
Abstract
Interest in the cerebellum is expanding given evidence of its contributions to cognition and emotion, and dysfunction in various psychopathologies. However, research into its genetic architecture and shared influences with liability for mental disorders is lacking. We conducted a genome-wide association study (GWAS) of total cerebellar volume and underlying cerebellar lobe volumes in 33,265 UK-Biobank participants. Total cerebellar volume was heritable (h2SNP = 50.6%), showing moderate genetic homogeneity across lobes (h2SNP from 35.4% to 57.1%; mean genetic correlation between lobes rg ≈ 0.44). We identified 33 GWAS signals associated with total cerebellar volume, of which 6 are known to alter protein-coding gene structure, while a further five mapped to genomic regions known to alter cerebellar tissue gene expression. Use of summary data-based Mendelian randomisation further prioritised genes whose change in expression appears to mediate the SNP-trait association. In total, we highlight 21 unique genes of greatest interest for follow-up analyses. Using LD-regression, we report significant genetic correlations between total cerebellar volume and brainstem, pallidum and thalamus volumes. While the same approach did not result in significant correlations with psychiatric phenotypes, we report enrichment of schizophrenia, bipolar disorder and autism spectrum disorder associated signals within total cerebellar GWAS results via conditional and conjunctional-FDR analysis. Via these methods and GWAS catalogue, we identify which of our cerebellar genomic regions also associate with psychiatric traits. Our results provide important insights into the common allele architecture of cerebellar volume and its overlap with other brain volumes and psychiatric phenotypes.
Similar content being viewed by others


Introduction
The cerebellum has historically been ascribed solely to a role in movement coordination, however, increasing evidence has underlined its relevance in cognition and emotion [1], with expansive functional connectivity with non-motor cortical regions [2,3,4] and activity during a wide-range of cognitive tasks [5]. Lesions during cerebellar development not only lead to motor alterations but also to cognitive and emotional deficits [6], and represent the second highest risk factor for autism spectrum disorder (ASD) [7]. Cerebellar anatomical alterations have also been identified in most other neurodevelopmental/psychiatric disorders, with particularly strong cerebellar-specific evidence in schizophrenia [8], but also in attention deficit hyperactivity disorder (ADHD) [9] and mood disorders [10], general liability to clinical mental disorders [11] and adolescent psychopathology [12].
Cerebellar volume reductions have also been reported in unaffected relatives of people with schizophrenia, bipolar disorder and depression, being also the only structure commonly reduced across all three disorders [13] and suggesting cerebellar volume reductions to be associated with genetic risk for mental disorders. Indeed, analysing shared altered genetic expression across these three disorders as well as ADHD and ASD shows strong cerebellar tissue enrichment [14].
Twin studies show cerebellar volume to be heritable (h2 = 33.6–86.4%) [15], but little is known about its polymorphic architecture. In this study, we aim to undertake an in-depth investigation of the common variant influences of total cerebellar volume, their association with altered cerebellar gene expression, genetic overlap with other cortical and subcortical anatomical phenotypes, and importantly, with several of these psychiatric disorders shown to be associated with cerebellar anatomical abnormalities (i.e. ASD, ADHD, schizophrenia, bipolar disorder and depression). While a recent omnibus-GWAS study [16] using an overlapping sample to ours, included 30 cerebellar volume metrics, none of these corresponded to total cerebellar volume and did not explore genetic association with mental disorders.
Methods
Total cerebellar volume measure generation
This study utilises T1-weghted structural brain magnetic resonance imaging (MRI) image derived phenotypes (IDPs) data for ~40,000 individuals from UK Biobank (http://www.ukbiobank.ac.uk/) (Supplementary Methods). The generation and semi-automated quality control of these IDPs by UK Biobank has been described previously [17]. Our research group accessed the data in two batches, each containing approximately half of the total sample (henceforth wave 1 and wave 2), which we analysed separately before being meta-analysed.
We generated a summated total cerebellar grey-matter volume measure from the 28 cerebellar lobule IDPs [18], aside from Crus I vermis due to its small size [19]. To explore the reliability of UK Biobank’s cerebellar volume measures, for the 1273 participants in our study who have been scanned twice by UK Biobank within a 5-year interval (with these second scans not included in our main analyses), between-scan intraclass correlation indicated a high test-retest reliability of our cerebellar volume metric (ICC = 0.92). Following outlier removal, we obtained residual total cerebellar volume values after correction for covariates of age, sex, head motion, date of scan and imaging centre attended, and head and table position in the scanner (see Supplementary Methods for details). We scaled these values, with beta values reflecting differences in standard deviations (SD) of residual cerebellar volume.
Genotyping and quality control
A description of UK-Biobank’s genetic-data collection, quality control and imputation processes can be found elsewhere (http://www.ukbiobank.ac.uk/scientists-3/genetic-data/). We applied additional quality controls independently to each wave’s genotypes (Supplementary Methods), including restriction to unrelated individuals of British/Irish ancestry (>96% sample) (Supplementary Fig. 1). Following local processing, from initial samples of 21,390 and 26,541 participants with genetic-data in wave 1 and wave 2,19,170 and 22,808 participants remained, with 7,003,604 and 6,935,580 genetic markers, respectively.
