
Abstract
The polycomb repressor complex 2 molecule EZH2 is now known to play a role in essential cellular processes, namely, cell fate decisions, cell cycle regulation, senescence, cell differentiation, and cancer development/progression. EZH2 inhibitors have recently been developed; however, their effectiveness and underlying molecular mechanisms in many malignancies have not yet been elucidated in detail. Although the functional role of EZH2 in tumorigenesis in neuroblastoma (NB) has been investigated, mutations of EZH2 have not been reported. A Kaplan–Meier analysis on the event free survival and overall survival of NB patients indicated that the high expression of EZH2 correlated with an unfavorable prognosis. In order to elucidate the functional roles of EZH2 in NB tumorigenesis and its aggressiveness, we knocked down EZH2 in NB cell lines using lentivirus systems. The knockdown of EZH2 significantly induced NB cell differentiation, e.g., neurite extension, and the neuronal differentiation markers, NF68 and GAP43. EZH2 inhibitors also induced NB cell differentiation. We performed a comprehensive transcriptome analysis using Human Gene Expression Microarrays and found that NTRK1 (TrkA) is one of the EZH2-related suppression targets. The depletion of NTRK1 canceled EZH2 knockdown-induced NB cell differentiation. Our integrative methylome, transcriptome, and chromatin immunoprecipitation assays using NB cell lines and clinical samples clarified that the NTRK1 P1 and P2 promoter regions were regulated differently by DNA methylation and EZH2-related histone modifications. The NTRK1 transcript variants 1/2, which were regulated by EZH2-related H3K27me3 modifications at the P1 promoter region, were strongly expressed in favorable, but not unfavorable NB. The depletion and inhibition of EZH2 successfully induced NTRK1 transcripts and functional proteins. Collectively, these results indicate that EZH2 plays important roles in preventing the differentiation of NB cells and also that EZH2-related NTRK1 transcriptional regulation may be the key pathway for NB cell differentiation.
Similar content being viewed by others


Introduction
Neuroblastoma (NB) is the second most common pediatric solid malignant tumor derived from the sympathetic nervous system. The overall prognosis of patients with NB has markedly improved; however, unlike many childhood malignancies for which survival has been improved by recent therapies, high-risk NB is still one of the most difficult tumors to cure, with only 40% of patients achieving long-term survival despite intensive multimodal therapy [1].
Multiple genetic abnormalities, such as aneuploidy, chromosomal gains (extra copies of 17q) and losses (allelic loss of 1p and/or 11q), amplification of chromosomal material (MYCN), specific mutations in ALK and ATRX, and recurrent genomic rearrangements affecting the chromosomal region at 5p15.33 proximal to the telomerase reverse transcriptase gene (TERT), appear to reflect the different clinical entities and have led to the better stratification of patients in therapy for NB [2,3,4]. Furthermore, epigenetic alterations have been shown to have an impact on NB tumorigenesis and aggressiveness, e.g., CASP8 [5, 6] promoter methylation and the CpG island methylator phenotype (CIMP) [7, 8].
We previously reported that MYCN-induced BMI1 positively regulates NB cell proliferation via the transcriptional suppression of tumor suppressors in NB cells [9]. These findings were supported by the subsequent study by Blasberg’s group showing a consistent positive correlation between BMI1 and MYCN expression in MYCN-amplified NBs [10]. EZH2 is a PcG member protein and H3K27 methylation is catalyzed by the SET domain of EZH2 and requires the presence of two additional proteins, embryonic ectoderm development (EED) and suppressor of zeste 12 (SUZ12). These proteins comprise the core components of polycomb repressive complex 2 (PRC2) [11]. The expression of EZH2 is upregulated in several cancers including prostate cancer, breast cancer, bladder, gastric, lung, and hepatocellular carcinomas [12, 13]. Functional EZH2 mutations have not been detected in NB [14]. The EZH2-mediated differentiation suppression and epigenetic regulation of CASZ1 was previously reported [15]. The canonical oncogenic function of EZH2 is dependent on the epigenetic silencing of tumor-suppressor genes by H3K27 methylation. However, several recent studies showed some additional functions of EZH2, such as the transcriptional activation of target genes, e.g., beta-catenin/TCF and NF-kB pathways [16, 17], and the methylation of non-histone proteins, e.g., STAT3 methylation in prostate cancer cells [18] and GATA4 methylation in fetal hearts [19]. Advances have recently been achieved in the development of EZH2 inhibitors. 3-Deazaneplanocin A hydrochloride (DZnep) is an inhibitor of S-adenosyl methionine-dependent methyltransferase, which targets the degradation of EZH2 and decreases PRC2 protein levels [20]. GSK126, a potent, highly selective, S-adenosyl-methionine-competitive, small-molecule inhibitor of EZH2 methyltransferase activity, decreases global H3K27me3 levels, and reactivates silenced PRC2 target genes [21, 22]. EPZ6438 is one of the most potent EZH2 inhibitors described to date and possesses significantly improved pharmacokinetic properties, including good oral bioavailability in animals [23].
In the present study, we demonstrated that the high expression of EZH2 correlated with the poor prognosis of NB patients. The depletion of EZH2 by lentivirus shRNAs significantly induced neuronal differentiation. The results of an expression microarray study indicated that NTRK1 is one of the EZH2-mediated epigenetic silencing targets in NB cells and regulates the EZH2-dependent undifferentiated status of NB. The complex transcriptional regulation of NTRK1 by epigenomic changes in NB cells was analyzed in detail using NB cell lines and clinical samples. The epigenomic molecular mechanisms responsible for the suppression of NTRK1 in unfavorable NB were also clarified.
Results
Knockdown of EZH2 promoted NB cell differentiation
We performed a Kaplan–Meier survival analysis using data in the R2: Genomics Analysis and Visualization Platform (http://r2.amc.nl). The results obtained suggested that the high expression of EZH2 correlated with the unfavorable outcomes of NB patients (Supplementary Table S1) and our previous findings indicated roles for PcGs in NB tumorigenesis and aggressiveness [9]. These results prompted us to examine the effects of EZH2 on NB cell biology and investigate the molecular mechanisms responsible for strong EZH2 expression-related phenotypes. We analyzed the expression of EZH2, BMI1, and RING1B in NB cell lines and found that MYCN-amplified cell lines possessed abundant amounts of the EZH2 protein and also that EZH2 mRNA levels were related to protein levels (Supplementary Figure S1). Furthermore, the EZH2 mRNA level was higher in the MYCN-amplified NB tumors than in the MYCN not-amplified tumors. These results prompted us to deplete EZH2 in NB-39-nu cells because they have high EZH2 mRNA/protein levels and several NB aggressiveness-related phenotypes [24, 25]. Additionally, we examined NB-differentiation-related gene expression and neurite extension in parental NB cell lines, as well as shRNA-infected NB-39-nu, NGP, SK-N-DZ, and TGW cells. We did not observe a significant difference in differentiation markers (GAP43, expression/neurite extension) between the parental NB cell lines and the mock-infected cells (Supplementary Figure S2); therefore, we used shCont- and shEZH2-transduced cells for the experiments.
