therapy resistance of cancer: implication in chemotherapy, radiotherapy, and immunotherapy. Front Oncol. 2020;10:612337.
Qi X, Wan Z, Jiang B, Ouyang Y, Feng W, Zhu H, et al. Inducing ferroptosis has the potential to overcome therapy resistance in breast cancer. Front Immunol. 2022;13:1038225.
Su J, Zhao Q, Zheng Z, Wang H, Bian C, Meng L, et al. Prospective application of ferroptosis in hypoxic cells for tumor radiotherapy. Antioxid (Basel). 2022;11:921.
Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, et al. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485:201–6.
Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3’ UTRs and near stop codons. Cell. 2012;149:1635–46.
Jiang X, Liu B, Nie Z, Duan L, Xiong Q, Jin Z, et al. The role of m6A modification in the biological functions and diseases. Signal Transduct Target Ther. 2021;6:74.
Jing FY, Zhou LM, Ning YJ, Wang XJ, Zhu YM. The biological function, mechanism, and clinical significance of m6A RNA modifications in head and neck carcinoma: a systematic review. Front Cell Dev Biol. 2021;9:683254.
Li W, Huang G, Wei J, Cao H, Jiang G. ALKBH5 inhibits thyroid cancer progression by promoting ferroptosis through TIAM1-Nrf2/HO-1 axis. Mol Cell Biochem. 2023;478:729–41.
Chen X, Chen L, Tang Y, He Y, Pan K, Yuan L, et al. Transcriptome-wide m(6)A methylome analysis uncovered the changes of m(6)A modification in oral pre-malignant cells compared with normal oral epithelial cells. Front Oncol. 2022;12:939449.
Xu Z, Peng B, Cai Y, Wu G, Huang J, Gao M, et al. N6-methyladenosine RNA modification in cancer therapeutic resistance: current status and perspectives. Biochem Pharm. 2020;182:114258.
Huang W, Chen TQ, Fang K, Zeng ZC, Ye H, Chen YQ. N6-methyladenosine methyltransferases: functions, regulation, and clinical potential. J Hematol Oncol. 2021;14:117.
Huang WM, Li ZX, Wu YH, Shi ZL, Mi JL, Hu K, et al. m6A demethylase FTO renders radioresistance of nasopharyngeal carcinoma via promoting OTUB1-mediated anti-ferroptosis. Transl Oncol. 2023;27:101576.
Taketo K, Konno M, Asai A, Koseki J, Toratani M, Satoh T, et al. The epitranscriptome m6A writer METTL3 promotes chemo- and radioresistance in pancreatic cancer cells. Int J Oncol. 2018;52:621–9.
Zhang L, Zhang J, Jin Y, Yao G, Zhao H, Qiao P, et al. Nrf2 is a potential modulator for orchestrating iron homeostasis and redox balance in cancer cells. Front Cell Dev Biol. 2021;9:728172.
Su LJ, Zhang JH, Gomez H, Murugan R, Hong X, Xu D, et al. Reactive oxygen species-induced lipid peroxidation in apoptosis, autophagy, and ferroptosis. Oxid Med Cell Longev. 2019;2019:5080843.
Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, et al. PEBP1 wardens ferroptosis by enabling lipoxygenase generation of lipid death signals. Cell. 2017;171:628–41.e26.
Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol. 2020;16:302–9.
Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, Dixon SJ, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell. 2017;171:273–85.
Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, et al. Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis. 2020;11:922.
Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature. 2020;585:603–8.
Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol. 2017;13:91–8.
Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell. 2014;156:317–31.
Koppula P, Zhuang L, Gan B. Cystine transporter SLC7A11/xCT in cancer: ferroptosis, nutrient dependency, and cancer therapy. Protein Cell. 2021;12:599–620.
Cozza G, Rossetto M, Bosello-Travain V, Maiorino M, Roveri A, Toppo S, et al. Glutathione peroxidase 4-catalyzed reduction of lipid hydroperoxides in membranes: the polar head of membrane phospholipids binds the enzyme and addresses the fatty acid hydroperoxide group toward the redox center. Free Radic Biol Med. 2017;112:1–11.
Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, et al. Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science. 2020;368:85–9.
Zheng J, Conrad M. The metabolic underpinnings of ferroptosis. Cell Metab. 2020;32:920–37.
Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem. 1999;274:11455–8.
Conrad M, Sato H. The oxidative stress-inducible cystine/glutamate antiporter, system x (c) (-) : cystine supplier and beyond. Amino Acids. 2012;42:231–46.
Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, et al. Ferroptosis as a p53-mediated activity during tumour suppression. Nature. 2015;520:57–62.
Wang W, Green M, Choi JE, Gijon M, Kennedy PD, Johnson JK, et al. CD8(+) T cells regulate tumour ferroptosis during cancer immunotherapy. Nature. 2019;569:270–4.
Fan Z, Wirth AK, Chen D, Wruck CJ, Rauh M, Buchfelder M, et al. Nrf2-Keap1 pathway promotes cell proliferation and diminishes ferroptosis. Oncogenesis. 2017;6:e371.
Rojo de la Vega M, Chapman E, Zhang DD. NRF2 and the hallmarks of cancer. Cancer Cell. 2018;34:21–43.
Zhang Y, Shi J, Liu X, Feng L, Gong Z, Koppula P, et al. BAP1 links metabolic regulation of ferroptosis to tumour suppression. Nat Cell Biol. 2018;20:1181–92.
Liu T, Jiang L, Tavana O, Gu W. The deubiquitylase OTUB1 mediates ferroptosis via stabilization of SLC7A11. Cancer Res. 2019;79:1913–24.
Ishimoto T, Nagano O, Yae T, Tamada M, Motohara T, Oshima H, et al. CD44 variant regulates redox status in cancer cells by stabilizing the xCT subunit of system xc(-) and thereby promotes tumor growth. Cancer Cell. 2011;19:387–400.
Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature. 2019;575:693–8.
Yang WS, Stockwell BR. Ferroptosis: death by lipid peroxidation. Trends Cell Biol. 2016;26:165–76.
Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, et al. Imidazole ketone erastin induces ferroptosis and slows tumor growth in a mouse lymphoma model. Cell Chem Biol. 2019;26:623–33.e9.
Lei G, Zhang Y, Hong T, Zhang X, Liu X, Mao C, et al. Ferroptosis as a mechanism to mediate p53 function in tumor radiosensitivity. Oncogene. 2021;40:3533–47.
Morgan MA, Lawrence TS. Molecular pathways: overcoming radiation resistance by targeting DNA damage response pathways. Clin Cancer Res. 2015;21:2898–904.
Wang HH, Wu ZQ, Qian D, Zaorsky NG, Qiu MH, Cheng JJ, et al. Ablative hypofractionated radiation therapy enhances non-small cell lung cancer cell killing via preferential stimulation of necroptosis in vitro and in vivo. Int J Radiat Oncol Biol Phys. 2018;101:49–62.
Okada H, Mak TW. Pathways of apoptotic and non-apoptotic death in tumour cells. Nat Rev Cancer. 2004;4:592–603.
Nehs MA, Lin CI, Kozono DE, Whang EE, Cho NL, Zhu K, et al. Necroptosis is a novel mechanism of radiation-induced cell death in anaplastic thyroid and adrenocortical cancers. Surgery. 2011;150:1032–9.
Chaurasia M, Bhatt AN, Das A, Dwarakanath BS, Sharma K. Radiation-induced autophagy: mechanisms and consequences. Free Radic Res. 2016;50:273–90.
Gebicki JM, Du J, Collins J, Tweeddale H. Peroxidation of proteins and lipids in suspensions of liposomes, in blood serum, and in mouse myeloma cells. Acta Biochim Pol. 2000;47:901–11.
Spitz DR, Azzam EI, Li JJ, Gius D. Metabolic oxidation/reduction reactions and cellular responses to ionizing radiation: a unifying concept in stress response biology. Cancer Metastasis Rev. 2004;23:311–22.
Shadyro OI, Yurkova IL, Kisel MA. Radiation-induced peroxidation and fragmentation of lipids in a model membrane. Int J Radiat Biol. 2002;78:211–7.
Azzam EI, Jay-Gerin JP, Pain D. Ionizing radiation-induced metabolic oxidative stress and prolonged cell injury. Cancer Lett. 2012;327:48–60.