Genome-wide association study (GWAS)
After merging genetic and cerebellar volume data, we conducted two separated GWASs using PLINK (v1.9) [20] including 17,818 participants in wave 1 (age mean[min,max]= 63[45,80]yrs, 53% female) and 15,447 participants in wave 2 (age mean [min, max] = 65 [48,81] yrs, 53% female) (Supplementary Table 1). The first ten genetic PCs were inputted in these analyses to account for any potential remaining population structure.
SNP-based heritability (h2 SNP)
Lower-bound estimates of narrow-sense single nucleotide polymorphism (SNP)-based heritability (h2SNP) for each wave were calculated using GCTA-GREML (Genome-wide complex trait analysis—genome-based restricted maximum likelihood) (64 bit; v1.26.0) [21, 22] on the raw genotypes and including covariates of the first 10 genetic principal components.
Identification of independent GWAS signals
Regional GWAS signals were refined using GCTA-COJO [21, 23] to identify independent index/lead SNPs (Supplementary Methods). Extended-LD regions are provided (r2 > 0.2 with index SNP and p < 0.05 association). LocusZoom [24] was used to visually inspect these signal peaks (Supplementary Fig. 2).
Comparison of GWASs from wave 1 and wave 2
Several methodologies were deployed to assess similarity between summary statistics from both waves, including between-wave SNP replication, LDSC [25] genetic correlation and PLINK [20] polygenic score analyses (Supplementary Methods).
Meta-analysis
We meta-analysed the two waves’ GWASs using METAL (2011-03-25 release) [26], weighting effect sizes by the inverse of the standard errors and retaining only the 6,193,476 markers present in both waves. Independent index SNP identification and SNP-based heritability estimates were calculated using the same methods as outlined above, creating a merged wave dataset for SNPs’ LD structure and estimates of h2SNP (GCTA-GREML).
Within cerebellum analysis—by lobe analysis
To investigate the homogeneity of cerebellar volume genetic architecture, we ascertained the GCTA-GREML h2SNP and LDSC between-lobe genetic correlation estimates for 7 cerebellar lobes based on demarcations of primary, horizontal and posterolateral fissures: anterior (I-V), superior posterior (VI-Crus I), inferior posterior (Crus II-IX) and flocculonodular (X) hemispheres and vermal regions of the latter three (Supplementary Methods).
Functional annotation and cerebellar gene expression
We physically mapped the extended-LD regions of each index SNP (r2 > 0.2 to Index SNP) to nearby transcripts and functionally annotated index SNPs and high LD proxy SNPs (r2 > 0.8 to index SNP) for SNP consequences using several methods (Supplementary Methods). We additionally mapped these proxy SNPs to GTEx-v7 expression quantitative trait loci (cis-eQTL), focusing on directly relevant cerebellar-labelled tissues, but also including analyses in other brain and whole-blood tissues. Use of Summary data-based Mendelian Randomisation (SMR) [27, 28] allowed for assessing mediation via altered cerebellar gene expression of our meta-GWAS identified SNP-cerebellar volume associations, and separation of pleiotropic associations from those caused by linkage within the genomic region (Supplementary Methods).
Genetic correlation analysis
We used LDSC to estimate genetic correlations between our total cerebellar volume meta-GWAS summary statistics and previously published GWAS summary statistics from two studies that included different sub-regional cerebellar measures [16, 29], cortical and subcortical anatomical measures [30,31,32], anthropomorphic traits (http://www.nealelab.is/uk-biobank/), and psychiatric disorders of schizophrenia, bipolar, major depression and ASD and ADHD [33,34,35,36,37] (Supplementary Methods). We additionally ascertained genetic overlap between cerebellar volume and these psychiatric disorders, irrespective of direction of effect, using conditional and conjunctional false discovery rate (FDR) analysis [38] (see Supplementary Methods). This included analysis of genetic enrichment in our cerebellar GWAS using stratified quantile–quantile (Q–Q) plots, and investigation of which of our COJO-identified GWAS signals contained SNPs showing evidence for a pleiotropic association with a psychiatric phenotype (conjunctional-FDR < 0.01). Finally, for our COJO proxy SNPs, we inspected GWAS catalogue for previous reports of associations with these psychiatric traits, as well as for any additional traits (Supplementary Methods).
Results
Between-wave results’ reliability and validity
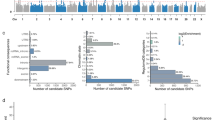
The GWASs of cerebellar volume identified 6 independent genome-wide significant index SNPs in each wave (Fig. 1; Supplementary Table 2A, B). Each showed high replication in the alternate wave, with all six wave 2 SNPs replicated in wave 1 (p < 0.0083{0.05/6}) and all but one wave 1 index SNPs replicated in wave 2. Four were genome-wide significant in both waves. SNP-based heritability estimates were similar across waves (wave 1 h2SNP[standard error (SE)] = 46.8 [3.4]% and wave 2 h2SNP [SE] = 45.3 [3.9]%; lambda GC 1.12 [intercept 1.01] and 1.10 [intercept 1.01], respectively), with a very strong between-wave genetic correlation (rg [SE] = 1.0 [0.1], p = 2.2 × 10−33). All polygenic scores derived from one wave significantly predicted total cerebellar volume in the opposing wave, with the most variance explained by wave 1 GWAS derived polygenic scores being at a SNP inclusion p-threshold (pT) <0.01 (19,210 SNPs, ΔR2 = 1.9%, p = 5.3 × 10−118) and at pT < 0.1 for wave 2 GWAS derived polygenic scores (146,489 SNPs, ΔR2 = 1.3%, p = 3.9 × 10−100) (Supplementary Table 3).