The knockdown of EZH2 by lentivirus-mediated shEZH2 transduction in NB-39-nu cells strongly promoted neurite extension, and was coupled with the upregulated expression of the neuronal cell differentiation markers, GAP43 and NF68 (Fig. 1a, b, d). In accordance with the depletion of EZH2, the H3K27 tri-methylation signal was markedly decreased (Fig. 1c).

Knockdown of EZH2 promotes neurite extension and induces neuronal markers. Results are representative of at least three independent experiments. a Images of EZH2 KD NB-39-nu cells under a contrast microscope. NB-39-nu cells were infected with the shCont- or shEZH2-lentivirus, as described in the Materials and Methods. Images were obtained 8 days after infection. b The percentages of neurite-extending cells in shCont- or shEZH2-expressing lentivirus-infected cells were counted 8 and 11 days after infection. Neurite extension was analyzed as described in the Materials and Methods. Error bars represent the standard deviation obtained with triplicate samples. c EZH2 and H3K27me3 levels were assessed using western blotting. After the infection with the shCont- or shEZH2-lentivirus, cells were collected on days 3 and 4 and subjected to a western blotting analysis. d The upregulation of the neuronal markers GAP43 and NF68 was confirmed by RT-PCR. e Tumor development in BALB/c AJcl nu/nu mice following the injection of NB-39-nu cells stably infected with shRNA against the control (shCont) or EZH2 (shEZH2-1). Tumor volumes were measured every 4 days. Data are presented as the mean ± SE of tumors in four mice
To evaluate tumorigenicity in vivo, EZH2-depleted NB-39-nu cells were injected subcutaneously into the backs of nude mice. EZH2 shRNA-infected cells (shEZH2-1) formed significantly smaller tumors than mock shRNA-infected cells (shCont, Fig. 1e).
Transcriptome analysis for EZH2-depleted cells
In order to elucidate the effects of the suppression of EZH2 on genome-wide gene expression profiles, we performed a transcriptome analysis using microarrays. Eighty-four upregulated genes (Supplementary Table S2) and 27 downregulated genes (Supplementary Table S3) were selected by the criteria described in the Materials and Methods (moderate t-test p < 0.05, fold change (FC) > 2.0) (Fig. 2a). Since EZH2 functions as a transcription suppressor mediating H3K27me3 modifications, the depression of EZH2 may result in the upregulated expression of its target genes. Therefore, we characterized upregulated genes using a pathway analysis (GeneSpring GX). The pathway analysis against the Wiki Pathway database [26] showed that several neuron-associated pathways such as “Spinal cord injury (WP1449 79963)” and “Regulation of the toll-like receptor signaling pathway (WP1449 81172)” as well as the cancer-related pathway “Integrated pancreatic cancer pathway (WP2377 71288)” were highly enriched in the selected upregulated genes (Fig. 2c). In addition, apoptosis-related pathways including “Extrinsic Pathway for Apoptosis”, “Ligand-dependent caspase activation”, and “Apoptosis Modulations by HSP70” were significantly enriched. Among these genes, we focused on NTRK1. NTRK1, a member of the neurotrophic tyrosine kinase receptor family, binds to nerve growth factor (NGF) to promote sensory neural differentiation and has been identified as a favorable clinical factor for NB. NTRK1 was the only overlapped gene when the 84 top-ranking upregulated genes were compared to previously reported “significantly higher expression in favorable NB” genes [27, 28] (Fig. 2b). Figure 2d–f show the confirmation of NTRK1 and several other upregulated genes in apoptosis-related/allograft rejection/integrated pancreatic cancer pathways by semi-quantitative and quantitative RT-PCR experiments. Previous studies indicated that there are the NTRK1 transcript variants 1/2/3 and alternative NTRK1 splice variant TrkAIII. TrkAIII has the deletion of exons 6, 7, and 9, and promoted tumorigenic NB cell behavior in vitro and in vivo (Supplementary Figure S3) [29]. Therefore, in order to detect the expression of NTRK1 in NB cells, we designed forward and reverse primers in exons 11 and 13, respectively (Supplementary Figure S3 and Supplementary Table S4). These primers amplify NTRK1 transcript variants 1–3 (Fig. 2e) and TrkAIII. The probe for NTRK1 transcripts in the Agilent Expression microarray was located on NTRK1 exons 12/13 (Supplementary Figure S3, A_24_P265506). We transduced NTRK1 cDNA (TrkAII, NM_001012331 with exons 6–7 and without exon 9) into NIH-3T3 cells as a control in the western blotting analysis using a monoclonal rabbit anti-NTRK1 antibody (clone 14G6) and detected ≈130- and 100-kDa signals. Since this antibody is produced by immunizing animals with a synthetic peptide surrounding Arg220 in exon 6, NTRK1 variants 1–3 were recognized. The depletion of EZH2 markedly increased these NTRK1 proteins (≈130 and 100 kDa, Fig. 2g) in NB cells, suggesting that NTRK1 variants 1–3 were induced by the depletion of EZH2 in NB cells (Fig. 2e, f). We examined NTRK1 de-repression in vivo using the xenograft tumors (Fig. 1e). The results of immunostaining for NTRK1 are shown along with the representative histology of NB-39-nu/shEZH2-1-treated tumors and NB-39-nu/shCont-treated tumors in Fig. 2h. Cells with an enlarged cytoplasm were observed in the EZH2 knockdown cells. NTRK1 expression was higher in the NB-39-nu/shEZH2-treated tumors than in the NB-39-nu/shCont-treated tumors.