Pearson AN, Carmicheal J, Jiang L, Lei YL, Green MD. Contribution of lipid oxidation and ferroptosis to radiotherapy efficacy. Int J Mol Sci. 2021;22:12603.
Lei G, Mao C, Yan Y, Zhuang L, Gan B. Ferroptosis, radiotherapy, and combination therapeutic strategies. Protein Cell. 2021;12:836–57.
Chen X, Kang R, Kroemer G, Tang D. Broadening horizons: the role of ferroptosis in cancer. Nat Rev Clin Oncol. 2021;18:280–96.
Chen Q, Zheng W, Guan J, Liu H, Dan Y, Zhu L, et al. SOCS2-enhanced ubiquitination of SLC7A11 promotes ferroptosis and radiosensitization in hepatocellular carcinoma. Cell Death Differ. 2023;30:137–51.
Li C, Zhang Y, Liu J, Kang R, Klionsky DJ, Tang D. Mitochondrial DNA stress triggers autophagy-dependent ferroptotic death. Autophagy. 2021;17:948–60.
Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6-methyladenosine in Cancer progression. Mol Cancer. 2020;19:88.
Shen C, Xuan B, Yan T, Ma Y, Xu P, Tian X, et al. m(6)A-dependent glycolysis enhances colorectal cancer progression. Mol Cancer. 2020;19:72.
Wang W, Shao F, Yang X, Wang J, Zhu R, Yang Y, et al. METTL3 promotes tumour development by decreasing APC expression mediated by APC mRNA N(6)-methyladenosine-dependent YTHDF binding. Nat Commun. 2021;12:3803.
Xu T, Zhang W, Chai L, Liu C, Zhang S, Xu T. Methyltransferase-like 3-induced N6-methyladenosine upregulation promotes oral squamous cell carcinoma by through p38. Oral Dis. 2023;29:639–48.
Liu L, Wu Y, Li Q, Liang J, He Q, Zhao L, et al. METTL3 promotes tumorigenesis and metastasis through BMI1 m(6)A methylation in oral squamous cell carcinoma. Mol Ther. 2020;28:2177–90.
Zhao W, Cui Y, Liu L, Ma X, Qi X, Wang Y, et al. METTL3 facilitates oral squamous cell carcinoma tumorigenesis by enhancing c-Myc stability via YTHDF1-mediated m(6)A modification. Mol Ther Nucleic Acids. 2020;20:1–12.
Ai Y, Liu S, Luo H, Wu S, Wei H, Tang Z, et al. METTL3 intensifies the progress of oral squamous cell carcinoma via modulating the m6A amount of PRMT5 and PD-L1. J Immunol Res. 2021;2021:6149558.
Gao M, Qi Z, Feng W, Huang H, Xu Z, Dong Z, et al. m6A demethylation of cytidine deaminase APOBEC3B mRNA orchestrates arsenic-induced mutagenesis. J Biol Chem. 2022;298:101563.
Zhou Y, Wang Q, Deng H, Xu B, Zhou Y, Liu J, et al. N6-methyladenosine demethylase FTO promotes growth and metastasis of gastric cancer via m(6)A modification of caveolin-1 and metabolic regulation of mitochondrial dynamics. Cell Death Dis. 2022;13:72.
Lv D, Ding S, Zhong L, Tu J, Li H, Yao H, et al. M(6)A demethylase FTO-mediated downregulation of DACT1 mRNA stability promotes Wnt signaling to facilitate osteosarcoma progression. Oncogene. 2022;41:1727–41.
Jeschke J, Collignon E, Al Wardi C, Krayem M, Bizet M, Jia Y, et al. Downregulation of the FTO m(6)A RNA demethylase promotes EMT-mediated progression of epithelial tumors and sensitivity to Wnt inhibitors. Nat Cancer. 2021;2:611–28.
Li J, Zhu L, Shi Y, Liu J, Lin L, Chen X. m6A demethylase FTO promotes hepatocellular carcinoma tumorigenesis via mediating PKM2 demethylation. Am J Transl Res. 2019;11:6084–92.
Li X, Xie X, Gu Y, Zhang J, Song J, Cheng X, et al. Fat mass and obesity-associated protein regulates tumorigenesis of arecoline-promoted human oral carcinoma. Cancer Med. 2021;10:6402–15.