Manhattan plots of associations with total cerebellar volume for (A) wave 1 data release (n = 17,818), (B) wave 2 data release (n = 15,447), and (C) wave 1 + wave 2 combined METAL meta-analysis.For the METAL plot, the 33 COJO-identified independent index SNPs are highlighted (diamond shape). In all cases, the dashed line indicates genome-wide significance at p < 5 × 10−8. Quantile–quantile (Q–Q) plots for each GWAS are provided next to the Manhattan plot. For all plots, points p > 5 × 10−3 (solid line) are removed for ease of interpretation.
Meta-analysis of GWAS results for wave 1 and wave 2
Given the high correlation between waves, we combined both waves’ summary statistics in a meta-GWAS (n = 33,265, SNPs = 6,193,476) (Fig. 1). The SNP-based heritability estimate in the combined sample was h2SNP [SE] = 50.6[2.0]% (lambda GC 1.18 [intercept 1.02]).
Cerebellar lobe analysis
SNP-based heritability estimates across individual lobes were similar to the overall cerebellar heritability, except for the lower vermal flocculonodular lobe heritability estimate (h2SNP [SE] = 35.4[1.9]%) (Supplementary Table 4). Between-lobe genetic correlation was moderate for most (between lobes mean rg ≈ 0.44) and all survived Bonferroni correction for the number of lobe-pairings tested (p < 0.0024 {0.05/21}), being strongest between the inferior posterior hemisphere and vermis (rg [95% CI] = 0.66 [0.60, 0.72], p = 1.4 × 10−103) and weakest between the flocculonodular hemisphere and vermis (rg [95% CI] = 0.19 [0.07, 0.30], p = 1.3 × 10−3) (Fig. 2; Supplementary Table 4).

Tile size and shade represent genetic correlation values (rg) between lobes calculated using LDSC regression analysis. Diagonal values of SNP-based heritability estimates calculated using GCTA-GREML. All correlations passed Bonferroni correction p < 0.0024{0.05/21}. v vermis.
Annotation and mapping of genome-wide significant regions from the meta-GWAS
We found 33 conditionally independent index SNPs associated with total cerebellar volume (Table 1; Supplementary Fig. 2). All index SNPs in each wave were present within the 33 meta-GWAS index SNPs, all 33 meta-GWAS index SNPs were at least nominally significant in each wave (p values ranging from 7 × 10−3 to 1.4 × 10−21 for wave 1 and from 5.3 × 10−3 to 9.5 × 10−16 for wave 2) and with all showing the same direction of effect across waves (Supplementary Table 5).
Functional annotation of the 33 independent GWAS signals (index SNPs and high LD partners r2 > 0.8) (Supplementary Tables 6, 7A, B) identified 5 containing non-synonymous SNPs leading to altered protein structure. Two of these were flagged as likely deleterious: the missense variants rs1800562 within HFE and rs13107325 within SLC39A8 transcripts. The other three non-synonymous SNPs were flagged as tolerated/benign, being within genes EIF2AK3, PPP2R4 (alias PTPA), and MYCL. A further synonymous annotated SNP located within PAPPA gene was within our strongest GWAS signal (rs72754248 Index SNP).
Six of the 33 GWAS signals mapped to genome-wide significant cis-eQTLs in GTEx-v7 cerebellum and cerebellar hemisphere tissue (index SNPs: rs7640903, rs55803832, rs546897, rs2572397, rs6984592 & rs3118634), associating with 14 gene transcripts: AF131216.5, AMT, CCDC71, GPX1, NCKIPSD, PPP2R4, PTK2, RP1-199J3.5, RP11-247A12.2, RP11-247A12.7, RP11-481A20.10, RP11-481A20.11, VCAN, and WDR6 (Supplementary Table 8A, B). When extending analyses to include all brain and whole-blood GTEx-v7 tissues, we found a further 3 GWAS signals mapping to whole-blood eQTLs (AP3B2 at rs62012045, CCDC53 at rs5742632 and REEP5 at rs3846716), moreover the marker rs2572397 revealed additional eQTLs for ALG1L11P (Basal Ganglia) and RP11-981G7.6 (Spinal Cord Cervical C1) (Supplementary Table 8C). SMR analysis found evidence for causal (or pleiotropic) relationships between GWAS and cerebellar gene expression associations for 3 GWAS signals namely at 5q14.2, 8p23.1 and 9q34.11 for 6 transcripts: PPP2R4, RP11-247A12.2, RP11-247A12.7, VCAN, FAM86B3P and FAM85B (Table 2). The strongest SMR association was observed for VCAN, showing a clear relationship between total cerebellar volume GWAS association signals and VCAN cerebellar gene expression (Supplementary Fig. 3).