EZH2 depletion induces NTRK1. NB-39-nu cells were infected with shEZH2-1 or shEZH2-2 lentiviruses as indicated in the Materials and Methods. Total RNAs were extracted 8 or 11 days after infection when neurite extension was evaluated. Results are representative of at least three independent experiments. a Summary for the transcriptome analysis in the volcano plot. We compared average gene expression levels between mock sample groups and EZH2 KD sample groups 8 and 11 days after infection. The threshold was set for p-values in the t-test (<0.05 after Benjamin–Hochberg corrections, y-axis) and fold changes (>2 in absolute fold changes, x-axis). b Genes upregulated by more than two-fold (p < 0.05) in EZH2 knocked down (by both shEZH2-1 and shEZH2-2) NB-39-nu cells were selected from samples on days 8 and 11 (84 genes in Supplementary Table S2, green dots in Fig. 2a positive fold changes). Venn diagram of the overlap among the upregulated genes in EZH2 KD samples, prognosis-related genes reported by Vermeulen et al. [27], and those reported by Ohira et al. [28]. NTRK1 was included in all data. c Results of a pathway analysis by GeneSpring GX 13.1 for genes upregulated by more than two-fold (p < 0.05) in EZH2 knocked down cells (by shEZH2-1 and shEZH2-2). Red indicates the pathway including NTRK1. d A heatmap generated from microarray data indicates the upregulation of some representative genes, which were involved in the top pathways in c. e The expression of genes indicated in d was confirmed by semi-quantitative RT-PCR. f qPCR analysis of NTRK1. g NTRK1 induction by EZH2 KD was confirmed by WB. h In the NB-39-nu/shEZH2-1-treated tumors (Fig. 1e), NTRK1-positive cells were distributed throughout the tumor tissue both as single cells and in clusters, while scattered NTRK1-positive cells were seen in the NB-39-nu/shCont-treated tumors
When we examined the effects of EZH2 KD on other MYCN-amplified NB cell lines, similar NB cell differentiation was observed; the depletion of EZH2 effectively induced neurite extension and NTRK1 de-repression along with the de-repression of the other differentiation-related molecules, GAP43 and RARB (Fig. 3a–d), suggesting that EZH2 is one of the key regulator molecules in NB cell differentiation and NTRK1 is a common EZH2 target in MYCN-amplified NB cell lines.

EZH2 depletion promotes differentiation and induces NTRK1 in NB cells. shEZH2-1 was introduced into NGP, SK-N-DZ, and TGW NB cells by a lentivirus system and total RNA collection/assessment of neurite extension were performed 5 (SK-N-DZ and TGW) or 8 (NGP) days after infection. The results are representative of at least three independent experiments. a NB cell differentiation-related gene expression was assessed by RT-PCR. b qPCR analysis of NTRK1. c, d Neurite extension was assessed by microscopy. The percentages of neurite-extending cells were counted. Data are presented as the mean ± s.d. from at least three independent experiments
EZH2 methylase inhibitors induced NB cell differentiation
In order to clarify whether EZH2 small-molecule inhibitors exert similar effects to EZH2 KD, we treated NB cells with two kinds of EZH2 inhibitors: EPZ6438 (Epizyme, Cambridge, MA, USA) [30] and GSK126 [21], which obstruct EZH2 H3K27 methylation activity without affecting the expression of EZH2 (Fig. 4a). The treatment with EZH2 inhibitors also induced the transcription of NTRK1 in NB cells lines (Fig. 4b, c, and Supplementary Figure S4) and we successfully detected ≈130- and 100-kDa NTRK1 proteins in EZH2 inhibitor-treated cells (Fig. 4d). We found that the effects of the treatments with EZH2 inhibitor EPZ6438 on the induction of neurite extension in NB cells were not as strong as those of EZH2 KD (Fig. 4e, f), suggesting that the role of EZH2 in NB cell de-differentiation was not fully dependent on its methylase activity. Importantly, NGFbeta treatment increased the effect of EPZ6438 on NB cell differentiation, suggesting the functional role of the induced NTRK1 by EZH2 inhibition (Fig. 4e–g).

Treatments with EZH2 inhibitors induce NTRK1 expression. NB-39-nu cells were treated with mock (0.1% (v/v) of DMSO), EPZ6438 (1 μM), or GSK126 (1 μM) for 4 days. Results are representative of at least three independent experiments. a Western blotting for H3K27me3 and EZH2 was performed using EPZ6438- or GSK126-treated cells. b, c NTRK1/GAPDH mRNA levels were assessed by semi-quantitative RT-PCR (b) and qPCR (c). d Western blots for NTRK1 in EZH2 inhibitor-treated NB-39-nu cells. e–g Photos (e) of and percentages (f) of neurite-extending NB-39-nu cells treated with EZH2 inhibitor EPZ6438 and/or NGF. Cells were treated with 1 μM of EPZ6438 for 2 days and cultured for 3 more days after the addition of NGFbeta (Sigma®,100 ng/ml). Neurite extension was assessed as indicated in Methods and GAP43 expression was studied by semi-quantitative RT-PCR (g)
NTRK1 contributes to EZH2-regulated NB cell differentiation
Since NTRK1 is known for its function in inducing neuronal cell differentiation and assuming that EZH2 represses some favorable prognostic factors to promote the progression of NB, we studied that NTRK1 plays an important role in the EZH2-dependent NB cell differentiation pathway (Fig. 5a, b). In order to establish whether NTRK1 contributes to EZH2-regulated NB cell differentiation, we used small interfering RNAs to suppress the expression of NTRK1 in EZH2 KD NB cells (Fig. 5a, b). The knockdown of NTRK1 significantly suppressed EZH2 KD-induced NB cell neurite extension (Fig. 5c). Furthermore, we analyzed the effects of NGF on the EZH2 KD NB cells, and found that neurite extension and GAP43 expression were increased in the EZH2 KD/NGF-treated NB cells (Fig. 5d). These results demonstrated that NTRK1 contributes to EZH2-regulated NB cell differentiation.

EZH2 KD-induced neurite extension is canceled by the depletion of NTRK1. a, b NB-39-nu cells were infected with a mock- or shEZH2-1-expressing lentivirus 2 and 3 days after NTRK1 siRNA transfection. EZH2 and NTRK1 expression was assessed by semi-quantitative RT-PCR (a) and qPCR (b). c Images of EZH2 and/or NTRK1 knockdown NB-39-nu cells. The percentages of neurite-extending cells were counted. Data are presented as the mean ± s.d. from at least three independent experiments. d Photos of and percentages of neurite-extending NB-39-nu cells treated with shEZH2 lentivirus and/or NGF. Cells were infected with shEZH2 lentivirus, cultured for 2 days, and cultured for 3 more days after the addition of NGFbeta (100 ng/ml). Neurite extension was assessed as described in Methods, and NTRK1 expression was analyzed by semi-quantitative RT-PCR
NTRK1 transcription is regulated by the PRC2 member EZH2 and/or DNA methylation
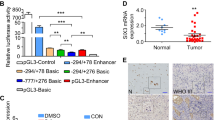
Although EZH2 is known for its function in gene silencing by its histone 3 lysine 27 (H3K27) tri-methylation activity, it currently remains unclear whether NTRK1 transcription is regulated by EZH2 through the H3K27 tri-methylation of the promoter regions. We reviewed the NTRK1 genome map and putative promoter regions. There were several alternative transcripts of NTRK1 (Supplementary Figure S3 and Fig. 6a); NTRK1 transcript variants 1, 2, and 3 were NM_001012331.1, NM_002529.3, and NM_001007792.1, respectively (Fig. 6a). The transcription of NTRK1 transcript variants 1 and 2 was regulated by the P1 promoter region (Fig. 6a). A combination of primer extension and RNase protection assays was used to identify the transcription-initiating region in NB cells upstream of the P1 promoter region. P1 promoter activity was confirmed by luciferase assays and the relevant cis-elements identified within the 1 kb proximal 5′-flanking region [31, 32].