Wang F, Liao Y, Zhang M, Zhu Y, Wang W, Cai H, et al. N6-methyladenosine demethyltransferase FTO-mediated autophagy in malignant development of oral squamous cell carcinoma. Oncogene. 2021;40:3885–98.
Yue B, Song C, Yang L, Cui R, Cheng X, Zhang Z, et al. METTL3-mediated N6-methyladenosine modification is critical for epithelial-mesenchymal transition and metastasis of gastric cancer. Mol Cancer. 2019;18:142.
Wang Q, Chen C, Ding Q, Zhao Y, Wang Z, Chen J, et al. METTL3-mediated m(6)A modification of HDGF mRNA promotes gastric cancer progression and has prognostic significance. Gut. 2020;69:1193–205.
Tang F, Chen L, Gao H, Xiao D, Li X. m(6)A: an emerging role in programmed cell death. Front Cell Dev Biol. 2022;10:817112.
Liu L, Li H, Hu D, Wang Y, Shao W, Zhong J, et al. Insights into N6-methyladenosine and programmed cell death in cancer. Mol Cancer. 2022;21:32.
Zhi Y, Zhang S, Zi M, Wang Y, Liu Y, Zhang M, et al. Potential applications of N(6) -methyladenosine modification in the prognosis and treatment of cancers via modulating apoptosis, autophagy, and ferroptosis. Wiley Interdiscip Rev RNA. 2022;13:e1719.
Cheung JCT, Deng G, Wong N, Dong Y, Ng SSM. More than a duologue: In-depth insights into epitranscriptomics and ferroptosis. Front Cell Dev Biol. 2022;10:982606.
Ma L, Chen T, Zhang X, Miao Y, Tian X, Yu K, et al. The m(6)A reader YTHDC2 inhibits lung adenocarcinoma tumorigenesis by suppressing SLC7A11-dependent antioxidant function. Redox Biol. 2021;38:101801.
Ma L, Zhang X, Yu K, Xu X, Chen T, Shi Y, et al. Targeting SLC3A2 subunit of system X(C)(-) is essential for m(6)A reader YTHDC2 to be an endogenous ferroptosis inducer in lung adenocarcinoma. Free Radic Biol Med. 2021;168:25–43.
Xu Y, Lv D, Yan C, Su H, Zhang X, Shi Y, et al. METTL3 promotes lung adenocarcinoma tumor growth and inhibits ferroptosis by stabilizing SLC7A11 m(6)A modification. Cancer Cell Int. 2022;22:11.
Liu L, He J, Sun G, Huang N, Bian Z, Xu C, et al. The N6-methyladenosine modification enhances ferroptosis resistance through inhibiting SLC7A11 mRNA deadenylation in hepatoblastoma. Clin Transl Med. 2022;12:e778.
Fan Z, Yang G, Zhang W, Liu Q, Liu G, Liu P, et al. Hypoxia blocks ferroptosis of hepatocellular carcinoma via suppression of METTL14 triggered YTHDF2-dependent silencing of SLC7A11. J Cell Mol Med. 2021;25:10197–212.
Ji FH, Fu XH, Li GQ, He Q, Qiu XG. FTO prevents Thyroid Cancer progression by SLC7A11 m6A methylation in a ferroptosis-dependent manner. Front Endocrinol (Lausanne). 2022;13:857765.
Sun S, Gao T, Pang B, Su X, Guo C, Zhang R, et al. RNA binding protein NKAP protects glioblastoma cells from ferroptosis by promoting SLC7A11 mRNA splicing in an m(6)A-dependent manner. Cell Death Dis. 2022;13:73.
Zou Y, Zheng S, Xie X, Ye F, Hu X, Tian Z, et al. N6-methyladenosine regulated FGFR4 attenuates ferroptotic cell death in recalcitrant HER2-positive breast cancer. Nat Commun. 2022;13:2672.
Song Z, Jia G, Ma P, Cang S. Exosomal miR-4443 promotes cisplatin resistance in non-small cell lung carcinoma by regulating FSP1 m6A modification-mediated ferroptosis. Life Sci. 2021;276:119399.
Yuan J, Lv T, Yang J, Wu Z, Yan L, Yang J, et al. HDLBP-stabilized lncFAL inhibits ferroptosis vulnerability by diminishing Trim69-dependent FSP1 degradation in hepatocellular carcinoma. Redox Biol. 2022;58:102546.