Genetic correlations
We found high positive genetic correlation above Bonferroni-corrected significance threshold (p < 0.0014 {0.05/35}) between our total cerebellum meta-GWAS summary statistics and those of previously published regional cerebellar measures (left & right hemispheres; IIV–V, VI–VII and VIII–IX vermal regions [29]: rg [95% CI] = 0.91 [0.84, 0.97] and 0.91 [0.84, 0.98]; 0.44 [0.28, 0.60], 0.45 [0.32, 0.57] and 0.56 [0.46, 0.65], respectively; left and right cerebellar regions [16]: rg [95% CI] = 0.88 [0.84, 0.93] and 0.99 [0.85, 0.93]; 27 cerebellar lobule regions excluding Crus I vermis [16], rg mean [min, max] = 0.65 [0.41, 0.80]) (Supplementary Table 9A). Of the 33 GWAS signals we identified, 28 reached genome-wide significance in these previous works while 5 were novel to the literature (Index SNPs rs6546070, rs6812830, rs3846716, rs3118634 and rs529059). We also found positive genetic correlation (p < 0.005 {0.05/10}) between our total cerebellar volume measure and brainstem, pallidum and thalamus volumes, as well as a trend towards a negative correlation with cerebral cortical surface area but which fell short of our Bonferroni-corrected significant threshold (Table 3A). We found no genetic correlations (p < 0.0083 {0.05/6}) with any anthropomorphic measure, confirming the results not to simply be reflecting general body size measures (Supplementary Table 9B).
We ascertained the genetic correlation between cerebellar volume and liability to psychiatric diagnoses. None showed significant consistent genetic correlation across the genome with cerebellar volume, even at nominal significance (Table 3B). Stratified Q–Q plots, however, suggested a clear enrichment of schizophrenia signal and, to a less degree, bipolar and ASD associations within our total cerebellar volume variants (Supplementary Fig. 4). No apparent relationship was seen with major depressive disorder or ADHD. Conjunctional-FDR analysis revealed 8 of the 33 GWAS signals showing evidence for a pleiotropic relationship with a psychiatric phenotype (5 with schizophrenia, 2 with bipolar, 1 with ASD, and 1 with ADHD), with one GWAS signal (index SNP rs2572397) associating with more than one psychiatric condition: being with decreased cerebellar volume, decreased schizophrenia and increased ASD risk liability (Supplementary Table 10). In total, the majority (7/9) of pleiotropic associations were in opposing directions of effect to that of cerebellar volume. Finally, we report 2 of our 33 COJO GWAS signals (rs13135092 and rs1935951) as being previously associated with psychiatric traits of schizophrenia, bipolar, ASD, and across- and between-psychiatric disorder diagnoses (Supplementary Table 11A, B).
Discussion
In this study we examined UK-Biobank brain imaging and genotype data of 33,265 individuals to investigate common allele influences on cerebellar volume. We found total cerebellar volume is moderately heritable in our sample (h2SNP = 50.6%), identifying 33 independent genome-wide significant signals (index SNPs and SNPs in LD) associated with this trait. We identified 6 within protein-coding sections of the genome while another 5 associated with cerebellar gene expression regulation. We found evidence for pleiotropy of identified variants with schizophrenia, bipolar and ASD. We did not, however, find significant genetic correlations across the whole genome, suggesting a smaller subset of pleiotropic regions and/or opposing direction of effects across these regions.
Our main GWAS of total cerebellar volume identified 33 index SNPs, of which 28 had been reported genome-wide significant (p < 5e−8) in previous GWASes of sub-regional cerebellar volume measures [16, 29]. The 5 other index SNPs had previously shown subthreshold associations with some of those sub-regional volumes, while reaching GWAS significance level for our composite total volume measure. This overlap suggests an important genetic homogeneity across cerebellar structures, as previously indicated by cerebellar gene expression research [39], and which is further substantiated by our findings of moderate-to-high genetic correlation between our results and those of previous sub-regional cerebellar GWASes, and also across the 7 cerebellar lobe volumes in which we divided the cerebellum following demarcations of primary, horizontal and posterolateral fissures.
We conducted follow-up analyses of each GWAS signal to identify likely causal SNPs. One signal contained the synonymous SNP rs35565319 in the IGF binging protein protease PAPPA transcript, with previous reports of possible cerebellar-specific interactional effects [40], high placenta expression and association with adverse pregnancy outcomes [41, 42] and neuronal survival [43]. Five other GWAS signals contained non-synonymous SNPs altering protein structure. Of the two labelled as likely deleterious, one was the rs13107325 variant within the metal cation symporter SLC39A8 transcript, being previously associated with a wide-range of traits including inferior posterior and flocculonodular lobule [44], striatum and putamen volumes [44, 45], schizophrenia [33, 45], neurodevelopmental outcomes and intelligence test performance [46, 47] and numerous other factors [44, 48,49,50] (http://www.nealelab.is/uk-biobank/). The other was the rs1800562 variant (alias Cys282Tyr) within the homoeostatic iron regulator HFE transcript, with associations with reduced putamen volume and striatal T2star signal [44], and iron and mineral regulation [44, 51, 52]. The other three non-synonymous SNPs included those within translation initiation factor kinase (EIF2AK3), proto-oncogene transcription factor (MYCL) and protein phosphatase 2A activator (PPP2R4 alias PTPA) protein-coding regions. The novel PTPA finding agrees with previous work of the role of phosphatase 2A controlling cell growth and division, regulating dendritic spine morphology [53] and whose dysfunction is a known cause of spinocerebellar ataxia [54].