Complex epigenetic regulation of NTRK1 promoters by EZH2 and/or DNA methylation. a The positions of NTRK1 variants were displayed in the UCSC genome browser (Human GRCh38/hg38, https://genome.ucsc.edu). ChIP qPCR site positions in the two promoters of NTRK1, Promoter 1 (P1) and Promoter 2 (P2). b qChIP assay for EZH2 in NTRK1 P1 and P2 promoters in NB-39-nu cells. Immunoprecipitation was performed by an anti-EZH2 antibody and control rabbit IgG. Primer locations are indicated in the diagrams in a. Primer sequences are shown in Supplementary Table S4. Results are presented as fold enrichment and are representative of at least three independent experiments. c qChIP assay for H3K27me3 in NTRK1 P1 and P2 promoters in mock- and EZH2 knocked down NB-39-nu cells by the shEZH2-1 lentivirus. Immunoprecipitation was performed by an anti-H3K27me3 antibody and control rabbit IgG. d qChIP assay for H3K27me3 and EZH2 in the NTRK1 P1 core promoter region in mock- and EPZ6438-treated NB-39-nu cells. Immunoprecipitation was performed with an anti-EZH2 antibody, an anti-H3K27me3 antibody, and control rabbit IgG. e RT-PCR for NTRK1 variants 1/2 and 3 in EZH2 knockdown NB cells. f Bisulfite-sequence analysis of NTRK1 P1 and P2 promoters. Red and gray asterisks indicate methylated and unmethylated CpG sites, respectively. CpG methylase-treated DNA was used as a positive control. g RT-PCR for NTRK1 variants 1/2 and 3 in 5 aza-dC (5 μM)-treated NB cells
Quantitative ChIP (qChIP) assay results showed that EZH2 specifically bound to the P1 promoter region of NTRK1, but not to the GAPDH control gene in NB-39-nu cells (Fig. 6b). The H3K27me3 mark at the NTRK1 P1 promoter region was significantly decreased by EZH2 KD (Fig. 6c). In contrast, EZH2 binding to the NTRK1 putative P2 promoter region was not clear and the H3K27me3 mark was not as obvious as that in the P1 promoter region (Fig. 6b,c). After EZH2 methylase inhibitor treatment, we examined H3K27me3 and EZH2 marks by qChIP analysis at the NTRK1 P1 core promoter region (Fig. 6d), and found that H3K27me3 marks were significantly reduced by the EZH2 methylase inhibitor. However, EZH2 marks were detected even after the inhibitor treatment, suggesting that methylase activity-inhibited EZH2 still binds to the NTRK1 promoter region and may be involved in the inhibition of NTRK1 transcription. Concordantly, EZH2 KD successfully induced NTRK1 transcript variants 1/2, but not 3 (Fig. 6e). These results strongly suggest that EZH2 directly regulates the transcription of NTRK1 by binding to its P1 promoter region in NB cells. In order to analyze the epigenetic status of the NTRK1 P1 and P2 promoter regions in more detail, we examined the DNA methylation status of the NTRK1 P1 and P2 promoter regions using the bisulfite sequencing method (Fig. 6f), indicating that the P1 promoter was fully de-methylated and the P2 promoter was completely methylated in NB-39-nu cells. In accordance with the genome methylation status, the 5-aza deoxycytidine treatment induced the NTRK1 variant 3 transcript, but not the variant 1 or 2 transcript (Fig. 6g).
Epigenetic regulation of NTRK1 in NB clinical samples
An integrative analysis was recently performed in order to investigate methylomes, transcriptomes, and copy number variations in 105 NB samples [33]. The genome-wide DNA methylation patterns of 105 NB samples were assessed using the Illumina 450k methylation array. We downloaded methylome data from the Gene Expression Omnibus (GEO) under accession ID GSE73518 [33] and compared them with those of 145 Japanese NB samples obtained by the same methylation array (Ohira M et al., manuscript in preparation). Strong DNA methylation was observed in the TSS1500 5′UTR region of NTRK1 promoter P2 (Fig. 7a, red), whereas the TSS1500 5′UTR region of the NTRK1 promoter P1 was not strongly methylated (Fig. 7a, blue). DNA methylation was assessed by the beta values of NB samples. High beta values were detected in the TSS1500 5′UTR region of NTRK1 promoter P2 in German and Japanese NBs (Fig. 7b), and higher beta values in promoter P2 were observed in MYCN-amplified NBs (Fig. 7b, NTRK1 variant 3 −1000, −500, and −300), suggesting MYCN-related DNA methylation in NTRK1 promoter P2. These results were consistent with those obtained from MYCN-amplified NB-39-nu cells (Fig. 6). We assessed NTRK1 expression levels in NB tumors with or without the amplification of MYCN. Microarray probes UKv4_A_23_P34804 and UKv4_A_24_P265506 were NTRK1 data obtained from the German Neuroblastoma Trial [33]. A_24_P265506 was data obtained from Japanese NB patients (Ohira M et al., manuscript in preparation). In accordance with previous findings [28, 34], the expression of NTRK1 was significantly higher in MYCN not-amplified NB samples than in MYCN-amplified samples (Fig. 7c). In order to confirm the results obtained in the NB cell line study (Fig. 6), we performed semi-quantitative RT-PCR for NTRK1 by using cDNAs obtained from the favorable and unfavorable subsets of NB [28]. We detected significantly high NTRK1 expression in favorable NB samples using primers for variants 1/2/3 (Fig. 7d). The high expression of NTRK1 was mainly observed for transcript variants 1/2, but not 3 (Fig. 7d).