Bu C, Hu S, Yu J, Li N, Gu J, Sheng Z, et al. Fear stress promotes glioma progression through inhibition of ferroptosis by enhancing FSP1 stability. Clin Transl Oncol. 2023;25:1378–88.
Zhang H, Liu J, Zhou Y, Qu M, Wang Y, Guo K, et al. Neutrophil extracellular traps mediate m(6)A modification and regulates sepsis-associated acute lung injury by activating ferroptosis in alveolar epithelial cells. Int J Biol Sci. 2022;18:3337–57.
Ye F, Wu J, Zhang F. METTL16 epigenetically enhances GPX4 expression via m6A modification to promote breast cancer progression by inhibiting ferroptosis. Biochem Biophys Res Commun. 2023;638:1–6.
Xu X, Cui J, Wang H, Ma L, Zhang X, Guo W, et al. IGF2BP3 is an essential N(6)-methyladenosine biotarget for suppressing ferroptosis in lung adenocarcinoma cells. Mater Today Bio. 2022;17:100503.
Wang K, Wang G, Li G, Zhang W, Wang Y, Lin X, et al. m6A writer WTAP targets NRF2 to accelerate bladder cancer malignancy via m6A-dependent ferroptosis regulation. Apoptosis. 2023;28:627–38.
Ye J, Chen X, Jiang X, Dong Z, Hu S, Xiao M. RNA demethylase ALKBH5 regulates hypopharyngeal squamous cell carcinoma ferroptosis by posttranscriptionally activating NFE2L2/NRF2 in an m(6) A-IGF2BP2-dependent manner. J Clin Lab Anal. 2022;36:e24514.
Lu Z, Yang H, Shao Y, Sun W, Jiang Y, Li J. IGF2BP3-NRF2 axis regulates ferroptosis in hepatocellular carcinoma. Biochem Biophys Res Commun. 2022;627:103–10.
Lin Y, Shen X, Ke Y, Lan C, Chen X, Liang B, et al. Activation of osteoblast ferroptosis via the METTL3/ASK1-p38 signaling pathway in high glucose and high fat (HGHF)-induced diabetic bone loss. FASEB J. 2022;36:e22147.
Zhuang S, Ma Y, Zeng Y, Lu C, Yang F, Jiang N, et al. METTL14 promotes doxorubicin-induced cardiomyocyte ferroptosis by regulating the KCNQ1OT1-miR-7-5p-TFRC axis. Cell Biol Toxicol. 2023;39:1015–35.
Chen S, Xia H, Sheng L. WTAP-mediated m6A modification on circCMTM3 inhibits hepatocellular carcinoma ferroptosis by recruiting IGF2BP1 to increase PARK7 stability. Dig Liver Dis. 2023;55:967–81.
Cao J, Chen X, Jiang L, Lu B, Yuan M, Zhu D, et al. DJ-1 suppresses ferroptosis through preserving the activity of S-adenosyl homocysteine hydrolase. Nat Commun. 2020;11:1251.
Shen M, Li Y, Wang Y, Shao J, Zhang F, Yin G, et al. N(6)-methyladenosine modification regulates ferroptosis through autophagy signaling pathway in hepatic stellate cells. Redox Biol. 2021;47:102151.
Gryzik M, Asperti M, Denardo A, Arosio P, Poli M. NCOA4-mediated ferritinophagy promotes ferroptosis induced by erastin, but not by RSL3 in HeLa cells. Biochim Biophys Acta Mol Cell Res. 2021;1868:118913.
Kang R, Zhu S, Zeh HJ, Klionsky DJ, Tang D. BECN1 is a new driver of ferroptosis. Autophagy. 2018;14:2173–5.
Shen M, Guo M, Li Y, Wang Y, Qiu Y, Shao J, et al. m(6)A methylation is required for dihydroartemisinin to alleviate liver fibrosis by inducing ferroptosis in hepatic stellate cells. Free Radic Biol Med. 2022;182:246–59.
Chen X, Yuan L, Zhang L, Chen L, He Y, Wang C, et al. GPX8 deficiency-induced oxidative stress reprogrammed m6A epitranscriptome of oral cancer cells. Epigenetics. 2023;18:2208707.