We also mapped 6 of GWAS signal regions with cis-eQTLs altering expression of 14 gene transcripts. Expanding the cis-eQTL analysis to additional brain regions and whole blood, we identified a further 3 GWAS signals mapping to 5 cis-eQTLs. SMR further investigated possible cerebellar expression mediation of SNP-trait associations for six gene transcripts at 3 GWAS signal regions, including again the PPP2R4/PTPA transcript. The strongest SMR association was with VCAN, encoding the extracellular matrix protein Versican, which plays crucial roles in nervous system development [55, 56]. The pseudogenes FAM86B3P and FAM85B were also identified from the SMR analysis, with FAM85B and the other non-coding gene cis-eQTLs for RP11-481A20.10 and RP11-481A20.11 in the same region having been indicated in mood instability and schizophrenia [57, 58]. While a higher confidence can be placed on SMR identified genes, its requirement for multiple cis-eQTL signals within a genomic region means genes with poorer coverage might be omitted, therefore both cis-eQTL-only and SMR identified genes should be considered for future follow-up work.
In total, therefore, from 732 unique gene transcripts overlapping with the extended-LD regions of our 33 index SNPs, functional annotation and cerebellar tissue gene expression mapping refined this to a list of 21 gene transcripts particularly warranting further interrogation (Supplementary Table 12).
Through inspection of GWAS Catalogue, we identified 2 GWAS signals (rs13135092 and rs1935951) previously associated with schizophrenia, and the former also with bipolar disorder, ASD and PGC cross-psychiatric disorder associations. Furthermore, using conjunctional-FDR analysis—leveraging genomic pleiotropy to indicate pleiotropic regions which might be below genome-wide significance for each psychiatric GWAS—we not only confirm psychiatric associations at these 2 GWAS signals with schizophrenia, but also identified 6 other GWAS signals with evidence for psychiatric pleiotropy (rs7530673 and rs1278519 with bipolar disorder; rs7640903 with ADHD; rs3118634 and rs62012045 with schizophrenia; rs2572397 with schizophrenia and ASD). Of these 8 GWAS signals, 6 followed the expected opposing direction of effect as would be predicted from case/control studies [8, 11], e.g. associating with increased psychiatric risk liability and decreased cerebellar size, whereas rs13135092 and rs2572397 showed the same direction of effect for both traits. Related to this, while we found evidence for enrichment of our cerebellar GWAS for schizophrenia, bipolar disorder and ASD using stratified Q–Q plots, in accordance with the majority of other structural brain phenotype GWASs [30, 32], we did not find a whole-genome level correlation when using LDSC, indicating regional heterogeneity of effect directions. These results highlight the benefit of using multiple methods to investigate genetic overlap between traits, as previously stressed [38, 59].
We found strong genetic correlation between our total cerebellar volume GWAS and those of the brainstem, pallidum and thalamus [32] but not other subcortical structures, cortical surface area or thickness [30,31,32]. These results agree with previous reports of a particular clustering of these three subcortical volumes [32, 60] and contrast to the significant phenotypic correlations amongst most subcortical volumes [32]. Importantly, the gene expression profile of cerebellar grey matter is quite distinct [39]. This shared common architecture, therefore, could be explained by cerebellar white matter connectivity between these regions. The major cerebellar input and output nuclei located within the brainstem and thalamus, respectively. Cerebellar-pallidal interactions are known to occur within the cortex, thalamus and via direct connections [61,62,63], with joint roles in sensorimotor regulation, learning and reward [61]. The common allele overlap found across these four brain structures, therefore, warrants further research into the neurobiological underpinnings of this potential network and its role in psychopathology, particularly given the association between cerebellothalamic and cerebellar-basal ganglia connectivity dysfunction in individuals with schizophrenia [64, 65].
There are several features of the study design to consider when interpreting the results presented. While the UK Biobank’s homogenous data collection and processing helps decrease methodological variation, the cohort does not represent the general UK population, deviating in important socioeconomic and demographic measures [66]. We further limited our analyses to participants with ancestry similar to a British and Irish reference (>96% sample), limiting the extrapolation of our results to other ancestries. Regarding the imaging data, while visual inspection of each segmentation was not possible due to the cohort size, we believe the UK Biobank’s semi-automated image artefact detection, our removal of outlier measures, confirmation of reliability of cerebellar measures in individuals with repeat scans, and correction for potential noise due to participants’ head motion and position within the scanner improve the validity of our cerebellar measures. The UK Biobank’s IDPs, however, are not optimised for the cerebellum, which can lead to poorer registration and segmentation of individuals lobules [67]. For this reason, as well as the high correlation between lobules and its conserved cytoarchitecture, our main analyses focused on total cerebellar volume. Lack of access to raw genotypes for the psychiatric phenotype GWASs prevented the use of methods such as bivariate GCTA-GREML which could have brought further insight into their genetic relationship with cerebellar volume.
In conclusion, we provide a genome-wide association study of the common genetic variation underlying human cerebellar volume. We find a moderate-to-high heritability for cerebellar volume, with relatively consistent heritability across lobes, and sharing common allele influences with brainstem, pallidal and thalamic volumes. We report enrichment for schizophrenia, bipolar and ASD signals, but not for major depression and ADHD. As a guide for future functional studies, we identify 33 independent index SNPs associated with cerebellar volume and 21 unique candidate genes for follow-up work: 6 protein-coding variants and 14 cerebellar tissue cis-eQTL associations, with 6 (4 common with the latter) showing potential causal relationships with gene expression. Overall, these results advance our knowledge on the common allele architecture of the cerebellum and pave the way to further research into the neurobiological basis of its anatomy, and associations with psychiatric conditions.
Data availability
Summary statistics from all GWAS analyses run are available from GWAS catalog (GCP000196).