Epigenetic regulation of NTRK1 P1 and P2 promoters in NB primary tumors. a A heatmap analysis of DNA methylation in clinical NB samples derived from Japan (JPN, n = 145, Ohira et al., manuscript in preparation) and Germany (GER, n = 105, [33]. Blue colors denote low levels of DNA methylation, red indicates high levels, and gray represents intermediate levels. Information on the probes is described in the Materials and Methods. b Comparison of DNA methylation levels on NTRK1 promoters in MYCN-amplified (MA) and not-amplified (MNA) samples. c Total NTRK1 expression in clinical NB samples derived from Japan (JPN) and Germany (GER). Gene expression levels of NTRK1 in NB patients with or without MYCN amplification were studied using Agilent expression microarrays. UKv4_A_23_P34804 (CTG GTA CCA GCT CTC CAA CAC GGA GGC AAT CGA CTG CAT CAC GCA GGG ACG TGA GTT GGA) and UKv4_A_24_A265506 (AGA ACC CAC AAT ACT TCA GTG ATG CCT GTG TTC ACC ACA TCA AGC GCCGG) were data from the German Neuroblastoma Trial [33]. A_24_P265506 was also studied in Japanese NB samples (probe sequence is same as the UKv4_A_24_A265506, Ohira et al., in preparation). The sample sizes of MYCN amplification and MYCN not-amplification NBs were shown. P-values were corrected by the Benjamin–Hochberg method. d Comparison of NTRK1 variant 1/2 and 3 expression levels in favorable NB (INSS stage I, and MYCN not-amplified) and unfavorable NB (INSS stage IV, age > 18 m, and MYCN not-amplified: samples 9–10; INSS stage IV, and MYCN amplified: samples 11–16) using RT-PCR
Discussion
A large number of genes were previously identified as targets by EZH2 enzymatic/non-enzymatic regulation in tumor biology [14]. In the present study, we performed a comprehensive analysis of EZH2-target genes and pathways using expression microarrays with NB-39-nu NB cells (Fig. 2). The MYCN gene was amplified and the PcGs EZH2/BMI1/RING1B were strongly expressed in NB-39-nu cells, suggesting its better suitability as a model for EZH2 depletion and the search for target genes (Supplementary Figure S1). We examined gene expression using Agilent expression microarrays by means of EZH2 depletion with two EZH2 shRNAs at several time points and found that 84 genes were reproducibly de-repressed (Fig. 2a, b, and Supplementary Table S2, genes with two-fold changes induced with p < 0.05 were selected). We compared de-repressed genes with prognosis-related genes in previous studies by Ohira et al. and Vermeulen et al. [27, 28]. The relationships among genes with significantly different expression profiles were visualized using a Venn diagram that NTRK1 is a candidate of the EZH2-targeted gene, which plays important roles in a favorable NB prognosis (Fig. 2b). The NTRK1 tyrosine kinase receptor was identified in 1991 and subsequently found to encode a high-affinity receptor for nerve growth factor (NGF) [35]. In NB, NTRK1 was expressed in favorable NB, and the amplification of MYCN strongly suppressed its expression [34, 36, 37]. The high expression of NTRK1 was closely associated with favorable risk factors and outcomes in a large, representative population of NB patients, including the MYCN status [38]. However, the exact mechanisms underlying the suppression of NTRK1 in MYCN-amplified NB cells has not yet been elucidated in detail. Although human NTRK1 maps to 1q21, no mutations or activating rearrangements have been identified in NB [3, 39, 40]. Several studies have been performed on the NTRK1 promoter. Chang et al. characterized the human TrkA (NTRK1) promoter in NB cells using transient transfection. They cloned NTRK1 cDNA (transcript variants 1/2) and the 5′ upstream genomic region (the promoter P1-containing region, Fig. 6). Cell-specific and variable transcription in NB cells was recapitulated by the transient transfection of NTRK1 P1 promoter-luciferase reporter constructs and the regulatory sequences mediating these processes were identified in a 138-basepair region upstream of the transcription initiation region of transcript variants 1/2 [31]. Iraci et al. investigated SP1/MIZ1/MYCN Repression Complex-related NTRK1 suppression in NB cells. The MYCN complex recruited HDAC1 to the NTRK1 P1 promoter region and suppressed the transcription of NTRK1 [41]. A recent study reported that MYCN drives neuroendocrine prostate cancer (NEPC) phenotypes and interacts with EZH2 and PRC2 molecules, resulting in an increase in H3K27me3 marks and the transcriptional suppression of androgen receptor-induced genes and PRC2/EZH2 target genes [42]. In an R2 database analysis, a significant correlation was observed between the expression of MYCN and EZH2 (Kocak-649-custom data set: r-value = 0.365 p-value = 7.8e−22; SEQC-498-RPM data set: r-value = 0.296 p-value = 1.5e−11). Our qPCR and western blot analyses also revealed the higher expression of EZH2 in MYCN-amplified NB cell lines than in the MYCN not-amplified SK-N-SH cells (Supplementary Figure S1).
The CpG island promoter methylation of NTRK1 (TRKA) was previously observed in the Promoter P2 region of NB samples by a customized Illumina GoldenGate methylation assay [43]. Furthermore, the genome-wide DNA methylation patterns of 105 NBs were assessed using the Illumina 450k methylation array and revealed that NTRK1 is one of the genes downregulated in high-risk patients with concomitant CpG hypermethylation (the P2 promoter region [33]). Our analysis of 145 NB tumor samples using the Infinium HumanMethylation450 BeadChip Kit also detected the methylation of the NTRK1 P2 promoter region, but not genome methylation in the NTRK1 P1 promoter region (Fig. 7). Our NB cell line-based experiments (Fig. 6) and NB clinical sample analysis (Fig. 7) clarified that NTRK1 transcript variants 1/2 are the NTRK1 transcripts expressed in favorable NBs and suppressed in MYCN-amplified unfavorable NBs. The transcription of variants 1/2 is regulated by the NTRK1 P1 promoter region, and the P1 promoter region was epigenetically regulated by EZH2-related histone modifications.
This study is the first to show the epigenetic regulation of NTRK1 expression via EZH2 in NB cells. Further analyses of EZH2-related gene repression in MYCN-amplified NB cells will be important for the development of effective therapies for unfavorable NB patients. NTRK1 transcription may be regulated epigenetically in NB cells by the combination of PRC molecules and genome DNA methylation. The development of EZH2 small-molecule inhibitors may be promising for NB differentiation induction therapy, e.g., in combination with DNA methylase inhibitors. Bate-Eya et al. recently reported that the depletion of EZH2 induced apoptosis in several NB cell lines [30]. Although the molecules responsible were not identified in that study, we detected FAS as an EZH2-depletion-induced molecule (Fig. 2). The study of apoptosis induction by the inhibition of EZH2 in combination with another inhibitor [33, 44] will be important for the development of new therapies for unfavorable NB patients.
Materials and methods
Ethics statements
The study was approved by the Ethics Committee of the Saitama Cancer Center (approved number 473), and written informed consent was obtained from all patients. All animals were maintained and used for experiments in accordance with the guidelines of the Institutional Animal Experiments Committee of the Saitama Cancer Center.