Feng D, Shi X, Xiong Q, Zhang F, Li D, Wei W, et al. A ferroptosis-related gene prognostic index associated with biochemical recurrence and radiation resistance for patients with prostate cancer undergoing radical radiotherapy. Front Cell Dev Biol. 2022;10:803766.
Pan X, Lin Z, Jiang D, Yu Y, Yang D, Zhou H, et al. Erastin decreases radioresistance of NSCLC cells partially by inducing GPX4-mediated ferroptosis. Oncol Lett. 2019;17:3001–8.
Zhang W, Sun Y, Bai L, Zhi L, Yang Y, Zhao Q, et al. RBMS1 regulates lung cancer ferroptosis through translational control of SLC7A11. J Clin Invest. 2021;131:e152067.
Zheng X, Liu B, Liu X, Li P, Zhang P, Ye F, et al. PERK regulates the sensitivity of hepatocellular carcinoma cells to high-LET carbon ions via either apoptosis or ferroptosis. J Cancer. 2022;13:669–80.
Emmanuel N, Li H, Chen J, Zhang Y. FSP1, a novel KEAP1/NRF2 target gene regulating ferroptosis and radioresistance in lung cancers. Oncotarget. 2022;13:1136–9.
Feng L, Zhao K, Sun L, Yin X, Zhang J, Liu C, et al. SLC7A11 regulated by NRF2 modulates esophageal squamous cell carcinoma radiosensitivity by inhibiting ferroptosis. J Transl Med. 2021;19:367.
Koppula P, Lei G, Zhang Y, Yan Y, Mao C, Kondiparthi L, et al. A targetable CoQ-FSP1 axis drives ferroptosis- and radiation-resistance in KEAP1 inactive lung cancers. Nat Commun. 2022;13:2206.
Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol. 2017;11:254–62.
Sun H, Cai H, Xu C, Zhai H, Lux F, Xie Y, et al. AGuIX nanoparticles enhance ionizing radiation-induced ferroptosis on tumor cells by targeting the NRF2-GPX4 signaling pathway. J Nanobiotechnol. 2022;20:449.
Zhang Y, Liu X, Zeng L, Zhao X, Chen Q, Pan Y, et al. Exosomal protein angiopoietin-like 4 mediated radioresistance of lung cancer by inhibiting ferroptosis under hypoxic microenvironment. Br J Cancer. 2022;127:1760–72.
Liu J, An W, Zhao Q, Liu Z, Jiang Y, Li H, et al. Hyperbaric oxygen enhances X-ray induced ferroptosis in oral squamous cell carcinoma cells. Oral Dis. 2022; Dec 10. https://doi.org/10.1111/odi.14461.
Zhang Z, Lu M, Chen C, Tong X, Li Y, Yang K, et al. Holo-lactoferrin: the link between ferroptosis and radiotherapy in triple-negative breast cancer. Theranostics. 2021;11:3167–82.
Luo H, Wang X, Song S, Wang Y, Dan Q, Ge H. Targeting stearoyl-coa desaturase enhances radiation induced ferroptosis and immunogenic cell death in esophageal squamous cell carcinoma. Oncoimmunology. 2022;11:2101769.
Xu Y, Wang Q, Li X, Chen Y, Xu G. Itraconazole attenuates the stemness of nasopharyngeal carcinoma cells via triggering ferroptosis. Environ Toxicol. 2021;36:257–66.
Tomita K, Fukumoto M, Itoh K, Kuwahara Y, Igarashi K, Nagasawa T, et al. MiR-7-5p is a key factor that controls radioresistance via intracellular Fe(2+) content in clinically relevant radioresistant cells. Biochem Biophys Res Commun. 2019;518:712–8.
Tomita K, Nagasawa T, Kuwahara Y, Torii S, Igarashi K, Roudkenar MH, et al. MiR-7-5p is involved in ferroptosis signaling and radioresistance Thru the generation of ROS in radioresistant HeLa and SAS Cell Lines. Int J Mol Sci. 2021;22:8300.
Domina EA, Philchenkov A, Dubrovska A. Individual response to ionizing radiation and personalized radiotherapy. Crit Rev Oncog. 2018;23:69–92.