Code availability
All genetic analyses used are open-source. Phenotype creation was performed by UK Biobank. Code for the creation of residual cerebellar values for genetic analyses (R), polygenic score (PLINK), or SNP lookup (R/GWAS catalog API) can be provided on request.
References
Buckner RL. The cerebellum and cognitive function: 25 years of insight from anatomy and neuroimaging. Neuron. 2013;80:807–15.
Seitzman BA, Gratton C, Marek S, Raut RV, Dosenbach NUF, Schlaggar BL, et al. A set of functionally-defined brain regions with improved representation of the subcortex and cerebellum. Neuroimage. 2020;206:116290.
Guell X, Schmahmann JD, Gabrieli JDE, Ghosh SS. Functional gradients of the cerebellum. eLife. 2018;7:e36652.
Buckner RL, Krienen FM, Castellanos A, Diaz JC, Yeo BT. The organization of the human cerebellum estimated by intrinsic functional connectivity. J Neurophysiol. 2011;106:2322–45.
King M, Hernandez-Castillo CR, Poldrack RA, Ivry RB, Diedrichsen J. Functional boundaries in the human cerebellum revealed by a multi-domain task battery. Nat Neurosci. 2019;22:1371–8.
Schmahmann JD. The role of the cerebellum in affect and psychosis. J Neurolinguist. 2000;13:189–214.
Van Overwalle F, Manto M, Cattaneo Z, Clausi S, Ferrari C, Gabrieli JDE, et al. Consensus paper: cerebellum and social cognition. Cerebellum. 2020;19:833–68.
Moberget T, Doan NT, Alnæs D, Kaufmann T, Córdova-Palomera A, Lagerberg TV, et al. Cerebellar volume and cerebellocerebral structural covariance in schizophrenia: A multisite mega-analysis of 983 patients and 1349 healthy controls. Mol Psychiatry. 2018;23:1512–20.
Wyciszkiewicz A, Pawlak MA, Krawiec K. Cerebellar volume in children with attention-deficit hyperactivity disorder (ADHD): replication study. J Child Neurol. 2017;32:215–21.
Lupo M, Siciliano L, Leggio M. From cerebellar alterations to mood disorders: a systematic review. Neurosci Biobehav Rev. 2019;103:21–8.
Romer AL, Knodt AR, Houts R, Brigidi BD, Moffitt TE, Caspi A, et al. Structural alterations within cerebellar circuitry are associated with general liability for common mental disorders. Mol Psychiatry. 2018;23:1084–90.
Moberget T, Alnæs D, Kaufmann T, Doan NT, Córdova-Palomera A, Norbom LB, et al. Cerebellar gray matter volume is associated with cognitive function and psychopathology in adolescence. Biol Psychiatry. 2019;86:65–75.
Zhang W, Sweeney JA, Yao L, Li S, Zeng J, Xu M, et al. Brain structural correlates of familial risk for mental illness: a meta-analysis of voxel-based morphometry studies in relatives of patients with psychotic or mood disorders. Neuropsychopharmacology. 2020;45:1369–79.
Hammerschlag AR, De Leeuw CA, Middeldorp CM, Polderman TJC. Synaptic and brain-expressed gene sets relate to the shared genetic risk across five psychiatric disorders. Psychol Med. 2020;50:1695–705.
Blokland GAM, De Zubicaray GI, McMahon KL, Wright MJ. Genetic and environmental influences on neuroimaging phenotypes: a meta-analytical perspective on twin imaging studies. Twin Res Hum Genet. 2012;15:351–71.
Smith SM, Douaud G, Chen W, Hanayik T, Alfaro-Almagro F, Sharp K, et al. An expanded set of genome-wide association studies of brain imaging phenotypes in UK Biobank. Nat Neurosci. 2021;24:737–45.
Alfaro-Almagro F, Jenkinson M, Bangerter NK, Andersson JLR, Griffanti L, Douaud G, et al. Image processing and Quality Control for the first 10,000 brain imaging datasets from UK Biobank. Neuroimage. 2018;166:400–24.
Diedrichsen J, Balsters JH, Flavell J, Cussans E, Ramnani N. A probabilistic MR atlas of the human cerebellum. Neuroimage. 2009;46:39–46.
Pezoulas VC, Zervakis M, Michelogiannis S, Klados MA. Resting-state functional connectivity and network analysis of cerebellum with respect to crystallized IQ and gender. Front Hum Neurosci. 2017;11:189.
Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7.
Yang J, Lee SH, Goddard ME, Visscher PM. GCTA: A tool for genome-wide complex trait analysis. Am J Hum Genet. 2011;88:76–82.
Yang J, Benyamin B, McEvoy BP, Gordon S, Henders AK, Nyholt DR, et al. Common SNPs explain a large proportion of the heritability for human height. Nat Genet. 2010;42:565–9.
Yang J, Ferreira T, Morris AP, Medland SE, Madden PAF, Heath AC, et al. Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat Genet. 2012;44:369–75.
Pruim RJ, Welch RP, Sanna S, Teslovich TM, Chines PS, Gliedt TP, et al. LocusZoom: Regional visualization of genome-wide association scan results. Bioinformatics. 2011;26:2336–7
Bulik-Sullivan B, Finucane HK, Anttila V, Gusev A, Day FR, Loh PR, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet. 2015;47:1236–41.