Cell culture and reagents
Human NB cell lines were obtained from official cell banks (American Type Culture Collection, Manassas, VA, USA and the RIKEN Bioresource Cell Bank, Tohoku University Cell Resource Center, Miyagi, Japan). The cells were cultured in RPMI1640 (Wako, Osaka, Japan) supplemented with 10% heat-inactivated FBS and 50 μg/ml penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA). Neurite-elongation of NB cells were scored for the presence of neurites longer than one cell in diameter. EPZ6438 and GSK126 were purchased from Funakoshi (Tokyo, Japan).
Western blotting analysis
Cells were lysed in EBC buffer [9] or CHAPS buffer (10 mM CHAPS, 50 mM pH 7.5 Tris-HCL, 100 mM NaCl, 10 mM MgCl2, and 1 mM EDTA) for the immunoblotting of NTRK1, and protease/phosphatase inhibitors were added to the buffers [9]. Western blotting experiments were performed as previously reported [45]. After being transferred to an Immobilon-P membrane (Millipore, Billerica, MA, USA), proteins were reacted with anti-EZH2 rabbit polyclonal antibody (Millipore 07–689), anti-BMI1 mouse monoclonal antibody (Millipore 229F6), anti-RING1B antibody [46], anti-trimethyl-Histone H3 rabbit polyclonal antibody (Millipore 07–449), histone H3 rabbit polyclonal antibody (Abcam ab1791, Cambridge, United Kingdom), anti-TrkA (NTRK1) rabbit monoclonal antibody (Cell Signaling Technology 14G6, Beverly, MA, USA), or anti-β-actin antibody (Sigma-Aldrich).
Lentiviral gene transduction
shRNA plasmids, including TRCN0000018365 (shEZH2-1), TRCN0000010475 (shEZH2-2), and the pLKO.1-puro control vector (shCont), were from the MISSION(TM) shRNA Library (Sigma-Aldrich). The method of shRNA/lentivirus preparation was described as previously [9]. Thereafter, 1 × 105 NB cells were seeded on each well of a six-well plate, and infected by lentiviral-conditioned media with 4 μg/ml polybrene (Sigma-Aldrich).
Semi-quantitative RT-PCR
A semi-quantitative RT-PCR analysis was carried out as described previously [45]. Total cellular RNA extraction was performed using ISOGEN II (Nippon Gene KK, Toyama, Japan). cDNA was synthesized from 2 μg total RNA. Primer sequences are described in Supplementary Table S4. RT-PCR results are representative of at least three independent experiments. Band intensities were quantified with ImageJ software (https://imagej.nih.gov/ij/index.html). Normalization was performed using the band intensity of the internal control GAPDH.
qPCR analysis
A qPCR analysis was performed as described previously [9]. The primers used for qPCR were designed by Primer Express® Software v3.0.1 (Thermo Fisher, Waltham, MA, USA). The primer sequence is indicated in Supplementary Table S4. Results are representative of at least three independent experiments.
Tumor formation in nude mice
For tumor formation, 5-week-old female athymic BALB/c AJcl nu/nu mice (CLEA Japan, Shizuoka, Japan) were injected with 1 × 106 NB-39-nu cells into the femur as described previously [9]. Tumor volume was evaluated every 4 days.
Pathological analysis
Pathological analysis was performed on formalin-fixed, paraffin-embedded tumor tissue sections. Immunostaining for NTRK1 was performed using rabbit monoclonal anti-NTRK1 antibody (EP1058Y; Abcam, 1:500 dilution) in Bond-Max autostainer (Leica Biosystems, Wetzler, Germany). Antigen retrieval was performed using Bond Epitope Retrieval Solution 2 (Leica Biosystems) at 100 °C for 10 min.
Transcriptome and pathway analyses
RNA was extracted at two time points, 8 and 11 days after shEZH2-expressing lentiviral infection, and a microarray analysis was performed using the Agilent platform of 8 × 60 K design ID G4851B (Agilent Single Color. 39494, Agilent Technologies, Santa Clara, CA, USA). Two hundred nanograms of total RNA was labeled with Cyanine3 using a Low Input Quick-Amp Labeling Kit (one color, Agilent Technologies) according to the manufacturer’s instructions. Differentially expressed genes (DEG) were defined by p-values in the moderated t-test of <0.05 after Benjamini–Hochberg corrections and fold changes of >2 or <0.5. Subsequent gene annotation was conducted by GeneSpringGX.
NTRK1 knockdown by siRNA
The small interfering RNAs targeting NTRK1, S9746 (siNTRK1-1), and S9747 (siNTRK1-2) were from Thermo Fischer. For transfection, Lipofectamine RNAiMAX Transfection Reagent (Invitrogen) was used according to the manufacturer’s protocol. Reverse transfection was performed when plating 5 × 104 NB-39-nu cells on six-well plates and, after a 24-h culture, forward transfection was performed. A total of 5 pmol siRNA was used for each transfection.
Quantitative ChIP assay
Quantitative ChIP assays were performed as described previously [47, 48]. Cross-linked chromatin prepared from the indicated cells was precipitated by normal rabbit IgG (Wako 148–09551), an anti-EZH2 rabbit polyclonal antibody (Millipore 07–689), or anti-trimethyl-Histone H3K27 rabbit polyclonal antibody (Millipore 07–449) with Protein G Sepharose 4 Fast Flow (GE Healthcare, Chicago, IL, USA). The primers used in the present study are indicated in Supplementary Table S4. Each series of experiments was conducted at least three times.
Bisulfite treatment and bisulfite sequencing
The bisulfate modification of genomic DNA was performed as previously described [49]. Primer sets for bisulfite sequencing in the promoter regions of the NTRK1 variants 1/2 and 3 were as follows: variants 1/2, 5′-ATTAGTTTTTTGTTGTTTTGGGTGT-3′ and 5′- TCAAATCTCAACTTCAATCCCTAAC-3′, and variant 3, 5′- GTAGTTTTTTTGTTTTTTTGTGGGT-3′ and 5′- CTAAATAAAACCCCTAACCCCTATC-3′.
DNA methylation assays
A DNA methylation analysis was performed using the Infinium HumanMethylation450 BeadChip Kit (Illumina, San Diego, CA, USA). Probes for the NTRK1 P1 region (−1000, −500, and −300) and P2 region (−1000, −500, and −300) were (cg25630380, cg10811945, and cg00626119) and (cg03438943, cg10981464, and cg26428727), respectively. We will submit the whole Infinium data of Japanese 145 NBs (Ohira et al., manuscript in preparation). β values in the NTRK1 region will be provided upon request.