Xiang M, Liu W, Tian W, You A, Deng D. RNA N-6-methyladenosine enzymes and resistance of cancer cells to chemotherapy and radiotherapy. Epigenomics. 2020;12:801–9.
Wang Y, Zhang L, Sun XL, Lu YC, Chen S, Pei DS, et al. NRP1 contributes to stemness and potentiates radioresistance via WTAP-mediated m6A methylation of Bcl-2 mRNA in breast cancer. Apoptosis. 2023;28:233–46.
Zhou S, Bai ZL, Xia D, Zhao ZJ, Zhao R, Wang YY, et al. FTO regulates the chemo-radiotherapy resistance of cervical squamous cell carcinoma (CSCC) by targeting beta-catenin through mRNA demethylation. Mol Carcinog. 2018;57:590–7.
Xiang Y, Laurent B, Hsu CH, Nachtergaele S, Lu Z, Sheng W, et al. RNA m(6)A methylation regulates the ultraviolet-induced DNA damage response. Nature. 2017;543:573–6.
Visvanathan A, Patil V, Arora A, Hegde AS, Arivazhagan A, Santosh V, et al. Essential role of METTL3-mediated m(6)A modification in glioma stem-like cells maintenance and radioresistance. Oncogene. 2018;37:522–33.
Xu X, Zhang P, Huang Y, Shi W, Mao J, Ma N, et al. METTL3-mediated m6A mRNA contributes to the resistance of carbon-ion radiotherapy in non-small-cell lung cancer. Cancer Sci. 2023;114:105–14.
He JJ, Li Z, Rong ZX, Gao J, Mu Y, Guan YD, et al. m(6)A reader YTHDC2 promotes radiotherapy resistance of nasopharyngeal carcinoma via activating IGF1R/AKT/S6 signaling axis. Front Oncol. 2020;10:1166.
Du H, Zou NY, Zuo HL, Zhang XY, Zhu SC. YTHDF3 mediates HNF1alpha regulation of cervical cancer radio-resistance by promoting RAD51D translation in an m6A-dependent manner. FEBS J. 2023;290:1920–35.
Deng X, Qing Y, Horne D, Huang H, Chen J. The roles and implications of RNA m(6)A modification in cancer. Nat Rev Clin Oncol. 2023;20:507–26.
Shen LT, Che LR, He Z, Lu Q, Chen DF, Qin ZY, et al. Aberrant RNA m(6)A modification in gastrointestinal malignancies: versatile regulators of cancer hallmarks and novel therapeutic opportunities. Cell Death Dis. 2023;14:236.
Bedi RK, Huang D, Eberle SA, Wiedmer L, Sledz P, Caflisch A. Small-molecule inhibitors of METTL3, the major human epitranscriptomic writer. ChemMedChem. 2020;15:744–8.
Moroz-Omori EV, Huang D, Kumar Bedi R, Cheriyamkunnel SJ, Bochenkova E, Dolbois A, et al. METTL3 inhibitors for epitranscriptomic modulation of cellular processes. ChemMedChem. 2021;16:3035–43.
Liao L, He Y, Li SJ, Zhang GG, Yu W, Yang J, et al. Anti-HIV drug elvitegravir suppresses cancer metastasis via increased proteasomal degradation of m6A methyltransferase METTL3. Cancer Res. 2022;82:2444–57.
Yankova E, Blackaby W, Albertella M, Rak J, De Braekeleer E, Tsagkogeorga G, et al. Small-molecule inhibition of METTL3 as a strategy against myeloid leukaemia. Nature. 2021;593:597–601.
Chen H, Pan Y, Zhou Q, Liang C, Wong CC, Zhou Y, et al. METTL3 inhibits antitumor immunity by targeting m(6)A-BHLHE41-CXCL1/CXCR2 axis to promote colorectal cancer. Gastroenterology. 2022;163:891–907.
Chen B, Ye F, Yu L, Jia G, Huang X, Zhang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc. 2012;134:17963–71.
He W, Zhou B, Liu W, Zhang M, Shen Z, Han Z, et al. Identification of a novel small-molecule binding site of the fat mass and obesity associated protein (FTO). J Med Chem. 2015;58:7341–8.
Qiao Y, Zhou B, Zhang M, Liu W, Han Z, Song C, et al. A novel inhibitor of the obesity-related protein FTO. Biochemistry. 2016;55:1516–22.