Willer CJ, Li Y, Abecasis GR. METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics. 2010;26:2190–1.
Zhu Z, Zhang F, Hu H, Bakshi A, Robinson MR, Powell JE, et al. Integration of summary data from GWAS and eQTL studies predicts complex trait gene targets. Nat Genet. 2016;48:481–7.
Pavlides JMW, Zhu Z, Gratten J, McRae AF, Wray NR, Yang J. Predicting gene targets from integrative analyses of summary data from GWAS and eQTL studies for 28 human complex traits. Genome Med. 2016;8:84.
Zhao B, Luo T, Li T, Li Y, Zhang J, Shan Y, et al. Genome-wide association analysis of 19,629 individuals identifies variants influencing regional brain volumes and refines their genetic co-architecture with cognitive and mental health traits. Nat Genet. 2019;51:1637–44.
Grasby KL, Jahanshad N, Painter JN, Colodro-Conde L, Bralten J, Hibar DP, et al. The genetic architecture of the human cerebral cortex. Science. 2020;367:eaay6690.
Hibar DP, Adams HHH, Jahanshad N, Chauhan G, Stein JL, Hofer E, et al. Novel genetic loci associated with hippocampal volume. Nat Commun. 2017;8:1–12.
Satizabal CL, Adams HHH, Hibar DP, White CC, Knol MJ, Stein JL, et al. Genetic architecture of subcortical brain structures in 38,851 individuals. Nat Genet. 2019;51:1624–36.
Pardiñas AF, Holmans P, Pocklington AJ, Escott-Price V, Ripke S, Carrera N, et al. Common schizophrenia alleles are enriched in mutation-intolerant genes and in regions under strong background selection. Nat Genet. 2018;50:381–9.
Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81.
Grove J, Ripke S, Als TD, Mattheisen M, Walters RK, Won H, et al. Identification of common genetic risk variants for autism spectrum disorder. Nat Genet. 2019;51:431–44.
Demontis D, Walters RK, Martin J, Mattheisen M, Als TD, Agerbo E, et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat Genet. 2019;51:63–75.
Stahl EA, Breen G, Forstner AJ, McQuillin A, Ripke S, Trubetskoy V, et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat Genet. 2019;51:793–803.
Andreassen OA, Djurovic S, Thompson WK, Schork AJ, Kendler KS, O’Donovan MC, et al. Improved detection of common variants associated with schizophrenia by leveraging pleiotropy with cardiovascular-disease risk factors. Am J Hum Genet. 2013;92:197–209.
Hawrylycz M, Miller JA, Menon V, Feng D, Dolbeare T, Guillozet-Bongaarts AL, et al. Canonical genetic signatures of the adult human brain. Nat Neurosci. 2015;18:1832–44.
Swindell WR, Masternak MM, Bartke A. In vivo analysis of gene expression in long-lived mice lacking the pregnancy-associated plasma protein A (PappA) gene. Exp Gerontol. 2010;45:366–74.
Morris RK, Bilagi A, Devani P, Kilby MD. Association of serum PAPP-A levels in first trimester with small for gestational age and adverse pregnancy outcomes: systematic review and meta-analysis. Prenat Diagn. 2017;37:253–65.
DiPrisco B, Kumar A, Kalra B, Savjani GV, Michael Z, Farr O, et al. Placental proteases PAPP-A and PAPP-A2, the binding proteins they cleave (IGFBP-4 and -5), and IGF-I and IGF-II: Levels in umbilical cord blood and associations with birth weight and length. Metabolism. 2019;100:153959.
Alassaf M, Daykin EC, Mathiaparanam J, Wolman MA. Pregnancy-associated plasma protein-aa supports hair cell survival by regulating mitochondrial function. Elife. 2019;8:e47061.
Elliott LT, Sharp K, Alfaro-Almagro F, Shi S, Miller KL, Douaud G, et al. Genome-wide association studies of brain imaging phenotypes in UK Biobank. Nature. 2018;562:210–6.
Luo Q, Chen Q, Wang W, Desrivières S, Quinlan EB, Jia T, et al. Association of a schizophrenia-risk nonsynonymous variant with putamen volume in adolescents: a voxelwise and genome-wide association study. JAMA Psychiatry. 2019;76:435–45.
Wahlberg KE, Guazzetti S, Pineda D, Larsson SC, Fedrighi C, Cagna G, et al. Polymorphisms in Manganese Transporters SLC30A10 and SLC39A8 Are Associated With Children’s Neurodevelopment by Influencing Manganese Homeostasis. Front Genet. 2018;9:664.
Hill WD, Marioni RE, Maghzian O, Ritchie SJ, Hagenaars SP, McIntosh AM, et al. A combined analysis of genetically correlated traits identifies 187 loci and a role for neurogenesis and myelination in intelligence. Mol Psychiatry. 2019;24:169–81.
Cabrera CP, Ng FL, Nicholls HL, Gupta A, Barnes MR, Munroe PB, et al. Over 1000 genetic loci influencing blood pressure with multiple systems and tissues implicated. Hum Mol Genet. 2019;28:151–61.
Mealer RG, Jenkins BG, Chen CY, Daly MJ, Ge T, Lehoux S, et al. The schizophrenia risk locus in SLC39A8 alters brain metal transport and plasma glycosylation. Sci Rep. 2020;10:1–15.