Availability of data and materials
The data sets supporting the conclusions of this article are available from the National Center for Biotechnology Information’s Gene Expression Omnibus (GEO) and are accessible through GEO accession numbers GSE98642.
References
Westermann F, Schwab M. Genetic parameters of neuroblastomas. Cancer Lett. 2002;184:127–47.
Peifer M, Hertwig F, Roels F, Dreidax D, Gartlgruber M, Menon R, et al. Telomerase activation by genomic rearrangements in high-risk neuroblastoma. Nature. 2015;526:700–4.
Pugh TJ, Morozova O, Attiyeh EF, Asgharzadeh S, Wei JS, Auclair D, et al. The genetic landscape of high-risk neuroblastoma. Nat Genet. 2013;45:279–84.
Kamijo T, Nakagawara A. Molecular and genetic bases of neuroblastoma. Int J Clin Oncol. 2002;17:190–5.
Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, et al. Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nat Med. 2000;6:529–35.
Haruta M, Kamijo T, Nakagawara A, Kaneko Y. RASSF1A methylation may have two biological roles in neuroblastoma tumorigenesis depending on the ploidy status and age of patients. Cancer Lett. 2014;348:167–76.
Abe M, Ohira M, Kaneda A, Yagi Y, Yamamoto S, Kitano Y, et al. CpG island methylator phenotype is a strong determinant of poor prognosis in neuroblastomas. Cancer Res. 2005;65:828–34.
Abe M, Westermann F, Nakagawara A, Takato T, Schwab M, Ushijima T. Marked and independent prognostic significance of the CpG island methylator phenotype in neuroblastomas. Cancer Lett. 2007;247:253–8.
Ochiai H, Takenobu H, Nakagawa A, Yamaguchi Y, Kimura M, Ohira M, et al. Bmi1 is a MYCN target gene that regulates tumorigenesis through repression of KIF1Bbeta and TSLC1 in neuroblastoma. Oncogene. 2010;29:2681–90.
Huang R, Cheung NK, Vider J, Cheung IY, Gerald WL, Tickoo SK, et al. MYCN and MYC regulate tumor proliferation and tumorigenesis directly through BMI1 in human neuroblastomas. FASEB J. 2011;25:4138–49.
Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 2010;7:299–313.
Chase A, Cross NC. Aberrations of EZH2 in cancer. Clin Cancer Res. 2011;17:2613–8.
Martinez-Garcia E, Licht JD. Deregulation of H3K27 methylation in cancer. Nat Genet. 2010;42:100–1. https://doi.org/10.1038/ng0210-100. PMID:20104248.
Kim KH, Roberts CW. Targeting EZH2 in cancer. Nat Med. 2016;22:128–34. https://doi.org/10.1038/nm.4036. PMID:26845405.
Wang C, Liu Z, Woo CW, Li Z, Wang L, Wei JS, et al. EZH2 mediates epigenetic silencing of neuroblastoma suppressor genes CASZ1, CLU, RUNX3, and NGFR. Cancer Res. 2012;72:315–24.
Lee ST, Li Z, Wu Z, Aau M, Guan P, Karuturi RK, et al. Context-specific regulation of NF-κB target gene expression by EZH2 in breast cancers. Mol Cell. 2011;43:798–810.
Xu K, Wu ZJ, Groner AC, He HH, Cai C, Lis RT, et al. EZH2 oncogenic activity in castration-resistant prostate cancer cells is Polycomb-independent. Science. 2012;338:1465–9.
Kim E, Kim M, Woo DH, Shin Y, Shin J, Chang N, et al. Phosphorylation of EZH2 activates STAT3 signaling via STAT3 methylation and promotes tumorigenicity of glioblastoma stem-like cells. Cancer Cell. 2013;23:839–52.
He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev. 2012;26:37–42.
Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007;21:1050–63.
McCabe MT, Ott HM, Ganji G, Korenchuk S, Thompson C, Van Aller GS, et al. EZH2 inhibition as a therapeutic strategy for lymphoma with EZH2-activating mutations. Nature. 2012;492:108–12.
Knutson SK, Wigle TJ, Warholic NM, Sneeringer CJ, Allain CJ, Klaus CR, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8:890–6.
Knutson SK, Kawano S, Minoshima Y, Warholic NM, Huang KC, Xiao Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13:842–54.
Niizuma H, Nakamura Y, Ozaki T, Nakanishi H, Ohira M, Isogai E, et al. Bcl-2 is a key regulator for the retinoic acid-induced apoptotic cell death in neuroblastoma. Oncogene. 2016;25:5046–55.
Satoh S, Takatori A, Ogura A, Kohashi K, Souzaki R, Kinoshita Y, et al. Neuronal leucine-rich repeat 1 negatively regulates anaplastic lymphoma kinase in neuroblastoma. Sci Rep. 2016;6:32682.
Kutmon M, Riutta A, Nunes N, Hanspers K, Willighagen EL, Bohler A, et al. WikiPathways: capturing the full diversity of pathway knowledge. Nucleic Acids Res. 2016;44:D488–94.
Vermeulen J, De Preter K, Naranjo A, Vercruysse L, Van Roy N, Hellemans J, et al. Predicting outcomes for children with neuroblastoma using a multigene-expression signature: a retrospective SIOPEN/COG/GPOH study. Lancet Oncol. 2009;10:663–71.
Ohira M, Oba S, Nakamura Y, Isogai E, Kaneko S, Nakagawa A, et al. Expression profiling using a tumor-specific cDNA microarray predicts the prognosis of intermediate risk neuroblastomas. Cancer Cell. 2005;7:337–50.
Tacconelli A, Farina AR, Cappabianca L, Desantis G, Tessitore A, Vetuschi A, et al. TrkA alternative splicing: a regulated tumor-promoting switch in human neuroblastoma. Cancer Cell. 2004;6:347–60.
Bate-Eya LT, Gierman HJ, Ebus ME, Koster J, Caron HN, Versteeg R, et al. Enhancer of zeste homologue 2 plays an important role in neuroblastoma cell survival independent of its histone methyltransferase activity. Eur J Cancer. 2017;75:63–72.
Chang BB, Persengiev SP, de Diego JG, Sacristan MP, Martin-Zanca D, Kilpatrick DL. Proximal promoter sequences mediate cell-specific and elevated expression of the favorable prognosis marker TrkA in human neuroblastoma cells. J Biol Chem. 1998;273:39–44.
Indo Y, Mardy S, Tsuruta M, Karim MA, Matsuda I. Structure and organization of the human TRKA gene encoding a high affinity receptor for nerve growth factor. Jpn J Hum Genet. 1997;42:343–51.