Huang Y, Yan J, Li Q, Li J, Gong S, Zhou H, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res. 2015;43:373–84.
Huang Y, Su R, Sheng Y, Dong L, Dong Z, Xu H, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell. 2019;35:677–91.e10.
Huff S, Tiwari SK, Gonzalez GM, Wang Y, Rana TM. m(6)A-RNA demethylase FTO inhibitors impair self-renewal in glioblastoma stem cells. ACS Chem Biol. 2021;16:324–33.
Su R, Dong L, Li Y, Gao M, Han L, Wunderlich M, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell. 2020;38:79–96.e11.
Selberg S, Seli N, Kankuri E, Karelson M. Rational design of novel anticancer small-molecule RNA m6A demethylase ALKBH5 inhibitors. ACS Omega. 2021;6:13310–20.
Mahapatra L, Andruska N, Mao C, Le J, Shapiro DJ. A novel IMP1 inhibitor, BTYNB, targets c-Myc and inhibits melanoma and ovarian cancer cell proliferation. Transl Oncol. 2017;10:818–27.
Muller S, Bley N, Busch B, Glass M, Lederer M, Misiak C, et al. The oncofetal RNA-binding protein IGF2BP1 is a druggable, post-transcriptional super-enhancer of E2F-driven gene expression in cancer. Nucleic Acids Res. 2020;48:8576–90.
Wallis N, Oberman F, Shurrush K, Germain N, Greenwald G, Gershon T, et al. Small molecule inhibitor of Igf2bp1 represses Kras and a pro-oncogenic phenotype in cancer cells. RNA Biol. 2022;19:26–43.
Liu Y, Guo Q, Yang H, Zhang XW, Feng N, Wang JK, et al. Allosteric regulation of IGF2BP1 as a novel strategy for the activation of tumor immune microenvironment. ACS Cent Sci. 2022;8:1102–15.
Weng H, Huang F, Yu Z, Chen Z, Prince E, Kang Y, et al. The m(6)A reader IGF2BP2 regulates glutamine metabolism and represents a therapeutic target in acute myeloid leukemia. Cancer Cell. 2022;40:1566–82.e10.
Feng P, Chen D, Wang X, Li Y, Li Z, Li B, et al. Inhibition of the m(6)A reader IGF2BP2 as a strategy against T-cell acute lymphoblastic leukemia. Leukemia. 2022;36:2180–8.
Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m(6)A/MYC/CEBPA signaling. Cell. 2018;172:90–105.e23.
Guo J, Xu B, Han Q, Zhou H, Xia Y, Gong C, et al. Ferroptosis: a novel anti-tumor action for cisplatin. Cancer Res Treat. 2018;50:445–60.
Chen L, Li X, Liu L, Yu B, Xue Y, Liu Y. Erastin sensitizes glioblastoma cells to temozolomide by restraining xCT and cystathionine-gamma-lyase function. Oncol Rep. 2015;33:1465–74.
Yuan S, Wei C, Liu G, Zhang L, Li J, Li L, et al. Sorafenib attenuates liver fibrosis by triggering hepatic stellate cell ferroptosis via HIF-1alpha/SLC7A11 pathway. Cell Prolif. 2022;55:e13158.
Wang Q, Bin C, Xue Q, Gao Q, Huang A, Wang K, et al. GSTZ1 sensitizes hepatocellular carcinoma cells to sorafenib-induced ferroptosis via inhibition of NRF2/GPX4 axis. Cell Death Dis. 2021;12:426.
Funding
This study was supported by the National Natural Science Foundation of China (No. 81873711, No. 82073378 and No. 31670788) and by the Open Fund of Guangdong Key Laboratory of Pharmaceutical Functional Genes (No. 2020B1212060031).
Author information
Authors and Affiliations
Contributions
XC, LZ, YH, SH, SC, WZ and DY wrote the manuscript, and all authors approved the submitted version.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Consent for publication
All authors agree to publish.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chen, X., Zhang, L., He, Y. et al. Regulation of m6A modification on ferroptosis and its potential significance in radiosensitization. Cell Death Discov. 9, 343 (2023). https://doi.org/10.1038/s41420-023-01645-1
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41420-023-01645-1
- Springer Nature Limited