Costas J. The highly pleiotropic gene SLC39A8 as an opportunity to gain insight into the molecular pathogenesis of schizophrenia. Am J Med Genet Part B Neuropsychiatr Genet. 2018;177:274–83.
Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408.
Sørensen E, Rigas AS, Didriksen M, Burgdorf KS, Thørner LW, Pedersen OB, et al. Genetic factors influencing hemoglobin levels in 15,567 blood donors: results from the Danish Blood Donor Study. Transfusion. 2019;59:226–31.
Wang J, Lou SSen, Wang T, Wu RJ, Li G, Zhao M, et al. UBE3A-mediated PTPA ubiquitination and degradation regulate PP2A activity and dendritic spine morphology. Proc Natl Acad Sci USA. 2019;116:12500–5.
Srivastava AK, Takkar A, Garg A, Faruq M. Clinical behaviour of spinocerebellar ataxia type 12 and intermediate length abnormal CAG repeats in PPP2R2B. Brain. 2017;140:27–36.
Theocharis AD. Versican in health and disease. Connect Tissue Res. 2008;49:230–4.
Rutten-Jacobs LCA, Tozer DJ, Duering M, Malik R, Dichgans M, Markus HS, et al. Genetic study of white matter integrity in UK Biobank (N=8448) and the overlap with stroke, depression, and dementia. Stroke. 2018;49:1340–7.
Ward J, Tunbridge EM, Sandor C, Lyall LM, Ferguson A, Strawbridge RJ, et al. The genomic basis of mood instability: identification of 46 loci in 363,705 UK Biobank participants, genetic correlation with psychiatric disorders, and association with gene expression and function. Mol Psychiatry. 2019;25:3091–9.
Ripke S, Neale BM, Corvin A, Walters JTR, Farh KH, Holmans PA, et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–7.
Smeland OB, Frei O, Shadrin A, O’Connell K, Fan CC, Bahrami S, et al. Discovery of shared genomic loci using the conditional false discovery rate approach. Hum Genet. 2020;139:85–94.
Eyler LT, Prom-Wormley E, Fennema-Notestine C, Panizzon MS, Neale MC, Jernigan TL, et al. Genetic patterns of correlation among subcortical volumes in humans: Results from a magnetic resonance imaging twin study. Hum Brain Mapp. 2011;32:641–53.
Bostan AC, Strick PL. The basal ganglia and the cerebellum: Nodes in an integrated network. Nat Rev Neurosci. 2018;19:338–50.
Hintzen A, Pelzer EA, Tittgemeyer M. Thalamic interactions of cerebellum and basal ganglia. Brain Struct Funct. 2018;223:569–87.
Milardi D, Arrigo A, Anastasi G, Cacciola A, Marino S, Mormina E, et al. Extensive direct subcortical cerebellum-basal ganglia connections in human brain as revealed by constrained spherical deconvolution tractography. Front Neuroanat. 2016;10:29.
Tarcijonas G, Foran W, Haas GL, Luna B, Sarpal DK. Intrinsic connectivity of the globus pallidus: an uncharted marker of functional prognosis in people with first-episode schizophrenia. Schizophr Bull. 2020;46:184–92.
Andreasen NC, Pierson R. The role of the cerebellum in schizophrenia. Biol Psychiatry. 2008;64:81–8.
Fry A, Littlejohns TJ, Sudlow C, Doherty N, Adamska L, Sprosen T, et al. Comparison of sociodemographic and health-related characteristics of UK biobank participants with those of the general population. Am J Epidemiol. 2017;186:1026–34.
Diedrichsen J. A spatially unbiased atlas template of the human cerebellum. Neuroimage. 2006;33:127–38.
Acknowledgements
This research was conducted using the UK-Biobank resource under project ref. 17044 and was supported by the Medical Research Council Programme grant ref. G08005009. TCh was supported by a Wellcome Trust Ph.D. scholarship (ref. 203770/Z/16/Z)
Author information
Authors and Affiliations
Contributions
TCh, XC and RJLA devised the project. TCh, VEP, SL, EB, XC and RJLA participated in imaging and genetic-data processing and outputs generation. TCh, RJLA, VEP, SL and EB performed the statistical analyses. TCh, KDS, JTRW, XC and RJLA interpreted the results and wrote the paper. All authors contributed on the discussion of results and revised and approved the final paper.
Corresponding author
Ethics declarations
Competing interests
JTRW has received grant funding from Takeda Pharmaceutical Company for research unrelated to this work. All other authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons license, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons license and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Chambers, T., Escott-Price, V., Legge, S. et al. Genetic common variants associated with cerebellar volume and their overlap with mental disorders: a study on 33,265 individuals from the UK-Biobank. Mol Psychiatry 27, 2282–2290 (2022). https://doi.org/10.1038/s41380-022-01443-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41380-022-01443-8
- Springer Nature Limited
This article is cited by
-
Insights into the genetic architecture of cerebellar lobules derived from the UK Biobank
Scientific Reports (2024)
-
Genetic architecture of the structural connectome
Nature Communications (2024)
-
Structural deviations of the posterior fossa and the cerebellum and their cognitive links in a neurodevelopmental deletion syndrome
Molecular Psychiatry (2024)
-
An adaptive and robust method for multi-trait analysis of genome-wide association studies using summary statistics
European Journal of Human Genetics (2024)
-
Genome-wide association study of cerebellar volume provides insights into heritable mechanisms underlying brain development and mental health
Communications Biology (2022)