Henrich KO, Bender S, Saadati M, Dreidax D, Gartlgruber M, Shao C, et al. Integrative genome-scale analysis identifies epigenetic mechanisms of transcriptional deregulation in unfavorable neuroblastomas. Cancer Res. 2016;76:5523–37.
Nakagawara A, Arima-Nakagawara M, Scavarda NJ, Azar CG, Cantor AB, Brodeur GM. Association between high levels of expression of the TRK gene and favorable outcome in human neuroblastoma. N Engl J Med. 1993;328:847–54.
Kaplan DR, Martin-Zanca D, Parada LF. Tyrosine phosphorylation and tyrosine kinase activity of the trk proto-oncogene product induced by NGF. Nature. 1991;350:158–60.
Nakagawara A, Arima M, Azar CG, Scavarda NJ, Brodeur GM. Inverse relationship between trk expression and N-myc amplification in human neuroblastomas. Cancer Res. 1992;52:1364–8.
Kogner P, Barbany G, Dominici C, Castello MA, Raschellá G, Persson H. Coexpression of messenger RNA for TRK protooncogene and low affinity nerve growth factor receptor in neuroblastoma with favorable prognosis. Cancer Res. 1993;53:2044–50.
de Souza DR, Sanabani SS, de Souza AC, Filho Odone V, Epelman S, Bendit I. Prognostic impact of MYCN, DDX1, TrkA, and TrkC gene transcripts expression in neuroblastoma. Pediatr Blood Cancer. 2011;56:749–56.
Scaruffi P, Cusano R, Dagnino M, Tonini GP. Detection of DNA polymorphisms and point mutations of high-affinity nerve growth factor receptor (TrkA) in human neuroblastoma. Int J Oncol. 1999;14:935–8.
Schramm A, Köster J, Assenov Y, Althoff K, Peifer M, Mahlow E, et al. Mutational dynamics between primary and relapse neuroblastomas. Nat Genet. 2015;47:872–7.
Iraci N, Diolaiti D, Papa A, Porro A, Valli E, Gherardi S, et al. A SP1/MIZ1/MYCN repression complex recruits HDAC1 at the TRKA and p75NTR promoters and affects neuroblastoma malignancy by inhibiting the cell response to NGF. Cancer Res. 2011;71:404–12.
Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, et al. N-Myc induces an EZH2-mediated transcriptional program driving neuroendocrine prostate cancer. Cancer Cell. 2016;30:563–77.
Lau DT, Hesson LB, Norris MD, Marshall GM, Haber M, Ashton LJ. Prognostic significance of promoter DNA methylation in patients with childhood neuroblastoma. Clin Cancer Res. 2012;18:5690–700.
Frumm SM, Fan ZP, Ross KN, Duvall JR, Gupta S, VerPlank L, et al. Selective HDAC1/HDAC2 inhibitors induce neuroblastoma differentiation. Chem Biol. 2013;20:713–25.
Kurata K, Yanagisawa R, Ohira M, Kitagawa M, Nakagawara A, Kamijo T. Stress via p53 pathway causes apoptosis by mitochondrial Noxa upregulation in doxorubicin-treated neuroblastoma cells. Oncogene. 2008;27:741–54.
Atsuta T, Fujimura S, Moriya H, Vidal M, Akasaka T, Koseki H. Production of monoclonal antibodies against mammalian Ring1B proteins. Hybridoma. 2001;20:43–6.
Orlando V, Strutt H, Paro R. Analysis of chromatin structure by in vivo formaldehyde cross-linking. Methods. 1997;11:205–14.
Fujimura Y, Isono K, Vidal M, Endoh M, Kajita H, Mizutani-Koseki Y, et al. Distinct roles of Polycomb group gene products in transcriptionally repressed and active domains of Hoxb8. Development. 2006;133:2371–81.
Sugawara W, Haruta M, Sasaki F, Watanabe N, Tsunematsu Y, Kikuta A, et al. Promoter hypermethylation of the RASSF1A gene predicts the poor outcome of patients with hepatoblastoma. Pediatr Blood Cancer. 2007;49:240–9.
Acknowledgements
We thank Ms. K Sakurai and Ms. N Kawanabe for their technical assistance and Mr. Daniel Mrozek (Medical English Service, Kyoto) for his English editorial assistance. This work was supported in part by Practical Research for Innovative Cancer Control (No. 16ck0106011j0003; No. 17ck0106332h0001) and the Tailor-made Medical Treatment Program (No. 15km0305013h0101) from the Japan Agency for Medical Research and Development, AMED, and JSPS KAKENHI Grant-in-Aid for Challenging Exploratory Research (17K19623).
Author contributions
Conceptualization: TK. Data curation: RPS, HA, and MO. Formal analysis: ZL, HT, ANS, SS, NA, MH, YS, KC, KM, AN1, JA, MO, and TK. Funding acquisition: AN2, MO, HA, and TK. Project administration: TK. Resources: AN2 and TK. Software: RS. Analyzed data: ZL, HT, RS, MO, and TK. Supervision: AN2 and TK. Validation: ZL, HT, ANS, NA, JA, MO, and TK. Visualization: ZL, HT, NA, MH, YS, KC, KM, MO, and TK. Writing—original draft: ZL, HT, AN1, RPS, MO, and TK. Writing—review and editing: TK. AN1: Atsuko Nakazawa, AN2: Akira Nakagawara.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. If you remix, transform, or build upon this article or a part thereof, you must distribute your contributions under the same license as the original. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/.
About this article

Cite this article
Li, Z., Takenobu, H., Setyawati, A.N. et al. EZH2 regulates neuroblastoma cell differentiation via NTRK1 promoter epigenetic modifications. Oncogene 37, 2714–2727 (2018). https://doi.org/10.1038/s41388-018-0133-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-018-0133-3
- Springer Nature Limited
This article is cited by
-
The transcriptional co-repressor Runx1t1 is essential for MYCN-driven neuroblastoma tumorigenesis
Nature Communications (2024)
-
NTRK1 knockdown induces mouse cognitive impairment and hippocampal neuronal damage through mitophagy suppression via inactivating the AMPK/ULK1/FUNDC1 pathway
Cell Death Discovery (2023)
-
Expression and activation of nuclear hormone receptors result in neuronal differentiation and favorable prognosis in neuroblastoma
Journal of Experimental & Clinical Cancer Research (2022)
-
Genome-wide analysis identifies critical DNA methylations within NTRKs genes in colorectal cancer
Journal of Translational Medicine (2021)
-
KDM6B promotes activation of the oncogenic CDK4/6-pRB-E2F pathway by maintaining enhancer activity in MYCN-amplified neuroblastoma
Nature Communications (2021)